
Abstract
Impaired nerve conduction, axonal degeneration, and synaptic alterations contribute to neurological disabilities in inflammatory demyelinating diseases. Cerebellar dysfunction is associated with demyelinating disorders, but the alterations of axon terminals in cerebellar gray matter during chronic demyelination are still unclear. We analyzed the morphological and ultrastructural changes of climbing fiber terminals in a mouse model of hereditary chronic demyelination. Three-dimensional ultrastructural analyses using serial block-face scanning electron microscopy and immunostaining for synaptic markers were performed in a demyelination mouse model caused by extra copies of myelin gene (PLP4e). At 1 month old, many myelinated axons were observed in PLP4e and wild-type mice, but demyelinated axons and axons with abnormally thin myelin were prominent in PLP4e mice at 5 months old. The density of climbing fiber terminals was significantly reduced in PLP4e mice at 5 months old. Reconstruction of climbing fiber terminals revealed that PLP4e climbing fibers had increased varicosity volume and enlarged mitochondria in the varicosities at 5-month-old mice. These results suggest that chronic demyelination is associated with alterations and loss of climbing fiber terminals in the cerebellar cortex, and that synaptic changes may contribute to cerebellar phenotypes observed in hereditary demyelinating disorders.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Many axons in the vertebrate nervous system are ensheathed by myelin, which is essential for fast saltatory conduction and for the integrity and survival of axons. Demyelination, which is the pathological loss of myelin, impairs nerve conduction and axonal survival and leads to neurological defects in myelin diseases. The alteration of axon terminals in gray matter has also been implicated in the pathophysiology of demyelinating diseases and animal models. For example, demyelination in the hippocampus is associated with synaptic loss and cognitive failure in multiple sclerosis, an inflammatory demyelinating disease [1]. Animal models, such as cuprizone-induced demyelination and experimental autoimmune encephalomyelitis, are associated with synaptic loss in the hippocampus and cerebellar cortex [2]. However, it is still unclear if synaptic changes are commonly associated with demyelination and involved in the pathophysiology of chronic demyelination in hereditary myelin disorders.
The cerebellum is a major brain region affected in demyelinating diseases, and cerebellar dysfunctions are commonly observed in myelin disorders that accompany demyelination in cerebellar white matter [3]. Cerebellar functions are maintained by Purkinje cells (PC), which receive excitatory inputs from climbing fibers (CF) [4,5,6]. CF, which originate from the inferior olivary nucleus, enter cerebellar white matter via the inferior cerebellar peduncle, pass through the granular layer, and directly form hundreds of excitatory synapses on targeted PC dendrites by postnatal day 20. Damage to the cerebellum leads to gait ataxia, poor hand coordination, and tremors, and contributes to disability [2, 3, 7], but it is unclear how CF terminals and related axonal organelles are affected under conditions of chronic demyelination in cerebellar gray matter.
In this study, we investigated alterations in the density and morphology of CF terminals and CF organelles, including mitochondria, in the cerebellum, using a chronic demyelination mouse model caused by extra-copies of the proteolipid protein (Plp1) gene, and found that structural alterations and loss of CF terminals are associated with chronic demyelination.
Materials and methods
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of the National Institutes for Physiological Sciences and performed in accordance with institute guidelines. Hemizygous transgenic (PLP4e/−) mice (PLP4e mice), which have two extra-copies of the Plp1 gene [8], were maintained by crossing with BDF1 mice. Transgenic (PLP4e/−) mice were produced by introducing a cosmid, which carried all seven exons of the mouse Plp gene, 20 kb of the 5′ flanking sequence and 4 kb of the 3′ flanking sequence, into the wild-type mouse germline by the microinjection method as described previously [8]. Male BDF1 mice, which are offspring of female C57BL/6 and male DBA/2, were purchased from SLC (Shizuoka, Japan).
