
Abstract
The redox-responsive regulator SurR in the hyperthermophilic archaea Pyrococcus furiosus and Thermococcus kodakarensis binds to the SurR-binding consensus sequence (SBS) by responding to the presence of elemental sulfur. Here we constructed a surR gene disruption strain (DTS) in T. kodakarensis, and identified the genes that were under SurR control by comparing the transcriptomes of DTS and parent strains. Among these genes, transcript levels of ferredoxin:NADP+ oxidoreductases 1 and 2 (FNOR1 and FNOR2) genes displayed opposite responses to surR deletion, indicating that SurR repressed FNOR1 transcription while enhancing FNOR2 transcription. Each promoter region contains an SBS upstream (uSBS) and downstream (dSBS) of TATA. In addition to in vitro binding assays, we examined the roles of each SBS in vivo. In FNOR1, mutations in either one of the SBSs resulted in a complete loss of repression, indicating that the presence of both SBSs was essential for repression. In FNOR2, uSBS indeed functioned to enhance gene expression, whereas dSBS functioned in gene repression. SurR bound to uSBS2 of FNOR2 more efficiently than to dSBS2 in vitro, which may explain why SurR overall enhances FNOR2 transcription. Further analyses indicated the importance in the distance between uSBS and TATA for transcriptional activation in FNOR2.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The basal transcription machinery in archaea consists of a multisubunit RNA polymerase (RNAP) and three basal transcription factors, TATA-binding protein (TBP), transcription factor E (TFE), and transcription factor B (TFB) (Reeve 2003; Jun et al. 2011; Werner and Grohmann 2011). TBP and TFB recognize archaeal promoters by binding to the TATA-Box and the TFB-responsive element (BRE), respectively, and then RNAP interacts with the TFB and TBP complex bound to the promoter to initiate transcription. TFE facilitates open complex formation during pre-initiation by interacting with the RNAP stalk and clamp and single-stranded DNA in the transcription bubble (Blombach et al. 2016; Schulz et al. 2016). The archaeal transcription machinery is similar to the eukaryotic systems involving RNAPII apparatus (Grohmann and Werner 2011; Werner and Grohmann 2011); however, the known transcriptional regulators are bacterial type (Geiduschek and Ouhammouch 2005; Lipscomb et al. 2009; Leyn and Rodionov 2015; Gindner et al. 2014). Repression by some transcriptional regulators is considered to occur through the formation of a protein–DNA supercomplex that interferes with the binding of TBP and/or TFB to the BRE/TATA-Box, as is the case for Lrs14 of Sulfolobus solfataricus (Bell and Jackson 2000), and/or the recruitment of RNAP, as is the mechanism proposed for both Thermococcus kodakarensis Tgr (Kanai et al. 2007) and Pyrococcus furiosus Phr (Vierke et al. 2003). The transcriptional activator TFB-RF1 acts as a TFB recruitment factor by binding just upstream of the BRE in P. furiosus (Ochs et al. 2012). Tgr of T. kodakarensis and XacR of Haloferax volcanii (Johnsen et al. 2015) act as a transcriptional activator and repressor depending on the location of the binding site relative to the TATA-Box. On the other hand, BarR in Sulfolobus tokodaii activates the expression of its own gene, even though the BRE/TATA-Box is located at the border between two barR-binding sites (Liu et al. 2014). Based on these reports, there seems to be a general tendency for these archaeal transcriptional regulating factors to act as activators when bound upstream of the TATA-Box and repressors when bound downstream, but there is still a need to further examine the individual promoters to understand the respective regulation mechanisms.
SurR, which is an ArsR-type transcription regulator, was identified as a key regulator responsible for molecular hydrogen production through an elemental sulfur (S0)-dependent redox switch of the CXXC motif in the hyperthermophilic archaeon P. furiosus (Lipscomb et al. 2009; Yang et al. 2010; Lipscomb et al. 2017), which can grow in the presence or absence of S0, depending on the available carbon source (Fiala and Stetter 1986). This transcriptional regulator is widely conserved among the order Thermococcales. A reduced form of SurR acts as a transcriptional activator through binding to the SurR-binding consensus sequence (SBS, GTTn3AAC) (Lipscomb et al. 2009). By contrast, an intramolecular disulfide bond is thought to form in the SurR CXXC motif in the presence of S0, resulting in the loss of its ability to bind to the SBS (Yang et al. 2010). The expression of genes involved in hydrogen-generating respiration, including the mbh gene cluster, which encodes the components of membrane-bound hydrogenase (MBH), is activated by binding of a reduced form of SurR to the SBS in the absence of S0. In turn, genes involved in H2S-generating respiration, including nsr, the gene encoding NAD(P)H sulfur reductase, are derepressed by loss of the ability of the oxidized form of SurR to bind to the SBS in the presence of S0. In Thermococcus onnurineus NA1, the redox state of SurR can be regulated by protein disulfide oxidoreductase and thioredoxin reductase couple (Lim et al. 2017). SurR is now considered as a master regulator of the primary electron flow pathways in the order Thermococcales (Lipscomb et al. 2017). This redox-responsive regulation dominated by a two cysteine-type thiol-based mechanism is also seen with several bacterial transcription factors, such as Escherichia coli OxyR and Bacillus subtilis Spx (Antelmann and Helmann 2011; Hillion and Antelmann 2015). Disruption of the surR gene of T. kodakarensis (Tk-surR) is lethal in the absence of S0, consistent with the fact that Tk-SurR activates the expression of genes involved in molecular hydrogen production (Santangelo et al. 2011).
Although a wealth of structural and biochemical evidence has been obtained, genetic studies on SurR have been rather limited to the responses of the cell brought about by surR gene disruption. In the present study, we took advantage of the genetic tools developed for T. kodakarensis (Sato et al. 2003, 2005; Matsumi et al. 2007; Santangelo et al. 2008). We first perform a comparative transcriptome analysis with a surR-deleted mutant (DTS) of T. kodakarensis grown in the presence of S0 to identify genes that are specifically under the control of Tk-SurR. We then proceed to examine the contributions of SBSs in the promoters of two genes under the control of SurR, ferredoxin:NADP+ oxidoreductase 1 and 2 (FNOR1 and FNOR2), both in vitro and in vivo. The promoters of FNOR1 and FNOR2 each contain two SBSs, upstream and downstream of the TATA-Box. Our transcriptome analysis indicated that the transcript levels of these two genes displayed opposite responses to surR deletion even though both promoters display SBS:TATA-Box:SBS structures. Our analyses indicated that the locations of SBSs and the binding affinities of Tk-SurR toward the individual SBSs are key determinants in its transcriptional regulation.
