
Abstract
Objectives
To determine the role of angiogenic factor with G-patch and FHA domain 1 (AGGF1) in inflammatory response of human dental pulp cells (DPCs) and the underneath mechanism and to explore its role in angiogenesis.
Materials and methods
The expression of AGGF-1 in human healthy and inflammatory pulp tissues was detected by immunohistochemistry. RT-qPCR and Western blot were used to evaluate the expression of AGGF1 in DPCs stimulated by lipopolysaccharide (LPS). After AGGF1 was knocked down, the expression of LPS-induced inflammatory cytokines in DPCs was quantified by RT-qPCR and ELISA. Immunofluorescence and Western blot were used to assess the activation of NF-κB signaling. Inflammatory cytokines were detected by RT-qPCR and ELISA in DPCs pretreated with NF-κB pathway inhibitors before LPS stimulation, and then the effect of AGGF1 on angiogenesis was also evaluated.
Results
AGGF1 expression increased in inflammatory dental pulp tissues. In DPCs stimulated by LPS, AGGF1 was upregulated in a dose-dependent manner (P < 0.05). In AGGF1 knockdown cells, the expression of IL-6, IL-8, and monocyte chemoattractant protein-1 (MCP-1/CCL-2) increased by LPS stimulation (P < 0.001). Nuclear translocation of p65 was promoted, and the addition of NF-κB inhibitors inhibited the expression of inflammatory factors. Meanwhile, knockdown of AGGF1 inhibited vascularization.
Conclusions
AGGF1 inhibited the synthesis of inflammatory cytokines through NF-κB signaling pathway and promoted the angiogenesis of DPCs.
Clinical relevance
This study might shed light in the treatment of pulpitis and regeneration of dental pulp tissues; however, more clinical trials are required to validate these findings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As the only soft tissue inside the tooth, the health of dental pulp tissues is of great significance for the normal function of teeth. Its function includes repair and regeneration, nutrition supply, and disease defense. After removal of the inflamed pulp tissue, the tooth vitality is lost and fragility increases, which can cause many complications [1], and pulpitis is the main disease leading to the removal of pulp tissues. Therefore, how to control dental pulp inflammation is an urgent problem.
Bacteria and their metabolites enter the pulp through deep caries, abrasion, fissures, apical foramen, and collateral root canals and then cause bacterial infection [2]. At the same time, a variety of immune cells, such as dental pulp cells (DPCs), macrophages, and dendritic cells, contained in dental pulp tissues, express toll-like receptors (TLRs) to recognize pathogen-associated molecular pattern molecules and initiate immune responses [3,4,5]. In pulpitis, the immune response mediates changes in blood vessels through cells expressing CD14, TLR-2, and TLR4 and promotes the expression of interleukin (IL)-1, IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1/CCL-2), and other inflammatory cytokines [6]. Gram-negative bacterial cell wall lipopolysaccharide (LPS) is the main ligand of TLR4. When TLR4 recognizes LPS, it activates inhibitor of nuclear factor kappa B (NF-κB) alpha (IκBα) kinase to phosphorylate IκBα, and NF-κB is released from the cytoplasmic NF-κB/IκBα complex. The nuclear localization domain is activated and exposed to form a p50/p65 dimer, which rapidly undergoes nuclear translocation, thereby initiating the expression of target genes, such as tumor necrosis factor α (TNF-α) and IL-1 [7]. Therefore, inhibiting the activation of NF-κB signaling pathway may be effective for the treatment of pulpitis.
Recent studies have found that angiogenic factor with G-patch and FHA domain 1 (AGGF1), as a new type of anti-inflammatory factor, can inhibit inflammation triggered with TNF-α in endothelial cells by weakening NF-κB signaling pathway, such as reducing promoter activity and NF-κB p65 phosphorylation [8]. AGGF1 is a vascular endothelial-derived protein with G-patch and FHA domains. It was first identified in patients with Klippel-Trenaunay syndrome [9]. A series of studies showed that AGGF1 participated in cardiovascular diseases, neurodegenerative diseases, cancer, aging, and other diseases by inhibiting neuroinflammation, apoptosis, and tumorigenesis, promoting angiogenesis, and inducing autophagy [10,11,12,13,14]. Therefore, we speculated that AGGF1 may also play a role in the pathogenesis of pulpitis, and no report on this respect has arisen in this field at present.