Serial block-face scanning electron microscopy (SBF-SEM)
PLP4e mice and WT littermates at 1 and 5 months old (mo) were anesthetized and transcardially perfused with phosphate buffered saline (PBS, pH 7.4) followed by 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). Cerebellar vermis areas were dissected, cut into small pieces (< 1 mm each) with razor blades in ice-cold fixative solution, and immersed in the same fixative at 4 °C overnight. All tissues were post-fixed, stained en bloc, and embedded in carbon-based conductive resin, as described previously [9, 10]. Prepared samples were observed with a field emission scanning electron microscope (Merlin or Sigma from Carl Zeiss, Germany) equipped with 3View, a system with a built-in ultramicrotome and a back-scattered electron detector (Gatan, CA, USA). The acquired serial images were processed by ImageJ with Fiji plugins (http://fiji.sc/wiki/index.php/Fiji), and segmentation, three-dimensional reconstruction, and image analyses were performed in TrakEM2 [11] and Amira (FEI Visualization Science Group, Hillsboro, OR, USA).
Serial electron microscope images were obtained at the resolution of 5–7 nm/pixel, a dwelling time of 1 µs, and the thickness of 70–80 nm. The G-ratio of each nerve fiber was obtained by dividing the axonal perimeter with the outer perimeter including the myelin sheath. As described previously [6, 12, 13], CFs were identified in the granular layer as fibers that run toward the molecular layer, contain many cytoskeletal elements including microtubules, and are ensheathed by myelin. In the molecular layer, CF varicosities were defined as axonal segments with increased diameter and the presence of synaptic vesicles and postsynaptic density on PC dendrites. Varicosity volume and mitochondria were measured by ImageJ with TrakEM2 plugins [11].
Immunohistochemistry
PLP4e and WT at 1 and 5 mo were anesthetized and transcardially perfused with PBS (pH 7.4) followed by 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Cerebella were dissected, post-fixed overnight in the same fixative at 4 °C and then incubated in PBS containing 30% sucrose for 48 h. Cerebella were embedded in O.C.T. compound (Sakura Finetek, Torrance, CA, USA) and floating sections (40-µm thick) were prepared by a Leica cryostat (CM 3050S series, Nussloch, Germany). Floating sections were washed three times with PBS, for 5 min each, microwaved for 5 min without boiling in 10 mM citrate buffer (pH 6.0), and permeabilized in PBS containing 3% bovine serum albumin (BSA, Wako, Japan) and 1% TritonX-100 (Wako, Japan) for 2 h at room temperature. After a brief wash with PBS, sections were incubated with primary antibodies in PBS containing 5% BSA and 0.1% TritonX-100 at 4 °C for 2 nights. The sections were washed three times with PBS for 5 min each, and incubated with secondary antibodies in PBS containing 5% BSA at 4 °C overnight. Sections were placed at room temperature for 3 h, washed three times with PBS for 5 min each, and embedded for image acquisition. The primary antibodies were mouse anti-calbindin-D28k (Swant, Switzerland, 1:4000), guinea-pig anti-vGluT2 (Frontier Institute, Japan, vGluT2-gp-Af810, 1:250) and rabbit anti-vGluT2 (Hokkaido University, Japan, rabbit anti-vesicular glutamate transporter 2, 1:900) [14]. Secondary antibodies were anti-mouse, guinea-pig and rabbit antibodies conjugated with Alexa 488, Alexa 568, or Cy5 (Abcam, Japan). Confocal images were obtained with Nikon A1R confocal laser scanning microscope equipped with NIS-Element software (Nikon, America), processed by ImageJ with Fiji plugins (http://fiji.sc/wiki/index.php/Fiji), and Adobe Photoshop CS6. Stacked images (60 or 100× objective lens with N.A. 1.40) were obtained at 0.5-µm intervals. Stacked images were processed by ImageJ with Fiji plugins. The density of CF presynaptic terminals was acquired by dividing the number of vGluT2-positive profiles with the area size selected along PC dendrites or the length of observed PC dendrites [15].
Statistics
Quantification was performed using data obtained from at least three mice in each group. All measurements were summarized with Microsoft Excel 2013. Statistical analyses were performed using Prism (GraphPad software, La Jolla, CA, America). For comparisons, we used a two-tailed Student’s t-test for normally distributed data and Mann–Whitney U test for others. Multiple comparisons were made with Bonferroni corrections. Graphs of the normally distributed data show mean and SD, and graphs of the other data are box (median and interquartile range) and whisker plots (min to max) or dot plots with a bar (median).