Materials and methods
Microorganisms and media
Thermococcus kodakarensis KU216 (∆pyrF) (Sato et al. 2005) and its derivatives (Table 1) were cultivated anaerobically in a nutrient-rich medium artificial seawater-yeast extract-tryptone (ASW-YT) (Atomi et al. 2004) containing 2.0 g L−1 S0 or in synthetic medium (Sato et al. 2005) containing 0.8 × artificial seawater, amino acids, and S0. For solid medium, 1% gelrite (Wako, Osaka, Japan) and 2 mL L−1 polysulfide solution (10 g of Na2S·9H2O and 3 g of sulfur flowers in 15 mL of H2O) were added. E. coli strains were cultivated at 37 °C in LB medium containing 50 µg mL−1 ampicillin.
Construction of the surR-deleted strain
The principles underlying specific gene disruption in T. kodakarensis have been described previously (Sato et al. 2005). The vector for disrupting the surR gene [954,255–954,965 (+) bp in the T. kodakarensis genome] through double-crossover homologous recombination was constructed as follows. A set of primers, TkSurR-Fw1/TkSurR-Rv1, was designed to amplify DNA fragments containing the surR gene along with its flanking regions (ca. 1000 bp each) by PCR using T. kodakarensis genomic DNA. The DNA fragment containing the surR gene was cloned into the BamHI/EcoRI sites of pUD2 (Sato et al. 2005), yielding the plasmid pUD2-SurR. The coding region of surR was then removed using inverse PCR with the primers Inv-TkSurR-Fw/Inv-TkSurR-Rv, and the resultant PCR fragment was 5′-phosphorylated and self-ligated. The resulting disruption vector, pUD2-∆SurR, was used for gene deletions in the host strain T. kodakarensis KU216 (∆pyrF). Gene deletions were confirmed by nucleotide sequencing with an ABI PRISM® 3130 Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA).
Microarray analysis
KU216 and the Tk-SurR-deleted strain DTS (∆pyrF, ∆surR) were individually cultivated at 85 °C in the presence and absence of S0. Cells were harvested in mid-log phase, and total RNA was extracted using the RNeasy mini kit (Qiagen, Venlo, The Netherlands). The microarray plate used in this study (Array Tko2) was manufactured at Takara Bio (Otsu, Japan) and covers all 2306 genes of the T. kodakarensis genome. Two identical sets (left and right) are loaded on each plate. Therefore, two sets of data were obtained from each microarray plate. The results are the average of two independent microarray slides, and error bars represent standard deviation. The experimental procedure for microarray analysis is described elsewhere (Kanai et al. 2007).
Construction of the FNOR1 and FNOR2 promoter region variants
The principles underlying the promoter-probe analysis with the plasmid pTKR have been described previously (Nagaoka et al. 2013). The plasmid pTKR was constructed from pTK01 (Santangelo et al. 2008), a derivative of pTN1 (Soler et al. 2007). The pTKR harbors a promoter-less catalase gene from Pyrobaculum calidifontis (Amo et al. 2002). A DNA fragment containing the 5′-flanking region of TK1326 or TK1685 was amplified by PCR from T. kodakarensis genomic DNA using the primer set TK1326-Fw/TK1326-Rv or TK1685-Fw/TK1685-Rv, respectively. The amplified fragments were cloned into the EcoRV/NdeI sites of pTKR (Nagaoka et al. 2013), yielding pTKRF1 and pTKRF2, respectively. The TK1326 promoter-probe plasmid variants (pTKRF1-1, pTKRF1-2, and pTKRF1-12) were obtained by QuikChange site-directed mutagenesis (Agilent Technologies, Santa Clara, CA) of pTKRF1 using specific primer sets (F1-∆uSBS1-Fw/F1-∆uSBS1-Rv for pTKRF1-1, F1-∆dSBS1-Fw/F1-∆dSBS1-Rv for pTKRF1-2, and both primer sets for pTKRF1-12). The TK1685 promoter-probe plasmid variants (pTKRF2-1, pTKRF2-2, pTKRF2-12) were obtained by site-directed mutagenesis of pTKRF2 using specific primer sets (F1-∆uSBS2-Fw/F1-∆uSBS2-Rv for pTKRF2-1, F1-∆dSBS2-Fw/F1-∆dSBS2-Rv for pTKRF2-2, and both primer sets for pTKRF2-12) and the other TK1685 promoter-probe plasmid variants (pTKRF2-3, pTKRF2-4, and pTKRF2-5) were obtained by the same way using specific primer sets (F2-uSBS2-Fw/F2-uSBS2-Rv for pTKRF2-3, F3-uSBS2-Fw/F3-uSBS2-Rv for pTKRF2-4, and F4-uSBS2-Fw/F4-uSBS2-Rv for pTKRF2-5).
Quantitative real-time PCR (qRT-PCR)
Total RNA was obtained from T. kodakarensis cells using the RNeasy mini kit, and qRT-PCR was performed using the Super-Script III RT/Platinum kit (Invitrogen, Carlsbad, CA, USA) and an ABI PRISM 7000 sequence detection system (Applied Biosystems, Foster City, CA, USA). Total RNA extracts were treated with DNase I following the manufacturer’s instructions (Roche Applied Science, Penzberg, Germany). The primers used to amplify the TK1326, TK1685, and 16S rRNA genes by qRT-PCR were a TK1326 sense primer/TK1326 antisense primer, a TK1685 sense primer/TK1685 antisense primer, and a tk_16S sense primer/tk_16S antisense primer, respectively. Each sequence-specific standard curve was generated with varying amounts of PCR products, which were separately amplified from the TK1326, TK1685, and 16S rRNA genes with the corresponding primer set used in qRT-PCR. The mRNA levels shown are an average of three independent technical replicates and were normalized against the level of 16S rRNA, which was set to 1.