Inflammation control is vital to preserve the vital pulp tissues of the affected teeth to the greatest extent, and regeneration of dental pulp tissues is also very important, of which revascularization is considered as a key part [15]. Previous studies on dental pulp revascularization were mainly achieved by stimulating dental pulp stem cells, root apical papilla stem cells, and bone marrow mesenchymal stem cells to differentiate into vascular endothelial cells to promote angiogenesis [16,17,18], or adding angiogenesis-related factors [19]. According to an earlier research, AGGF1 was highly expressed in vascular endothelial cells and was closely related to the angiogenesis [20].
Based on the abovementioned studies, AGGF1 can play a dual role in inhibiting inflammation and promoting angiogenesis in endothelial cells [10, 20]. Therefore, our present study was to investigate the effect of AGGF1 on DPC inflammation induced by LPS and explore the underneath mechanism. Meanwhile, the role of AGGF in pulp angiogenesis was also evaluated.
Materials and methods
Dental pulp sample collection
A total of 6 patients, who came to the Department of Oral and Maxillofacial Surgery, School of Stomatology, Shandong University, for third molar extraction, were recruited in this study. All the patients were general healthy with no systemic diseases. One wisdom tooth was obtained from each patient (n = 6; 5 males, 1 female; aged 20 to 30). Three patients had healthy third molars, and the other three were diagnosed with symptomatic irreversible pulpitis. Patients in the symptomatic irreversible pulpitis group presented a history of sensitivity to hot and cold tests with spontaneous pain. This research protocol was approved by the Medical Ethics Committee of Shandong University School of Stomatology (Protocol Number: GR201801). The teeth were immediately rinsed with a phosphate buffer solution (PBS; Hyclone, Logan, UT, USA) after extraction. Then, they were split longitudinally into two halves with a dental diamond disc, and the pulp was clamped with forceps and placed in 4% paraformaldehyde and later paraffin embedded for hematoxylin and eosin (H&E) staining and immunohistochemistry analyses. Another five participants (aged 15 to 24) who sought for third molar extraction were also recruited in this study. Informed consents were obtained from these patients, and their third molars without caries and periodontal diseases were collected after the extraction. This research protocol was approved by the Medical Ethics Committee of School of Stomatology, Shandong University (Protocol Number: GR201801). The extracted teeth were then rinsed with PBS and split longitudinally into two halves with a dental diamond disc, and the pulps were clamped with forceps. The pulp tissues were used for primary cell culture.
Histology analysis
Five-micrometer-thick serial sections were prepared from each specimen and placed on a glass slide. Then, 5 slices were selected for each sample, baked at 60 °C for 2 h, and dewaxed in xylene. After rehydrated in gradient alcohol, the slices were stained with hematoxylin (Solarbio, Beijing, China) for 2 min and then with eosin (Solarbio) for 4 min. Histopathological changes within the tissue were observed under a light microscope (OLYMPUS BX51, Tokyo, Japan).
Immunohistochemistry
After rehydration, the tissue sections were placed in antigen retrieval solution and incubated with polyclonal rabbit anti-AGGF1 (1:100; Proteintech, Chicago, IN, USA) at 4 °C overnight. Immunohistochemical detection was performed by using a streptavidin peroxidase (SP) kit (Zhongshanjinqiao, Beijing, China) according to the manufacturer’s instructions. The specific reaction was visualized using diaminobenzidine substrate kit (DAB; Zhongshanjinqiao). Nucleus were counterstained with hematoxylin for 5 s. Yellow-brown staining in the cells was recorded as AGGF1–positive immunostaining. Three slides from each specimen were randomly selected to calculate staining intensity of AGGF1 using ImageJ 1.44 software (NIH, Bethesda, Maryland, USA).
Cell culture
Pulp tissues were washed three times with PBS and then cut into small pieces and digested with 3 mg/mL collagenase I (Sigma-Aldrich Chemie, Schnelldorf, Germany) and 4 mg/mL Dispase II (Sigma) for 1 h in a 37 °C shaker. Afterwards, single-cell suspension was inoculated into a 25-cm2 air-permeable flask, cultured with alpha minimal essential medium (α-MEM; Hyclone) supplemented with 20% fetal bovine serum (FBS; BioInd, Kibbutz, Israel), and placed at 37 °C in a humidified 5% CO2 incubator. The medium was changed every three days, and the cells were passaged with 0.25% trypsin-EDTA (Solarbio) solution until 80–90% confluence. Passages 3–5 were used for subsequent experiments.