Results
Chronic demyelination in cerebellar white matter of aged PLP4e mice
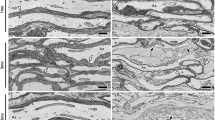
It has been reported that myelin degeneration and chronic demyelination begin around 3 mo in PLP4e mice [8, 16]. To investigate myelination and demyelination in cerebellar white matter of PLP4e mice and WT littermates at 1 and 5 mo, we used ultrastructural analyses of serial electron microscope images obtained with SBF-SEM. CF pass through cerebellar white matter, penetrate into the granular layer, enter the molecular layer, and make synapses along PC dendrites (Fig. 1a) [6, 12, 13]. The electron microscope images also showed myelinated segments of CF in the granular layer and PC layer (Fig. 1b1, b2), and the unmyelinated segment of CF in the molecular layer (Fig. 1b3). Myelinated segments of CF were not observed in the molecular layer. In the cerebellar white matter, the myelin sheath (asterisk in Fig. 1c–e, blue color in insets), formed electron dense layers around axons, and wrapped most axons (yellow color in insets in Fig. 1c–f) in WT and PLP4e mice at 1 mo (Fig. 1c, d) and in WT mice at 5 mo (Fig. 1e). In contrast, we observed many demyelinated axons in the white matter of PLP4e cerebellum at 5 mo (Fig. 1f). The G-ratio, calculated by dividing the axonal perimeter with the outer perimeter including the myelin sheath, in PLP4e mice was higher than that in WT mice at 5 mo (Fig. 1g), which suggests that the myelin sheath was thinner in PLP4e axons compared with age-matched WT axons. Axons in SBF-SEM images are identified by the presence of abundant and evenly distributed cytoskeletal elements throughout the axoplasm and also organelles including endoplasmic reticulum enriched beneath the axolemma, and many axons in PLP4e mice were without myelin ensheathment at 5 mo (Fig. 1h). These results revealed myelination and chronic demyelination in cerebellar white matter of PLP4e mice.

Chronic demyelination is induced in cerebellar white matter of aged PLP4e mice. A scheme (a) describes the inputs onto Purkinje cells (PC, black) from climbing fibers (CF, red) and mossy fibers (m, green) through granule cells (light blue). ML molecular layer, PCL Purkinje cell layer, GL granular layer, WM white matter, Pa PC axon. An electron microscope image of an adult wild-type (WT) mouse shows the mouse cerebellar cortex (b1), and the red arrowhead indicates CF in GL. The image shows a myelinated segment of CF in the PCL (b2, red arrowhead) and another image shows a CF in ML (b3, red arrowhead). Electron microscope images of cerebellar white matter show myelinated axons of WT (c) and PLP4e (d) mice at 1 month old (1 mo) and a WT mouse (e) at 5 months old (5 mo), and demyelinated axons of a PLP4e mouse at 5 mo (f). Single axons (ax or yellow color in insets) with myelin sheath (asterisks or blue color in insets) and axonal mitochondria (arrowheads or red color in insets) are marked in the panels or colored in the insets (c–f, insets). Bars: 25 µm (b1), 4 µm (b2), or 2 µm (b3, c–f). The G-ratio in PLP4e mice at 5 mo are higher than those in PLP4e mice at 1 mo and WT mice (g). N = 125 axons (g). ****p < 0.0001 by a Mann–Whitney U test. The percentage of axons without myelin in PLP4e mice at 5 mo are higher than those in PLP4e mice at 1 mo and WT mice (h). N = 20 images (in each WT group, h) or N = 40 images (in each PLP4e group, h). ****p < 0.0001, n.s. no significance by a Mann–Whitney U test
Decrease of CF synaptic terminals in the cerebellar molecular layer of aged PLP4e mice
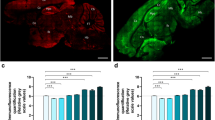
To evaluate the effect of chronic demyelination on the density of CF presynaptic terminals, we performed immunohistochemical analyses of cerebellar gray matter in WT and PLP4e mice at 1 and 5 mo. Calbindin-D28k, a member of cytosolic calcium (Ca2+) buffer proteins including parvalbumins, calbindin-D9k, calbindin-D28k and calretinin, is a specific marker of PC in cerebellum and used in order to visualize distribution and length of PC dendrites. Light microscope images of calbindin-D28k revealed no obvious difference in the morphology of PC soma and dendrites in PLP4e and WT mice. To investigate the density of presynaptic terminals, we examined the immunolocalization of a marker of CF-PC synapses, vesicle glutamate transporter 2 (vGluT2, Fig. 2a–l). The density of CF terminals was significantly decreased in PLP4e at 5 mo compared with that in age-matched WT (Fig. 2m, n). By contrast, the density of vGluT2-positive synaptic puncta was similar at 1 mo in WT and PLP4e mice (Fig. 2m, n). These results revealed a decreased number of vGluT2-positive CF terminals under chronic demyelination.