Expression and purification of Tk-SurR
The primers used to clone surR were TkSurR-Fw and TkSurR-Rv (Table 1). The amplified DNA fragment encoding surR was cloned into the NdeI/EcoRI sites of pET21a, yielding the plasmid pSurR. E. coli BL21-CodonPlus (DE3)-RIL cells harboring pSurR were grown in LB medium containing 100 µg mL−1 ampicillin at 37 °C for 6 h. After induction by treatment with 1 mM isopropyl-β-d-thiogalactopyranoside for 4 h, cells were harvested by centrifugation at 10,000×g for 30 min, resuspended in Buffer A (20 mM Tris–Cl, 1 mM EDTA, 1 mM 2-mercaptoethanol, and 0.2 mM phenylmethylsulfonyl fluoride, pH 7.5), and disrupted by sonication. After removing cell debris by centrifugation at 23,000×g for 60 min, the supernatants were used for purification. The supernatant was incubated at 80 °C for 30 min and centrifuged (23,000×g for 60 min), and the resultant supernatant was applied to a HiTrap Q anion-exchange column (GE Healthcare, Little Chalfont, UK). Tk-SurR was eluted with a linear gradient of NaCl (0–1.0 M) prepared in Buffer A. Fractions containing Tk-SurR were confirmed by SDS-PAGE and collected. The protein concentration was determined by the Bradford assay using bovine serum albumin as a standard (Bradford 1976).
Circular dichroism (CD) spectroscopy
CD spectroscopic experiments were performed on a J-820 CD spectropolarimeter (JASCO) at 20 °C. Far-UV CD spectra in the 215–260 nm region were measured using a quartz cuvette (pathlength of 0.2 cm) at 0.05 nm steps over the wavelength range, with a scan speed of 50 nm min−1. The experiments were performed with 1.8 µM Tk-SurR prepared in 50 mM potassium phosphate buffer (pH 7.4) in the presence of 5 mM dithiothreitol or 5 mM diamide. All samples were preincubated at room temperature for 30 min before each scan. All spectra were corrected for the contributions of buffer to the signals.
Gel-shift assay
The promoter regions of the TK1326 and TK1685 genes were amplified by PCR with the primer set TK1326-Fw2/IRD700-Rv (an IRD700-labeled primer) from the pTKRF1 plasmid and its derivatives or with the primer set TK1685-Fw2/IRD700-Rv from the pTKRF2 plasmid and its derivatives, respectively, producing IRD700-labeled DNA probes (Table 1). The complex between Tk-SurR and each free probe was detected by a mobility shift in a non-denaturing 5% polyacrylamide gel. Various concentrations of Tk-SurR were incubated with 2 nM IRD700-labeled DNA probe in 20 μL of binding buffer containing 20 mM Tris–HCl (pH 7.5), 15 mM KCl, 0.2% Tween 20, and 63 ng mL−1 salmon sperm for 30 min at 60 °C. After adding 0.25% bromophenol blue dye containing 40% sucrose to the reaction mixture, the complex was separated by electrophoresis on a 5% polyacrylamide gel in Tris–borate–EDTA electrophoresis buffer (45 mM Tris–borate, pH 8.0, and 1 mM EDTA). Signals were visualized with an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA). Fractional saturation was determined at each protein concentration by densitometry of free DNA using NIH ImageJ software. The data were obtained from three independent experiments and fitted to the Hill equation (fraction bound = [protein]n/([protein]n + K d n), where n is the Hill coefficient and K d is the apparent dissociation constant, using Kaleida Graph software (Adelbeck Software, Reading, PA, USA).
Immunoblotting analysis
Cell extracts (20 µg of each) obtained by sonication were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (ATTO, Tokyo, Japan). Immunodetection was performed with an antibody raised against Pc-Kat in rabbit and Alexa Fluor 700 goat anti-rabbit IgG (Invitrogen). Signals were visualized with an Odyssey infrared imaging system (LI-COR Biosciences).
Results
Transcriptome analysis
The Tk-surR gene-deleted strain, named T. kodakarensis DTS, was obtained by homologous recombination (Supplemental Fig. 1). Consistent with the growth property of the T. kodakarensis TS1101 strain (∆pyrF, ∆trpE::pyrF, ∆surR) reported previously (Santangelo et al. 2011), the DTS strain did not grow in the absence of S0. The reduced form of Tk-SurR is presumed to activate genes involved in hydrogen production by sequence-specific binding. In the present study, to identify genes whose expression is dependent on the regulation of Tk-SurR in the presence of S0, a comparative transcriptome analysis was carried out using total RNA extracted from the T. kodakarensis DTS and KU216 strains. The transcriptome data are available under the number GSE71984 in the gene expression omnibus (GEO) database at NCBI (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE71984). The genes whose transcript levels increased or decreased more than fourfold in the DTS strain in two independent experiments are listed in Tables 2 and 3, respectively. The transcript levels of eight genes were more than fourfold higher in the DTS strain than in the KU216 strain, including TK1326, which encodes a component of FNOR1 (Table 2). Among the eight genes, TK0164–TK0166 and TK1024–TK1025 constitute a gene cluster. On the other hand, the transcript levels of 37 genes were more than fourfold lower in the DTS strain than in the KU216 strain (Table 3). Among these down-shifted genes in the DTS strain, TK2076–TK2077, TK2073–TK2075, TK2070–TK2072, TK2080–TK2093 (TK2090 and TK2092 were not detected in the analysis), TK0038–TK0044, and TK0119–TK0121 are present in a gene cluster. The TK2076–TK2077 genes, which are predicted to encode formate:ferredoxin oxidoreductase, were the most down-shifted in the DTS strain. The transcripts of TK2080–TK2093, which encode the components of MBH, decreased to less than one-sixteenth of the level observed in the KU216 strain. In addition, the expression levels of four genes, TK1209, TK1481, TK0675, and TK1685, which encode a transmembrane electron transport protein, an NADH:polysulfide oxidoreductase, a hypothetical protein belonging to the radical S-adenosylmethionine superfamily, and a component of FNOR2, respectively, decreased dramatically in the DTS strain.
SBSs (GTTn3AAC) were found upstream of the TK2076–TK2077, TK2080–TK2093, TK1481, TK1209, TK0675, TK1325, and TK1685 genes (Table 4). Furthermore, two SBSs were found in the upstream regions of the TK0675, TK1325, and TK1685 genes. Based on a comprehensive identification of each transcription start site in T. kodakarensis obtained by differential RNA-sequencing analysis (Jäger et al. 2014), we predicted the TATA-Box sequence in the promoter region of each gene. According to the analysis, 18 bp is the average distance between the 3′-end of the nearest upstream SBS and the 5′-start point of the TATA-Box sequence in the five down-shifted genes, i.e., TK2076–TK2077, TK1481, TK1209, TK0675, and TK1685. The distance in the upstream region of the TK2080–TK2093 gene cluster was exceptionally long at 47 bp. For efficient SurR binding, an extended motif has been proposed, which includes a second palindromic half-site with a 5 bp gap: GTTn3AACn5GTT (Lipscomb et al. 2009). This extended motif was also found in the promoter regions of the TK2080–TK2093 gene cluster and the TK1481, TK1209, TK0675, and TK1685 genes. We also identified a number of genes that although responding to Tk-surR deletion, did not harbor SBSs in their promoter regions at least up to 200 bp. These were the TK2073–TK2075, TK0038–TK0044, and TK0119–TK0121 gene clusters, and the TK2079, TK0631, and TK0842 genes. These genes are most likely regulated by factors that are under the control of Tk-SurR and not directly recognized by Tk-SurR itself.