Cell viability assay
The effect of LPS on the viability of DPCs was evaluated by CCK-8 cell counting kit (CCK-8; Dojindo Laboratories, Kumamoto, Japan). The DPCs were seeded into 96-well plates and cultured in a 37 °C incubator overnight. The cells were then stimulated with 0, 0.1, 1, 5, and 10 μg/mL LPS (Solarbio) for 24 and 48 h and then were incubated with α-MEM containing 10 μL test reagents for 2.5 h at 37 °C. The optimal absorbance at 450 nm was determined using a microplate reader (SPECTROstar Nano; BMG Labtech, Offenburg, Germany).
Real-time quantitative polymerase chain reaction
To analyze the effect of LPS on the expression of AGGF1, DPCs were stimulated with 0, 0.01, 0.1, 1, or 5 μg/mL LPS for 2, 6, 12, and 24 h. RNAs were isolated by TRIzol (Takara, Kusatsu, Japan), messenger RNA (mRNA) concentrations were measured by NanoDrop 2000 ultramicro spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and mRNA was reverse-transcribed to complementary DNA (cDNA) by a PrimeScript™ RT kit (Takara). Real-time quantitative polymerase chain reaction (RT-qPCR) experiments were then performed with LightCycler 96 Real-Time PCR System (Roche, Basel, Switzerland) to detect the gene level of AGGF1. The expression of AGGF1 was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and its relative level was calculated using the 2− (ΔCt) method. RT-qPCR primers were listed in Table 1.
Transfection with siRNA and effects of AGGF1 knockdown on inflammatory cytokine expression
The DPCs were seeded in a 24-well plate at a density of 3 × 105 cells per well and achieved a 40–60% confluence after incubation at 37 °C overnight. Then, the cells were transfected with 10, 30, and 50 nM fluorescence small interfering RNA (FAM-siRNA; GenePharma, Shanghai, China) using Lipofectamine 2000 (Invitrogen, Cal, USA) to determine the transfection efficiency. To obtain the best AGGF1 gene knockdown effect, three different si-AGGF1 sequences (469, 827, 911) were designed. The sequences for all siRNA were listed in Table 2. After 6 h transfection, the reduced serum medium (Opti-MEM; Gibco, Grand Island, NY, USA) was replaced with 10% FBS α-MEM and cultured for an additional 24 or 48 h. The sequence with the strongest knockdown effect was detected by RT-qPCR and selected for the follow-up experiment.
The DPCs were transfected with 30 nM AGGF1 siRNA using Lipofectamine 2000 (5 μL/well). After transfection for 6 h, the DPCs were then stimulated with LPS in serum-free medium for 2, 6, 12, and 24 h. Cell supernatants were collected to determine the protein levels of IL-6, IL-8, and CCL-2 by enzyme-linked immunosorbent assay (ELISA) kits (BioLegend, San Diego, CA, USA). Gene expression of IL-6, IL-8, and CCL-2 was detected by RT-qPCR. The RT-qPCR primers for IL-6, IL-8, and CCL2 were listed in Table 1.
Enzyme-linked immunosorbent assay
The conditional medium collected from RT-qPCR assay was used to measure the protein levels of IL-6, IL-8, and CCL-2 by ELISA kits (BioLegend). The absorbance values at 450 and 570 nm were measured with a microplate reader (SPECTROstar Nano), and the protein levels of IL-6, IL-8, and CCL-2 were calculated according to the standard curve.
Immunofluorescence
The DPCs were seeded in 24-well plates at a density of 5 × 104 cells per well and then were transfected with AGGF1 siRNA for 6 h followed by LPS stimulation for 1.5 h. The cells were fixed with 4% paraformaldehyde for 15 min and then permeabilized with 0.5% Triton X-100 (Solarbio) for 10 min. The cells were blocked with 10% goat serum and then incubated with primary antibodies against NF-κB RelA/p65 (1:400; Cell Signaling Technology, MA, USA) at 4 °C overnight. On the next day, the cells were incubated with a secondary antibody (Alexa Fluor 594-conjugated goat anti-rabbit IgG at 1:500; Proteintech) in the dark at 37 °C for 1 h. Nuclei were stained with 2-(4-amidinophenyl)-6-indolecarba-midine dihydrochloride (DAPI; Proteintech). Images were observed under a fluorescence microscope (OLYMPUS IX73, Tokyo, Japan) in a dark room and captured by a camera and imaging software (OLYMPUS cellSens Standard 1.17).