A decrease of climbing fiber (CF) synaptic terminals in the molecular layer of the cerebellum in aged PLP4e mice. Double immunofluorescence (a–l) for vGluT2 (green) and calbindin-D28k (red) in wild-type (WT, a–c, g–i) and PLP4e mice (d–f, j–l) at 1 month old (1 mo, a–f) and 5 months old (5 mo, g–l). The density of CF terminals was significantly decreased in 5 mo PLP4e mice when compared with that in age-matched WT mice (m, n). N = 23, 20, 23, and 20 dendritic segments of Purkinje cell (PC) (m) or 24, 27, 42, and 40 areas on individual PC dendrites (n) for 1 mo WT, 1 mo PLP4e, 5 mo WT, and 5 mo PLP4e mice, respectively. ***p < 0.001, ****p < 0.0001, n.s. no significance by a Student’s t test. Bars: 50 µm
Enlargement of CF presynaptic terminals in cerebellar gray matter of aged PLP4e mice
Structural alterations of synaptic terminals are associated with elimination of synapses [4]. To further investigate structural changes of CF-PC synapses and organelles in cerebellar gray matter, we used SBF-SEM for 3D ultrastructural analyses. In the serial electron microscope images, the soma and dendrites of PC were readily identified. PC soma are large, and locate between the molecular and granular layers (Fig. 1b1). PC dendrites are characterized by thick and frequently branching processes densely packed with organelles such as mitochondria and endoplasmic reticulum (Fig. 3a). CF form varicosities with synapses on the PC dendrites which are characterized by accumulation of synaptic vesicles, presynaptic and postsynaptic membrane attachment and presynaptic mitochondria (Figs. 3b1, b2). 3D reconstruction of serial images revealed that the number of CF varicosities per unit length of PC dendrite in PLP4e mice at 5 mo was less than that in WT littermates (Fig. 3c, d). The volume of each varicosity in PLP4e mice at 5 mo also significantly increased compared with that in WT littermates (Fig. 3e–g). The volume of individual mitochondria in the CF presynaptic terminal of PLP4e mice was larger than that of WT mice at 5 mo (Fig. 3e, f, h) and the percentage volume occupied by mitochondria was similar in PLP4e and WT mice at 5 mo (Fig. 3i). These results revealed that the loss of CF synaptic terminals in chronic demyelination accompanies synaptic alterations involving enlargement of synaptic varicosities and mitochondria in the varicosities.