The different expression profiles of two FNOR genes
Our transcriptome analysis showed that the transcript level of TK1685 decreased, whereas that of TK1326 increased in the DTS strain. Both genes encode FNOR isozymes (Santangelo et al. 2011). TK1326–TK1325 and TK1685–TK1684 encode the β- and α-subunits of two FNORs, designated here as FNOR1 and FNOR2, respectively. FNOR1 and FNOR2 are 60% identical to each other. We measured the mRNA levels of these two genes in the KU216 strain grown in the presence or absence of S0 and in the DTS strain grown in the presence of S0 using qRT-PCR (Fig. 1). The transcript levels of TK1326 and TK1685 were about fivefold and 1.6-fold higher in the KU216 strain grown in the presence of S0 than in that grown in the absence of S0, respectively. The transcript levels of TK1326 (FNOR1) in the DTS strain were twofold higher than in the KU216 strain, whereas those of TK1685 (FNOR2) were twofold lower in the DTS strain than in the KU216 strain (Fig. 1a, b). The results indicate that in strain KU216, transcripts of both FNOR1 (TK1326) and FNOR2 (TK1685) increase in the presence of S0 (Fig. 1), but the genes respond differently to Tk-surR disruption. Although the tendencies were the same, the mean intensity ratios [TK1326 of log2(DTS/KU216) = 2.12; TK1685 of log2(DTS/KU216) = −2.06] obtained by our DNA microarray analysis differed from the fold changes calculated by qRT-PCR analysis (Tables 2, 3; Fig. 1). This may be due to low levels of transcripts of the oxidoreductase genes, resulting in low hybridization efficiency and non-specific hybridization and/or cross-hybridization in the DNA microarray analysis (Koltai and Weingarten-Baror 2008). Quantification of the transcripts achieved by qRT-PCR analysis was used for further study, as it is regarded as the more accurate method for quantification. Two SBSs were found upstream and downstream of the TATA-Box in both the FNOR1 and FNOR2 (hereafter named uSBS1 and dSBS1 for FNOR1, and uSBS2 and dSBS2 for FNOR2) (Table 4). The distance between the uSBS and the TATA-Box was 19 bp in the FNOR2, and 4 bp in the FNOR1.

qRT-PCR analysis of FNORs expression in the KU216 and Tk-SurR-deleted strains. Relative transcript abundances of TK1326 for FNOR1 (a) and TK1685 for FNOR2 (b). a The relative mRNA expression levels of TK1326 and TK1685 were determined by qRT-PCR using total RNA prepared from the KU216 strain cultivated in ASW-YT media containing S0 or pyruvate (−S0) and the DTS strain cultivated in medium containing S0 at mid-logarithmic phase. Error bars represent standard deviations from three independent experiments. The mRNA levels were normalized to the 16S rRNA level, which was set to 1
In vivo analyses of the SBSs of FNOR1 and FNOR2 genes
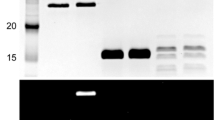
To examine the Tk-SurR-mediated transcriptional regulation of FNOR1 and FNOR2 in vivo, we carried out a reporter analysis using the Pc-kat gene, which encodes a thermostable catalase from P. calidifontis (Nagaoka et al. 2013). The promoter regions of the FNOR1 and FNOR2 were individually fused directly upstream of the Pc-Kat gene in the plasmid pTKR (Fig. 2a, b). The resulting plasmids were introduced into the T. kodakarensis DAD strain (∆pyrF, ∆pdaD) (Fukuda et al. 2008). This strain exhibits agmatine auxotrophy, which is lethal in medium lacking agmatine because an arginine decarboxylase gene is disrupted. By contrast, the DAD strain supplied with the pdaD in trans grows in the absence of agmatine, enabling agmatine-based selection of transformants. The expression levels of Pc-Kat in DAD cells harboring the corresponding plasmid cultivated in the presence or absence of S0 are shown in Fig. 2. In DAD cells harboring pTKRF1, FNOR1 promoter activity was more than fivefold higher in cells grown in the presence of S0 than in those grown in the absence of S0, consistent with the corresponding transcript abundance ratio obtained by qRT-PCR (Figs. 1a, 2a). We next introduced point mutations into the individual SBSs to impair binding of Tk-SurR. pTKRF1-1 has a defect in uSBS1, pTKRF1-2 in dSBS1, and pTKRF1-12 has defects in both. The transcript levels observed from the variant promoters were comparable to that from pTKRF1 when the cells were grown in the presence of S0. However, when the cells were grown in the absence of S0, disturbing the SBSs resulted in transcript levels fivefold higher than that from pTKRF1 grown under the same condition, and equivalent to the levels observed in cells grown with S0. These results indicated that both consensus sequences (uSBS1 and dSBS1) in the FNOR1 promoter function as an operator mediating repression by Tk-SurR. Interestingly, disturbing a single SBS led to complete derepression, indicating that the individual SBSs cannot repress gene expression alone and that the presence of both SBSs are necessary for repression in vivo.