Protein isolation and Western blot analysis
To analyze the effect of LPS on the expression of AGGF1, DPCs were stimulated with 0, 0.1, 1, or 5 μg/mL LPS for 12 h. To detect the effect of AGGF1 knockdown on NF-κB signaling pathway, DPCs were transfected with AGGF1 siRNA for 6 h and then stimulated with LPS for 1.5 h. The plate was washed thrice with pre-chilled PBS, and then cells were lysed on ice with a mixture of RIPA lysis buffer (Solarbio) and 1% phosphatase inhibitor (Boster, Wuhan, Hubei, China) at a ratio of 100:1. After sonication, the cells were centrifuged at 12,000 r/min for 15 min. The supernatant was discarded, and then the protein concentration was measured by BCA protein detection kit (KeyGEN BioTECH, Nanjing, Jiangsu, China). Equal loading quantity of protein (20 μg/lane) was electrophoresed and separated by 10% gels for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto polyvinylidene fluoride membranes (PVDF; Millipore, Billerica, MA, USA). After being steeped in 5% skimmed milk powder for 1 h, PVDF membranes were incubated with primary antibodies overnight at 4 °C. The primary antibodies were rabbit anti-AGGF1 (1:500), rabbit anti-NF-κB p65 (1:1000; Cell Signaling Technology), rabbit anti-phosphorylated NF-κB p65 (1:1000; Cell Signaling Technology), and GAPDH (1:10000, Proteintech). The membranes were incubated with horseradish peroxidase-conjugated secondary antibody (1:10000; Proteintech) for 1 h at room temperature. Immunoreactive bands were visualized by enhanced chemiluminescence reagents (Millipore) and scanned by an extra-sensitive imager (Amersham Imager 600; GE Healthcare Life Sciences, Pittsburgh, PA, USA). ImageJ was used to quantify the fold expression of the target proteins.
Effects of NF-κB signaling inhibitor on inflammatory cytokine expression
After AGGF1 knockdown, DPCs were incubated in α-MEM containing 5 μM BAY11-7082 (Abcam, Cambridge, UK) for 1.5 h, and then LPS with serum-free medium was applied for another 24 h. Cell supernatants were collected to determine the protein levels of IL-6, IL-8, and CCL-2 by ELISA. Gene expression of IL-6, IL-8, and CCL-2 was detected by RT-qPCR.
In vitro Matrigel angiogenesis assay and effects of AGGF1 knockdown on vascular endothelial growth factor expression
The DPCs were transfected with siRNA for 6 h and then incubated with 10% FBS α-MEM for another 1, 3, and 7 d. Cell supernatants were collected to determine the protein level of vascular endothelial growth factor (VEGF) by ELISA kits (BioLegend), and then the mRNA expression of VEGF was detected by RT-qPCR. The 48-well plates were coated with 120 μL Matrigel (Corning Life Sciences, Kennebunkport, ME) per well on ice and then placed in a 37 °C incubator for 30 min to solidify the Matrigel. Afterwards, cells transfected with siAGGF1 or siNC were added onto Matrigel and incubated at 37 °C for 2, 4, 8, 12, and 24 h. Images were observed under a fluorescence microscope (OLYMPUS IX73), and ImageJ was used to quantify the segments, nodes, meshes, and tubule length.
Statistical analysis
All data were represented as mean ± standard deviation (SD). Multiple comparisons were analyzed by one-way ANOVA, and variance between two groups was compared by two-way t test with GraphPad Prism software (version 6, by MacKiev Software, Boston, MA, USA). Value of P < 0.05 was considered statistically significant.
Results
Histological assessment of healthy and inflammatory pulp tissues
Healthy dental pulp tissue presented as a loose connective tissue with fibers, and a large number of star-shaped fibroblasts could be seen in the tissue (Fig. 1a). However, the inflammatory dental pulp tissue showed obvious infiltration of inflammatory cells, and the pulpal blood vessels expanded and congested (Fig. 1b).