Enlargement of climbing fiber (CF) presynaptic terminals in the cerebellar gray matter of aged PLP4e mice. A low magnification electron microscope image (a) and a thematic sketch shows the mouse cerebellar cortex (inset in a). The soma of a Purkinje cell (PC) is labeled. ML molecular layer, PCL Purkinje cell layer, GL granular layer. High magnification images (b1, b2) show a CF varicosity (yellow) on a PC dendrite with two small spines (light blue), presynaptic and postsynaptic membrane attachment (red), and mitochondria (arrowheads, purple). Three-dimensional reconstruction images of PC dendrites (light blue) with the same length and associated CF (yellow) in wild-type (WT, c, e) and PLP4e (d, f) mice at 5 months old (5 mo) show different numbers and sizes of CF varicosities (yellow swellings) containing mitochondria (purple in the varicosities). Three-dimensional image of a PLP4e mouse at 5 mo shows a CF varicosity (f) that appears larger than others (e). The volume of CF varicosities (g) and mitochondrial volume in the varicosities (h) of 5 mo PLP4e mice were larger than those of age-matched WT mice, but were similar in WT and PLP4e mice at 1 month old (1 mo). The percent mitochondrial volume in the bouton (Mito Vol/Varicosity Vol, i) was similar in age-matched WT and PLP4e mice. N = 43, 54, 38, and 30 boutons (g, i) or 111, 199, 113, and 161 mitochondria (h) for 1 mo WT, 1 mo PLP4e, 5 mo WT, and 5 mo PLP4e mice, respectively. *p < 0.05, **p < 0.01, ****p < 0.0001, n.s. no significance by a Mann–Whitney U test. Bars: 20 µm (a), 1 µm (b1, b2), 5 µm (c, d), 2 µm (e, f)
Discussion
We examined the alterations of CF terminals in the cerebellar cortex of chronically demyelinated PLP4e mice. In aged PLP4e mice, demyelinated axons were prominent in the cerebellar white matter, and the number of CF terminals was significantly reduced in the cerebellar cortex. The 3D ultrastructural analyses of the molecular layer revealed that the volume of CF varicosities and mitochondrial volume in the varicosities were both increased in the cerebellar cortex under chronic demyelination. These results revealed that the loss of synaptic terminals, coupled with hypertrophy in the remaining varicosities and enlargement of presynaptic mitochondria, were associated with chronic demyelination.
Genetic mutations of Plp1 cause various symptoms ranging from the severe Pelizaeus–Merzbacher disease (PMD) to the mild spastic paraplegia 2 (SPG2) in humans, and increased copies of PLP are a frequent cause of PMD [17,18,19,20]. Since the severity of the disease depends on the dosage of PLP1 protein, PLP4e mice used here with mild overexpression of PLP show normal myelination followed by abnormal progressive demyelination [8, 21, 22]. The clinical symptoms of PMD include impairment of movement, ataxia, intention tremors, and altered perception, which are symptoms partly shared with other demyelinating diseases such as multiple sclerosis [20, 23,24,25,26,27]. Because axons are relatively remained in PMD brains, impaired myelination and nerve conduction are involved in the neurological defects of PMD [28]. However, our results indicated that synaptic alterations are also associated with the pathophysiology of PMD, as observed in other demyelinating diseases and their models [1, 2, 29, 30]. The loss of synapses may be commonly observed in different types of demyelinating diseases and their models, although future studies are necessary to address if PMD and/or SPG2 caused by different mutations or expression of PLP also involve synaptic alterations.
Synaptic elimination during development has been associated with the complement system and phagocytic removal of synaptic components [31,32,33] and inflammatory demyelination and neurodegeneration [31]. In MS brains and a pharmacological animal model, a decrease in synapses was associated with decreased expression of neuronal molecules, which was mediated by specific miRNA [1, 29]. Developmentally, the selection and maintenance of CF terminals are mediated by C1q-like family member, C1ql1, on CF terminals and brain-specific angiogenesis inhibitor 3 (Bai3) expressed on PC [34]. Because the decreased expression of C1ql1 in adulthood results in a decreased number of CF-PC synapses, it is possible that these molecules are involved in a decrease of CF terminals [34]. Further studies are needed to determine the mechanisms that contribute to the progression of chronic demyelination diseases.
The synaptic alterations in the PMD model included volume changes of synaptic varicosities and mitochondria. The presynaptic boutons or varicosities of CF are dynamic structures that can be affected by synaptic transmission [35, 36]. The volume of varicosities is associated with presynaptic terminal activity, which is modulated by conduction impairment and ectopic firing of demyelinated axons [37, 38]. The loading of neurotransmitters into vesicles and the fusion of vesicle membrane with the plasma membrane are energy-dependent processes [39, 40]. Neurotransmission induces Ca2+ influx [41, 42], and mitochondria have a central role in the maintenance of presynaptic homeostasis by supplying ATP and buffering Ca2+ during synaptic activity [43,44,45]. Therefore, the alterations of CF terminal structures, including mitochondrial morphology, may represent enhanced synaptic transmission of the remaining CF boutons and mitochondrial support for energy metabolism and Ca2+ regulation, but future studies are needed to evaluate the physiological roles of varicosity enlargement and increase in mitochondrial volume in CF terminals.