The roles of SBSs of each FNOR promoter region in transcriptional regulation by Tk-SurR. A-a Structural features of the FNOR promoter are shown. +1 indicates the transcriptional start point. The predicted TATA-Box and SBSs are shown in white letters and bold letters, respectively. A-b Representative Pc-Kat expression patterns in T. kodakarensis DAD cells harboring pTKR, pTKRF1, pTKRF1-1, pTKRF1-2, and pTKRF1-12 are shown. Crude extracts (20 µg) of T. kodakarensis DAD cells harboring the corresponding plasmids were separated by SDS-PAGE and detected by western blotting using antisera against Pc-Kat (upper) and Coomassie brilliant blue staining (lower) as a loading control. Lane M, molecular mass markers. A-c The values shown indicate the expression level of the Pc-kat gene under the control of the native FNOR1 promoter relative to cells grown in the presence of S0 (set to 100%). Black and gray bars represent the Pc-Kat levels in cells harboring the corresponding plasmid grown in the presence and absence of S0, respectively. B-a Structural features of the FNOR2 promoter region are shown. B-b The representative Pc-Kat expression patterns in T. kodakarensis DAD cells harboring pTKR, pTKRF2, pTKRF2-1, pTKRF2-2, and pTKRF2-12 are shown. B-c The values shown indicate the expression level of the Pc-kat gene under the control of the native FNOR2 promoter relative to cells grown in the presence of S0 (set to 100%). All the experiments were performed in triplicate
In the case of FNOR2, promoter activity was higher in DAD cells harboring pTKRF2 grown in the presence of S0 than in cells grown in the absence of S0, in agreement with the corresponding transcript abundance ratio obtained by qRT-PCR (Figs. 1b, 2b). As in the case of FNOR1, we introduced mutations into uSBS2 (pTKRF2-1), dSBS2 (pTKRF2-2), or both (pTKRF2-12). Expression from pTKRF2-1 was not detected, irrespective of the presence or absence of S0, suggesting that uSBS2 functions in a positive manner, enhancing transcription of FNOR2. The fact that disturbing uSBS2 still resulted in a decrease in transcript levels in cells grown with S0 indicates that the reduced form of Tk-SurR with binding ability is still present under these growth conditions, at least at a concentration sufficient to bind to uSBS2 and enhance transcription. Expression from pTKRF2-2 was more than 2.5-fold greater than that from pTKRF2 in cells grown in the absence of S0. This indicates that dSBS2 is involved in repressing the gene expression of FNOR2 via Tk-SurR binding. As the levels of Pc-Kat in cells grown with and without S0 were equivalent, this suggests that the intracellular concentration of the reduced form of Tk-SurR is sufficient to trigger maximum activation even in cells grown with S0, and that the differences in expression levels observed in the presence/absence of S0 is controlled by the degree of repression brought about by dSBS2. In cells harboring pTKRF2-12, which lacked both SBS2s, a moderate level of expression was observed that showed little response to the presence/absence of S0. Taken together, the data indicate that uSBS2 functions as an upstream activating site that enhances FNOR2 transcription, whereas dSBS2 functions as an operator for gene repression. The results also suggest that the activating function of uSBS2 is rather constitutive, and that the overall response of gene expression towards the presence/absence of S0 is governed by dSBS2.
To further investigate whether the uSBS2 location is essential for the transcription activation, we introduced a uSBS2 cassette (GTTCTAAACCTTTGGTT) 5 bases (pTKRF2-3) or 10 bases (pTKRF2-4) upstream or 5 bases downstream (pTKRF2-5) of the original location of the uSBS2 in FNOR2 (Fig. 3a). As in the promoter assays described above, the transcription activities of the promoter variants were estimated with Pc-Kat in the presence and absence of S0. As a result, expression from pTKRF2-3, pTKRF2-4, and pTKRF2-5 were not detected, similar to the results of pTKRF2-1 (Fig. 3). This indicates that the location of uSBS2 is crucial for its function in transcriptional activation.

The importance of the uSBS2 location in FNOR2 promoter region. a Structural variants of the FNOR2 promoter region are shown. b The representative Pc-Kat expression patterns in T. kodakarensis DAD cells harboring pTKRF2, pTKR, pTKRF2-3, pTKRF2-4, and pTKRF2-5 are shown. c The values shown indicate the expression level of the Pc-kat gene under the control of the native FNOR2 promoter relative to cells grown in the presence of S0 (set to 100%). All the experiments were performed in triplicate
Structural analysis of Tk-SurR
Tk-SurR was purified to near homogeneity under aerobic conditions (Fig. 4a). To investigate the binding ability of Tk-SurR in response to redox conditions, we carried out a gel-mobility shift assay using a DNA probe amplified from the promoter region in pTKRF2. The band corresponding to the free DNA probe displayed an upward shift with increasing concentrations of Tk-SurR reduced by DTT (Fig. 4b), indicating that Tk-SurR bound to the promoter of the FNOR2, consistent with the finding that the reduced form of P. furiosus SurR binds to the SBS (Yang et al. 2010). By contrast, binding was not observed when the DNA probe was incubated with Tk-SurR oxidized by the thiol-specific oxidant diamide (Kosower and Kosower 1995). A previous report estimated that approximately 80% of P. furiosus SurR is in the reduced state, even when purified aerobically (Yang et al. 2010). Next, we monitored the conformational change between the reduced and oxidized forms of Tk-SurR by CD spectrum analysis. The far-UV CD spectra of Tk-SurR reduced and oxidized by DTT and diamide, respectively, both showed minimal values at 222 nm, typical for an α-helix (Fig. 4c). However, the value of molecular ellipticity at 222 nm of the reduced form was lower than that of the oxidized form, suggesting that a conformational change of Tk-SurR occurred in response to the redox conditions, most likely via the switch of the CXXC motif.

Binding abilities of the reduced and oxidized forms of Tk-SurR to SBS. a A 5 μg sample of purified recombinant Tk-SurR was separated, and the gel was stained with Coomassie brilliant blue. Lane M molecular mass markers. b The electrophoretic mobility shift assay showing the ability of the reduced (upper panel) and oxidized (lower panel) forms of Tk-SurR to bind to the native promoter region of the FNOR2 gene. The protein concentrations used are indicated above each lane. The position of bands corresponding to free DNA [F] and bound DNA [B] are shown. c Far-UV CD spectra of Tk-SurR (1.8 µM) oxidized by 5 mM diamide (black line) or reduced by 5 mM DTT (gray line) at 20 °C
Gel-shift assay
To determine the dissociation constants of Tk-SurR bound to the SBSs of the FNOR1 and FNOR2 promoters, a gel-mobility shift assay was performed using DNA probes amplified from the promoter regions in pTKRF1, pTKRF2, and their variants. This allowed us to examine Tk-SurR binding with promoters harboring both SBSs (native in Fig. 5), single SBSs (uSBS, dSBS) and no SBSs (SBS-less). The DNA probe containing the native FNOR1 promoter displayed a shift with increasing concentrations of Tk-SurR (Fig. 5a), indicating that Tk-SurR bound to the FNOR1 promoter as was observed with the FNOR2 promoter. Next, we determined the apparent dissociation constants by quantitative titrations at a fixed concentration of the probe DNA. The increase in the level of bound DNA with increasing concentrations of Tk-SurR was fitted to the Hill equation, yielding apparent dissociation constants of K d = 283 ± 9 nM for Tk-SurR:FNOR1 promoter and K d = 189 ± 9 nM for Tk-SurR:FNOR2 promoter (Fig. 5b). The binding affinity of Tk-SurR with the FNOR1 promoter was lower than that with the FNOR2 promoter. The binding isotherms of Tk-SurR to the promoters of the FNOR1 and FNOR2 showed a sigmoidal dependence on the protein concentration, with a Hill coefficient of 4.9 ± 0.6 and 3.3 ± 0.4, respectively, indicating that the binding of Tk-SurR to each promoter was highly cooperative. No band shifts were observed with the SBS-less probes, confirming that Tk-SurR specifically recognized the SBSs in each promoter. When we examined Tk-SurR binding with individual SBSs, the dissociation constants were 748 ± 34 nM (Tk-SurR:uSBS1), 272 ± 5 nM (Tk-SurR:dSBS1), 81 ± 5 nM (Tk-SurR:uSBS2) and 116 ± 6 nM (Tk-SurR:dSBS2). As predicted from the in vivo experiments, the affinity of Tk-SurR for uSBS2 was slightly, but consistently higher than that for dSBS2 in vitro. This supports the assumption that increased binding of Tk-SurR to dSBS2 via its reduction governs the regulation of gene expression in FNOR2.