Comparison of the expression of AGGF-1 in human healthy and inflammatory dental pulp tissues. a Representative H&E images of healthy tissue. b Representative H&E image of inflammatory tissue. c Representative immunohistochemistry image of healthy tissue. d Representative immunohistochemistry image of inflammatory tissue (magnification ×200, scale bar 100 μm). e Quantitative analysis of AGGF-1 expression in two groups. ***P < 0.001
Expression of AGGF1 in healthy and inflammatory human dental pulp tissues
In order to investigate the correlation between AGGF1 and pulpitis, we first detected the expression of AGGF1 in dental pulp tissues by immunohistochemical staining. The results showed that AGGF1 was mainly located in the cytoplasm (Fig. 1c, d), and the rate of AGGF1 positive area in inflammatory pulp tissues was much higher than that in healthy pulp tissues (P < 0.001) (Fig. 1e).
Effect of LPS on the expression of AGGF1
To investigate whether AGGF1 was involved in LPS-induced inflammation, DPCs were stimulated with different concentrations of LPS. The results of CCK-8 showed that 0.1, 1, 5, and 10 μg/mL LPS had no significant effect on cell viability for 24 and 48 h (Fig. 2a). When DPCs were stimulated with 0.01, 0.1, 1, or 5 μg/mL LPS for 2, 6, 12, and 24 h, the gene level of AGGF1 was significantly upregulated in a concentration-dependent manner from 0.01 to 1 μg/mL (P < 0.05) (Fig. 2b). Furthermore, the protein level of AGGF1 was significantly upregulated when the concentration of LPS was 1 μg/mL (P < 0.001) (Fig. 2c). Thus, 1 μg/mL LPS was used in the subsequent experiments.

Effect of LPS stimulation on the production of AGGF1. a The cytotoxic effect of 0.1, 1, 5, and 10 μg/mL LPS on DPCs was detected by CCK-8 assay after treatment for 24 and 48 h. b mRNA expression of AGGF-1 in DPCs treated with LPS. c The protein level of AGGF1 was determined by Western blot. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001
SiRNA transfection and AGGF1 knockdown efficiency selection
To reversely verify the role of AGGF1 in LPS-induced inflammation, AGGF1 was knocked down with siRNA in DPCs. According to the results of FAM-siRNA transfection, the transfection concentration of 30 nM could obtain the optimal transfection efficiency (Fig. 3a–c), and then siRNA (30 nM) was selected for the subsequent experiments. To select the sequence with optimal silencing efficiency for AGGF1, RT-qPCR was used to evaluate the expression levels at 24 and 48 h after transfection. The results indicated that sequence 827 showed the highest knockdown efficiency (P < 0.01) (Fig. 3d, e). Therefore, this sequence was selected for the subsequent experiments.

Effects of AGGF1 knockdown. a DPCs transfected with 30 nM FAM-siRNA were observed under an inverted microscope. b Representative image under a fluorescence microscope after transfection. c Representative merged images of a and b (magnification ×100, scale bar 100 μm). d, e DPCs were transfected with siRNA for 6 h, and total RNA were extracted after 24 and 48 h, the relative expression of AGGF1. **P < 0.01, ***P < 0.001, and ****P < 0.0001
Effects of AGGF1 knockdown on inflammatory cytokine expression in LPS-induced DPCs
RT-qPCR results revealed that AGGF1 knockdown significantly promoted IL-6, IL-8, and CCL-2 expression in gene level after 2, 6, 12, and 24 h of LPS treatment (P < 0.0001) (Fig. 4a–c). As for the ELISA result, AGGF1 knockdown strongly promoted the secretion of IL-6 after 6, 12, and 24 h of LPS stimulation (P < 0.01), but it had no obvious difference at 2 h (Fig. 4d). As for IL-8 and CCL-2 secretion, AGGF1 silencing significantly upregulated protein level of the two cytokines (P < 0.001) (Fig. 4e, f). Therefore, AGGF1 knockdown could significantly promote the expression of inflammatory cytokines at both gene and protein levels in DPCs induced by LPS.