References
Dutta R, Ph D, Chomyk AM, Chang A, Ribaudo MV, Deckard SA, Doud MK, Edberg DD, Bai B, Li M, Baranzini SE, Fox RJ, Staugaitis SM, Macklin WB, Trapp BD (2013) Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann Neurol 73:637–645. https://doi.org/10.1002/ana.23860.Hippocampal
Mandolesi G, Musella A, Gentile A, Grasselli G, Haji N, Sepman H, Fresegna D, Bullitta S, De Vito F, Musumeci G, Di Sanza C, Strata P, Centonze D (2013) Interleukin-1 alters glutamate transmission at Purkinje cell synapses in a mouse model of multiple sclerosis. J Neurosci 33:12105–12121. https://doi.org/10.1523/JNEUROSCI.5369-12.2013
Mandolesi G, Grasselli G, Musella A, Gentile A, Musumeci G, Sepman H, Haji N, Fresegna D, Bernardi G, Centonze D (2012) GABAergic signaling and connectivity on Purkinje cells are impaired in experimental autoimmune encephalomyelitis. Neurobiol Dis 46:414–424. https://doi.org/10.1016/j.nbd.2012.02.005
Watanabe M, Kano M (2011) Climbing fiber synapse elimination in cerebellar Purkinje cells. Eur J Neurosci 34:1697–1710. https://doi.org/10.1111/j.1460-9568.2011.07894.x
Hashimoto K, Ichikawa R, Kitamura K, Watanabe M, Kano M (2009) Translocation of a “winner” climbing fiber to the purkinje cell dendrite and subsequent elimination of “losers” from the soma in developing cerebellum. Neuron 63:106–118. https://doi.org/10.1016/j.neuron.2009.06.008
Xu-Friedman MA, Harris KM, Regehr WG (2001) Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J Neurosci 21:6666–6672
Kleim J, Freeman JH, Bruneau R, Nolan BC, Cooper NR, Zook A, Walters D (2002) Synapse formation is associated with memory storage in the cerebellum. Proc Natl Acad Sci USA 99:13228–13231. https://doi.org/10.1073/pnas.202483399
Kagawa T, Ikenaka K, Inoue Y, Kuriyama S, Tsujii T, Nakao J, Nakajima K, Aruga J, Okano H, Mikoshiba K (1994) Glial cell degeneration and hypomyelination caused by overexpression of myelin proteolipid protein gene. Neuron 13:427–442. https://doi.org/10.1016/0896-6273(94)90358-1
Nguyen HB, Thai TQ, Saitoh S, Wu B, Saitoh Y, Shimo S, Fujitani H, Otobe H, Ohno N (2016) Conductive resins improve charging and resolution of acquired images in electron microscopic volume imaging. Sci Rep 6:23721. https://doi.org/10.1038/srep23721
Thai TQ, Nguyen HB, Saitoh S, Wu B, Saitoh Y, Shimo S, Elewa YHA, Ichii O, Kon Y, Takaki T, Joh K, Ohno N (2016) Rapid specimen preparation to improve the throughput of electron microscopic volume imaging for three-dimensional analyses of subcellular ultrastructures with serial block-face scanning electron microscopy. Med Mol Morphol 49:154–162. https://doi.org/10.1007/s00795-016-0134-7
Cardona A, Saalfeld S, Schindelin J, Arganda-Carreras I, Preibisch S, Longair M, Tomancak P, Hartenstein V, Douglas RJ (2012) TrakEM2 software for neural circuit reconstruction. PLoS One 7:e38011. https://doi.org/10.1371/journal.pone.0038011
Hámori J, Szentágothai J (1966) Identification under the electron microscope of climbing fibers and their synaptic contacts. Exp Brain Res 1:65–81. https://doi.org/10.1007/BF00235210
Palay SL, Chan-Palay V (1974) Cerebellar cortex cytology and organization. Springer, Berlin, pp 253–285
Miyazaki T, Fukaya M, Shimizu H, Watanabe M (2003) Subtype switching of vesicular glutamate transporters at parallel fibre-Purkinje cell synapses in developing mouse cerebellum. Eur J Neurosci 17:2563–2572. https://doi.org/10.1046/j.1460-9568.2003.02698.x
Choo M, Miyazaki T, Yamazaki M, Kawamura M, Nakazawa T, Zhang J, Tanimura A, Uesaka N, Watanabe M, Sakimura K, Kano M (2017) Retrograde BDNF to TrkB signaling promotes synapse elimination in the developing cerebellum. Nat Commun 8:195. https://doi.org/10.1038/s41467-017-00260-w