Binding affinity of Tk-SurR to each SBS located in the upstream region of the FNOR1 and FNOR2. a The interaction between Tk-SurR and different fragments encompassing either the native promoter region of the FNOR1 (left panel), the FNOR2 (right panel), or their SBS mutants (uSBS1, dSBS1, and both SBS1s; uSBS2, dSBS2, and both SBS2s) observed by electrophoretic mobility shift assays. The protein concentrations used are indicated above each lane. b Binding isotherm; the ratios of the bound DNA fractions quantified by densitometry from the above gel-mobility shift assays are plotted against the concentrations of the Tk-SurR dimer. The bound fraction levels shown are from the native promoter region of the FNOR1 gene (open circle), dSBS1 (open square), and uSBS1 (open diamond) (left panel), and that of the FNOR2 gene (open circle), dSBS2 (open square), and uSBS2 (open diamond) (right panel). Error bars represent one standard deviation for each point derived from triplicate experiments
Discussion
Here we have carried out in vitro and in vivo analyses on Tk-SurR to better understand how the protein regulates gene expression. A conformational change of SurR from P. furiosus is considered to occur due to formation of an intramolecular disulfide bond in the CXXC motif in the presence of S0, thereby forming a DNA-unbound state of the regulator (Yang et al. 2010). Our present CD data also confirmed a structural change of Tk-SurR depending on the presence of DTT and diamide (Fig. 4c), and as expected, the oxidized form of Tk-SurR did not bind to SBS (Fig. 4b).
Our transcriptome analysis between the DTS strain and KU216 strain revealed a large number of genes under the control of Tk-SurR. A much larger number of genes displayed lower levels of transcripts upon surR disruption than the number of those that increased (Tables 2, 3). This was also the case with genes that actually harbor an SBS as a cis-regulatory element (Table 4). FNOR1 (TK1326) was the only promoter among the up-shifted genes in the DTS strain that contained an SBS. The response of the mbh gene cluster to the presence/absence of S0 has previously been examined, and transcription was rapidly terminated by due to a conformational change of Tk-SurR in response to the addition of S0 (Jäger et al. 2014). In this study, we show that a further decrease in transcript levels of mbh was observed with surR disruption, even when cells were grown with S0 (Table 3). In addition, transcripts of FNOR2 were also detected in the KU216 strain in the presence of S0 (Fig. 1a). These results suggest that the reduced form of Tk-SurR exists to some extent in the cytoplasm even in the presence of S0, activating (albeit weakly) the expression of genes positively regulated by Tk-SurR.
Our results indicate that both uSBS1 and dSBS1 in the FNOR1 promoter function as an operator mediating repression by Tk-SurR. As is the case for S. solfataricus Lrs14 (Bell and Jackson 2000) and/or T. kodakarensis Tgr (Kanai et al. 2007) and P. furiosus Phr (Vierke et al. 2003), the Tk-SurR-DNA supercomplex can be presumed to interfere with the binding of TBP and/or TFB to the BRE/TATA-Box and/or the recruitment of RNAP. In the case of FNOR1, a single SBS disruption led to complete derepression, indicating that the individual SBSs cannot repress gene expression alone and that the presence of both SBSs is essential for repression in vivo. Disruption of a single SBS (pTKRF1-1 and pTKRF1-2) also abolished the response of gene expression towards S0 (Fig. 2a), indicating that both uSBS1 and dSBS1 are needed for the response. The cooperative behaviors of the two SBSs in vivo are also supported by the binding assays carried out in vitro. If the two sites were recognized completely independently, we should observe band shifts in two concentration ranges in Fig. 5a (FNOR1 promoter, left panel), as Tk-SurR binding to dSBS1 (300 nM) and uSBS1 (500 nM) occur at different concentration ranges. However, the band shift with the native promoter (with dSBS1 and uSBS1) predominantly occurs at the same concentration range as that with the promoter with only dSBS1. Furthermore, this band displays lower mobility than the shifted bands observed with probes with single SBSs (data not shown), indicating that the band represents a probe bound by Tk-SurR at both SBSs. Taken together, we can presume that Tk-SurR binding to dSBS1 promotes binding of Tk-SurR to uSBS1 in a cooperative manner.
In the case of FNOR2, our data indicate that uSBS2 functions as an upstream activating site that enhances FNOR2 transcription, whereas dSBS2 functions as an operator for gene repression. The results also suggest that the activating function of uSBS2 is rather constitutive, and that the overall response of gene expression towards the presence/absence of S0 is regulated by the binding or release of Tk-SurR to dSBS2. Tk-SurR bound to uSBS2 of FNOR2 more efficiently than to dSBS2 in vitro, which may explain why Tk-SurR overall enhances FNOR2 transcription. We also revealed that the position of uSBS2 is important for the site to act as an activating element. Shifting uSBS2 5 bp upstream or downstream completely abolished the activating effect of the SBS. As the double helix of B-DNA is right-handed with about 10–10.5 base pairs per turn, a 5-base shift in location can be considered to change the accessible surface for Tk-SurR. However, a 10-base shift upstream also abolished the activating effect. Although further analyses are necessary to determine the importance of the orientation of the uSBS2 relative to the TATA-Box, our results indicate that the position of uSBS2 is important for the transcriptional activation. A kinetic study using a gel-shift assay showed that the dissociation constant of Tk-SurR was slightly lower for uSBS2 (K d = 81 ± 5 nM) than for dSBS2 (K d = 116 ± 6 nM) (Fig. 5b). These results suggest that preferential binding of a small amount of the reduced form of Tk-SurR to uSBS2 enables induction of FNOR2 expression, whereas expression is gradually repressed by binding of Tk-SurR to dSBS2 as the concentration of reduced Tk-SurR increases (Fig. 6). A transcriptional activator, TFB-RF1, supports the transcription of a gene with a weak BRE for efficient activation. The BRE sequence is considered to be not only a determinant of the direction of transcription, but also a key factor in stabilizing the TFB–TBP–RNA polymerase complex (Ochs et al. 2012). We speculate that Tk-SurR recruits TFB to the BRE sequence of the FNOR2 to stabilize the transcription initiation complex to activate transcription. The results of this study raise the possibility that the location of the SBS as well as the binding affinity of Tk-SurR for the SBS are key determinants for controlling the expression of genes under Tk-SurR control.

A proposed model of FNORs expression regulation by Tk-SurR. a The conformations of Tk-SurR in the presence and absence of S0. The two cysteine residues in the CXXC motif of Tk-SurR form an intramolecular disulfide bond in the presence of S0. A reduced form of SurR in the absence of S0 binds to the SBS (GAAn3TTC). b At a low S0 concentration or in the absence of S0, a large amount of reduced Tk-SurR (shaded circles) would bind to SBS to regulate gene expression. The reduced form is gradually oxidized (white circles) with an increasing amount of S0 in the medium, resulting in the formation of a DNA-unbound state. However, a small amount of the reduced form is still involved in gene regulation even in the presence of S0. In transcriptional regulation of FNOR1 expression, both uSBS1 and dSBS1 function as an operator for gene repression upon Tk-SurR binding. The binding of Tk-SurR to uSBS1 will trigger strong repression of FNOR1 expression. On the other hand, uSBS2 and dSBS2 act as the upstream activating site and operator for gene activation and repression upon Tk-SurR binding, respectively, in the regulation of FNOR2 expression. The binding of Tk-SurR to dSBS2 reduces excessive expression of the FNOR2
It is unclear why two FNORs are present on T. kodakarensis genome. Most archaea belonging to the order Thermococcales contain two kinds of FNORs on the genomes, and SBSs are located in their predicted promoter regions (Fig. 7). These archaea are all S0-reducing organisms and possess Tk-SurR orthologs. It is likely that the expressions of FNORs are mainly under the regulation of Tk-SurR in response to S0, as in the case of T. kodakarensis.

Distribution of FNOR orthologues in the order Thermococcales. Clustering of FNOR α- and β-subunit genes in the genomes of the order Thermococcales. White and gray arrows indicate the ORFs for FNOR α- and β-subunits, respectively. Protein accession numbers of GenBank or NCBI reference sequence are displayed below the ORFs. SBSs (GTTn3AAC) located within 100 bp upstream of each start codon sequence are shown in circle
References
Amo T, Atomi H, Imanaka T (2002) Unique presence of a manganese catalase in a hyperthermophilic archaeon, Pyrobaculum calidifontis VA1. J Bacteriol 184:3305–3312. doi:10.1128/JB.184.12.3305-3312.2002
Antelmann H, Helmann JD (2011) Thiol-based redox switches and gene regulation. Antioxid Redox Signal 14:1049–1063. doi:10.1089/ars.2010.3400
Atomi H, Fukui T, Kanai T, Morikawa M, Imanaka T (2004) Description of Thermococcus kodakaraensis sp. nov., a well studied hyperthermophilic archaeon previously reported as Pyrococcus sp. KOD1. Archaea 1:263–267. doi:10.1155/2004/204953
Bell SD, Jackson SP (2000) Mechanism of autoregulation by an archaeal transcriptional repressor. J Biol Chem 275:31624–31629. doi:10.1074/jbc.M005422200
Blombach F, Smollett KL, Grohmann D, Werner F (2016) Molecular mechanisms of transcription initiation—structure, function, and evolution of TFE/TFIIE-like factors and open complex formation. J Mol Biol 428:2592–2606. doi:10.1016/j.jmb.2016.04.016
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254. doi:10.1016/0003-2697(76)90527-3
Fiala G, Stetter KO (1986) Pyrococcus furiosus sp. nov. represents a novel genus of marine heterotrophic archaebacteria growing optimally at 100 °C. Arch Microbiol 145:56–61. doi:10.1007/BF00413027
Fukuda W, Morimoto N, Imanaka T, Fujiwara S (2008) Agmatine is essential for the cell growth of Thermococcus kodakaraensis. FEMS Microbiol Lett 287:113–120. doi:10.1111/j.1574-6968.2008.01303.x
Geiduschek EP, Ouhammouch M (2005) Archaeal transcription and its regulators. Mol Microbiol 56:1397–1407. doi:10.1111/j.1365-2958.2005.04627.x
Gindner A, Hausner W, Thomm M (2014) The TrmB family: a versatile group of transcriptional regulators in Archaea. Extremophiles 18:925–936. doi:10.1007/s00792-014-0677-2
Grohmann D, Werner F (2011) Recent advances in the understanding of archaeal transcription. Curr Opin Microbiol 14:328–334. doi:10.1016/j.mib.2011.04.012