Ma J, Tanaka KF, Shimizu T, Bernard CCA, Kakita A, Takahashi H, Pfeiffer SE, Ikenaka K (2011) Microglial cystatin F expression is a sensitive indicator for ongoing demyelination with concurrent remyelination. J Neurosci Res 89:639–649. https://doi.org/10.1002/jnr.22567
Willard HF, Riordan JR (1985) Assignment of the gene for myelin proteolipid protein to the X chromosome: implications for X-linked myelin disorders. Science 230:940–942. https://doi.org/10.1126/science.3840606
Saugier-Veber P, Munnich A, Bonneau D, Rozet J-M, Le Merrer M, Gil R, Boespflug-Tanguy O (1994) X-linked spastic paraplegia and Pelizaeus–Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat Genet 6:257–262
Inoue K (2005) PLP1-related inherited dysmyelinating disorders: Pelizaeus–Merzbacher disease and spastic paraplegia type 2. Neurogenetics 6:1–16. https://doi.org/10.1007/s10048-004-0207-y
Garbern JY (2007) Pelizaeus–Merzbacher disease: genetic and cellular pathogenesis. Cell Mol Life Sci 64:50–65. https://doi.org/10.1007/s00018-006-6182-8
Readhead C, Schneider A, Griffiths I, Nave KA (1994) Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron 12:583–595. https://doi.org/10.1016/0896-6273(94)90214-3
Wolf NI, Sistermans EA, Cundall M, Hobson GM, Davis-Williams AP, Palmer R, Stubbs P, Davies S, Endziniene M, Wu Y, Chong WK, Malcolm S, Surtees R, Garbern JY, Woodward KJ (2005) Three or more copies of the proteolipid protein gene PLP1 cause severe Pelizaeus–Merzbacher disease. Brain 128:743–751. https://doi.org/10.1093/brain/awh409
Sarret C, Lemaire JJ, Tonduti D, Sontheimer A, Coste J, Pereira B, Feschet F, Roche B, Boespflug-Tanguy O (2016) Time-course of myelination and atrophy on cerebral imaging in 35 patients with PLP1-related disorders. Dev Med Child Neurol 58:706–713. https://doi.org/10.1111/dmcn.13025
Nevin ZS, Factor DC, Karl RT, Douvaras P, Laukka J, Windrem MS, Goldman SA, Fossati V, Hobson GM, Tesar PJ (2017) Modeling the mutational and phenotypic landscapes of Pelizaeus–Merzbacher disease with human iPSC-derived oligodendrocytes. Am J Hum Genet 100:617–634. https://doi.org/10.1016/j.ajhg.2017.03.005
Laukka JJ, Kamholz J, Bessert D, Skoff RP (2017) Novel pathologic findings in patients with Pelizaeus–Merzbacher disease. Neurosci Lett 627:222–232. https://doi.org/10.1016/j.neulet.2016.05.028
Koeppen AH, Robitaille Y (2002) Pelizaeus–Merzbacher disease. J Neuropathol Exp Neurol 61:747–759
Charzewska A, Wierzba J, Iżycka-Świeszewska E, Bekiesińska-Figatowska M, Jurek M, Gintowt A, Kłosowska A, Bal J, Hoffman-Zacharska D (2016) Hypomyelinating leukodystrophies—a molecular insight into the white matter pathology. Clin Genet 90:293–304. https://doi.org/10.1111/cge.12811
Seitelberger F (1995) Neuropathology and genetics of Pelizaeus–Merzbacher disease. Brain Pathol 5:267–273. https://doi.org/10.1111/j.1750-3639.1995.tb00603.x
Dutta R, Chang A, Doud MK, Kidd GJ, Ribaudo MV, Young EA, Fox RJ, Staugaitis SM, Trapp BD (2011) Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol 69:445–454. https://doi.org/10.1002/ana.22337
Jürgens T, Jafari M, Kreutzfeldt M, Bahn E, Brück W, Kerschensteiner M, Merkler D (2016) Reconstruction of single cortical projection neurons reveals primary spine loss in multiple sclerosis. Brain 139:39–46. https://doi.org/10.1093/brain/awv353
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AMM, Lambris JD, Smith SJ, John SWM, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. https://doi.org/10.1016/j.cell.2007.10.036
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT (2011) Development synaptic pruning by microglia is necessary for normal brain synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1459. https://doi.org/10.1126/science.1202529
Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, Barres BA (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504:394–400. https://doi.org/10.1038/nature12776
Kakegawa W, Mitakidis N, Miura E, Abe M, Matsuda K, Takeo YH, Kohda K, Motohashi J, Takahashi A, Nagao S, Muramatsu SI, Watanabe M, Sakimura K, Aricescu AR, Yuzaki M (2015) Anterograde C1ql1 signaling is required in order to determine and maintain a single-winner climbing fiber in the mouse cerebellum. Neuron 85:316–330. https://doi.org/10.1016/j.neuron.2014.12.020
Nishiyama H, Fukaya M, Watanabe M, Linden DJ (2007) Axonal motility and its modulation by activity are branch-type specific in the intact adult cerebellum. Neuron 56:472–487. https://doi.org/10.1016/j.neuron.2007.09.010
Becker N, Wierenga CJ, Fonseca R, Bonhoeffer T, Nägerl UV (2008) LTD induction causes morphological changes of presynaptic boutons and reduces their contacts with spines. Neuron 60:590–597. https://doi.org/10.1016/j.neuron.2008.09.018
Felts PA, Kapoor R, Smith KJ (1995) A mechanism for ectopic firing in central demyelinated axons. Brain 118:1225–1231. https://doi.org/10.1093/brain/118.5.1225
Waxman SG (2008) Mechanisms of disease: sodium channels and neuroprotection in multiple sclerosis—current status. Nat Clin Pract Neurol 4:159–169. https://doi.org/10.1038/ncpneuro0735
Chavan V, Willis J, Walker SK, Clark HR, Liu X, Fox MA, Srivastava S, Mukherjee K (2015) Central presynaptic terminals are enriched in ATP but the majority lack mitochondria. PLoS One 10:1–19. https://doi.org/10.1371/journal.pone.0125185
Khatri N, Man HY (2013) Synaptic activity and bioenergy homeostasis: implications in brain trauma and neurodegenerative diseases. Front Neurol 4:1–11. https://doi.org/10.3389/fneur.2013.00199
Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL, Mutani R, Giordana MT, Cavalla P (2007) Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol 66:732–739. https://doi.org/10.1097/nen.0b013e31812571b0
Matute C, Domercq M, Sánchez-Gómez M-V (2006) Glutamate-mediated glial injury: mechanisms and clinical importance. Glia 53:212–224. https://doi.org/10.1002/glia.20275
Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH (2013) Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep 4:413–419. https://doi.org/10.1016/j.celrep.2013.06.040
MacAskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT (2009) Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61:541–555. https://doi.org/10.1016/j.neuron.2009.01.030
Billups B, Forsythe ID (2002) Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci 22:5840–5847. https://doi.org/10.1523/JNEUROSCI.22-14-05840.2002
Acknowledgements
We thank Drs. M Yuzaki, A Kakegawa (Keio University) and M Watanabe (Hokkaido University) for helpful discussion and support. This work is partly supported by JSPS KAKENHI Grant number 16K12345 (to N.O.) and Grants-in-Aid for Scientific Research on Innovative Areas “glial assembly” (no. 25117005 to K.I.), and Research Grant from National Center of Neurology and Psychiatry (no. 30-5 to N.O.) and Novartis Pharma, Cooperative Research Program of “Network Joint Research Center for Materials and Devices” and Cooperative Study Programs of National Institute for Physiological Sciences (to N.O.). We would like to thank Setsuro Fujii Memorial, Osaka Foundation for Promotion of Fundamental Medical Research for providing the support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Nguyen, H.B., Sui, Y., Thai, T.Q. et al. Decreased number and increased volume with mitochondrial enlargement of cerebellar synaptic terminals in a mouse model of chronic demyelination. Med Mol Morphol 51, 208–216 (2018). https://doi.org/10.1007/s00795-018-0193-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00795-018-0193-z