Hillion M, Antelmann H (2015) Thiol-based redox switches in prokaryotes. Biol Chem 396:415–444. doi:10.1515/hsz-2015-0102
Jäger D, Förstner KU, Sharma CM, Santangelo TJ, Reeve JN (2014) Primary transcriptome map of the hyperthermophilic archaeon Thermococcus kodakarensis. BMC Genom 15:684. doi:10.1186/1471-2164-15-684
Johnsen U, Sutter JM, Schulz AC, Tästensen JB, Schönheit P (2015) XacR—novel transcriptional regulator of d-xylose and l-arabinose catabolism in the haloarchaeon Haloferax volcanii. Environ Microbiol 17:1663–1676. doi:10.1111/1462-2920.12603
Jun SH, Reichlen MJ, Tajiri M, Murakami KS (2011) Archaeal RNA polymerase and transcription regulation. Crit Rev Biochem Mol Biol 46(1):27–40. doi:10.3109/10409238.2010.538662
Kanai T, Akerboom J, Takedomi S, van de Werken HJ, Blombach F, van der Oost J, Murakami T, Atomi H, Imanaka T (2007) A global transcriptional regulator in Thermococcus kodakaraensis controls the expression levels of both glycolytic and gluconeogenic enzyme-encoding genes. J Biol Chem 282:33659–33670. doi:10.1074/jbc.M703424200
Koltai H, Weingarten-Baror C (2008) Specificity of DNA microarray hybridization: characterization, effectors and approaches for data correction. Nucleic Acids Res 36:2395–2405. doi:10.1093/nar/gkn087
Kosower NS, Kosower EM (1995) Diamide: an oxidant probe for thiols. Methods Enzymol 251:123–133. doi:10.1016/0076-6879(95)51116-4
Leyn SA, Rodionov DA (2015) Comparative genomics of DtxR family regulons for metal homeostasis in Archaea. J Bacteriol 197:451–458. doi:10.1128/JB.02386-14
Lim JK, Jung HC, Kang SG, Lee HS (2017) Redox regulation of SurR by protein disulfide oxidoreductase in Thermococcus onnurineus NA1. Extremophiles 21:491–498. doi:10.1007/s00792-017-0919-1
Lipscomb GL, Keese AM, Cowart DM, Schut GJ, Thomm M, Adams MW, Scott RA (2009) SurR: a transcriptional activator and repressor controlling hydrogen and elemental sulphur metabolism in Pyrococcus furiosus. Mol Microbiol 71:332–349. doi:10.1111/j.1365-2958.2008.06525.x
Lipscomb GL, Schut GJ, Scott RA, Adams MWW (2017) SurR is a master regulator of the primary electron flow pathways in the order Thermococcales. Mol Microbiol. doi:10.1111/mmi.13668
Liu H, Orell A, Maes D, van Wolferen M, Lindås AC, Bernander R, Albers SV, Charlier D, Peeters E (2014) BarR, an Lrp-type transcription factor in Sulfolobus acidocaldarius, regulates an aminotransferase gene in a β-alanine responsive manner. Mol Microbiol 92:625–639. doi:10.1111/mmi.12583
Matsumi R, Manabe K, Fukui T, Atomi H, Imanaka T (2007) Disruption of a sugar transporter gene cluster in a hyperthermophilic archaeon using a host-marker system based on antibiotic resistance. J Bacteriol 189:2683–2691. doi:10.1128/JB.01692-06
Nagaoka E, Hidese R, Imanaka T, Fujiwara S (2013) Importance and determinants of induction of cold-induced DEAD RNA helicase in the hyperthermophilic archaeon Thermococcus kodakarensis. J Bacteriol 195:3442–3450. doi:10.1128/JB.00332-13
Ochs SM, Thumann S, Richau R, Weirauch MT, Lowe TM, Thomm M, Hausner W (2012) Activation of archaeal transcription mediated by recruitment of transcription factor B. J Biol Chem 287:18863–18871. doi:10.1074/jbc.M112.365742
Reeve JN (2003) Archaeal chromatin and transcription. Mol Microbiol 48:587–598. doi:10.1046/j.1365-2958.2003.03439.x
Santangelo TJ, Cubonová L, Reeve JN (2008) Shuttle vector expression in Thermococcus kodakaraensis: contributions of cis elements to protein synthesis in a hyperthermophilic archaeon. Appl Environ Microbiol 74:3099–3104. doi:10.1128/AEM.00305-08
Santangelo TJ, Cuboňová L, Reeve JN (2011) Deletion of alternative pathways for reductant recycling in Thermococcus kodakarensis increases hydrogen production. Mol Microbiol 81:897–911. doi:10.1111/j.1365-2958.2011.07734.x
Sato T, Fukui T, Atomi H, Imanaka T (2003) Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J Bacteriol 185:210–220. doi:10.1128/JB.185.1.210-220.2003
Sato T, Fukui T, Atomi H, Imanaka T (2005) Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl Environ Microbiol 71:3889–3899. doi:10.1128/AEM.71.7.3889-3899.2005
Schulz S, Gietl A, Smollett K, Tinnefeld P, Werner F, Grohmann D (2016) TFE and Spt4/5 open and close the RNA polymerase clamp during the transcription cycle. Proc Natl Acad Sci USA 113:E1816–E1825. doi:10.1073/pnas.1515817113
Soler N, Justome A, Quevillon-Cheruel S, Lorieux F, Le Cam E, Marguet E, Forterre P (2007) The rolling-circle plasmid pTN1 from the hyperthermophilic archaeon Thermococcus nautilus. Mol Microbiol 66:357–370. doi:10.1111/j.1365-2958.2007.05912.x
Vierke G, Engelmann A, Hebbeln C, Thomm M (2003) A novel archaeal transcriptional regulator of heat shock response. J Biol Chem 278:18–26. doi:10.1074/jbc.M209250200
Werner F, Grohmann D (2011) Evolution of multisubunit RNA polymerases in the three domains of life. Nat Rev Microbiol 9:85–98. doi:10.1038/nrmicro2507
Yang H, Lipscomb GL, Keese AM, Schut GJ, Thomm M, Adams MW, Wang BC, Scott RA (2010) SurR regulates hydrogen production in Pyrococcus furiosus by a sulfur-dependent redox switch. Mol Microbiol 77:1111–1122. doi:10.1111/j.1365-2958.2010.07275.x
Acknowledgements
This study was supported by a Grant from the Japan Society for the Promotion of Science (JSPS) KAKENHI (26292045) to S.F. and KAKENHI (17H05027) to R.H. Bioinformatic analysis was supported by a Grant for Individual Special Research, provided by Kwansei-Gakuin University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Albers.
Electronic supplementary material
Below is the link to the electronic supplementary material.
792_2017_952_MOESM1_ESM.eps
Supplemental Fig. 1. Targeted disruption of the surR gene by homologous recombination. PCR analysis of the DNA region containing the disrupted surR in the DTS strain. The sizes of the PCR fragments obtained from the 5′ and 3′ flanking regions of surR with the primers TkSurR-Fw1 and TkSurR-Rv1 are 2.7 kbp for the KU216 strain and 2.0 kbp for the DTS strain. (EPS 1186 kb)
Rights and permissions
About this article

Cite this article
Hidese, R., Yamashita, K., Kawazuma, K. et al. Gene regulation of two ferredoxin:NADP+ oxidoreductases by the redox-responsive regulator SurR in Thermococcus kodakarensis . Extremophiles 21, 903–917 (2017). https://doi.org/10.1007/s00792-017-0952-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-017-0952-0