Effects of AGGF-1 knockdown on LPS-induced IL-6, IL-8, and CCL-2 expression. a–c mRNA expression of IL-6, IL-8, and CCL2 was detected by RT-qPCR. d–f The protein levels of IL-6, IL-8, and CCL2 were detected by ELISA. *P < 0.05 and ****P < 0.0001 compared with NC group; ##P < 0.01, ###P < 0.001, and ####P < 0.0001 compared with LPS alone group. NC, negative control; Si, Si-AGGF1; Si + LPS, Si-AGGF1 + LPS
Effects of AGGF1 knockdown on NF-κB signaling pathway
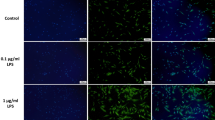
To investigate the molecular mechanism under which AGGF1 inhibited inflammation in DPCs, the activation of NF-κB signaling pathway was examined. The activation of NF-κB signaling pathway was dependent upon the phosphorylation of p50/p65 and the subsequent translocation of NF-κB p65 from cytoplasm to nucleus. The results of Western blot indicated that AGGF1 knockdown significantly promoted phosphorylation of NF-κB p65 (p-p65) (P < 0.0001) (Fig. 5a). Immunofluorescence was used to detect nuclear translocation of NF-κB p65. It was found that NF-κB p65 of untreated cells was predominantly localized in cytoplasm, and when stimulated with LPS, NF-κB p65 was partially transferred to the nucleus. However, when AGGF1 was knocked down, more NF-κB p65 was transferred to the nucleus (Fig. 5b). To further confirm the involvement of NF-κB signaling pathway, 5 μM p65 inhibitor (BAY11-7082) was used to determine its effect on the expression of inflammatory cytokines. The results of RT-qPCR and ELISA showed that BAY11-7082 significantly inhibited mRNA and protein expressions of IL-6, IL-8, and CCL-2 in AGGF1 knockdown DPCs with LPS stimulation (P < 0.05) (Fig. 5c, d).

Effects of AGGF1 knockdown on LPS-induced activation of NF-κB pathway. a The protein level of p-p65 was determined by Western blot. b The nuclear translocation of NF-κB p65 was evaluated by immunofluorescence staining (NF-κB p65, green fluorescent signals; DAPI, blue signals; magnification: ×200, scale bar 50 μm). c mRNA expression of IL-6, IL-8, and CCL-2 after stimulated with BAY11-7082. d The protein levels of IL-6, IL-8, and CCL-2 after stimulated with BAY11-7082. ****P < 0.0001 compared with NC group; ####P < 0.0001 compared with LPS alone group; $P < 0.05 and $$P < 0.0001 compared with Si-AGGF1 + LPS group
Knockdown of AGGF1 inhibited the angiogenesis of DPCs
After DPCs were seeded onto solidified Matrigel, cells began to attach, migrate, align, and form tubule-like structures. After 4 h incubation, the formation of vascular-like structures could be clearly seen in untreated DPC group, whereas AGGF1 knockdown group formed less vessel-like structures (Fig. 6a). After 6 h culture, the tubule-like structures remained relatively stable in cells but began to degrade after 24 h (Fig. 6a). Compared with untreated DPCs, when incubated for 4 h, the length of tubules (P < 0.001) became shorter and the number of meshes (P < 0.01), segments (P < 0.001) and nodes (P < 0.001) decreased after AGGF1 was knocked down (Fig. 6b). The results of RT-qPCR and ELISA showed that the mRNA and protein expressions of VEGF were significantly lower in AGGF1 knockdown group than in untreated DPC group (P < 0.01) (Fig. 6c, d).

Effects of AGGF1 knockdown on the angiogenesis of DPCs. a Micrographs of vessel formation on Matrigel after 2, 4, 8, 12, and 24 h incubation (magnification: ×40, scale bar 200 μm). b Quantification of segments, nodes, meshes, and tubule length after 4 h incubation. c mRNA expression of VEGF was detected by RT-qPCR. d The protein level of VEGF was detected by ELISA. **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with NC group
Discussion
Surrounded by hard tissues, pulps are connected to the periapical soft tissues only through a small apical foramen. Once pulp tissues are damaged, it is difficult to recover by themselves [21]. Teeth with perfect root canal treatment are still easier to lose even with their life span prolonged [1]. Therefore, regeneration of dental pulp tissues is the dream of generations of dentists. The principles of tissue engineering and biological methods were applied to dental pulp regeneration to change dental pulp from an irreversible disease state to a healthy state where it can function normally [22]. There are two key steps in the regeneration of dental pulp tissues. The first step is the control of inflammation at different levels of the tissues [23]. The second step is the regeneration of dental pulp tissues, including vascular regeneration, nerve regeneration, and dentin regeneration [24]. The purpose of our study is to provide a strategy for pulp tissue regeneration by controlling inflammation and promoting vascular regeneration.
Pulpitis is a typical inflammation of pulp tissues, when the integrity of the enamel or cementum is damaged, bacteria and their metabolites invade the pulpal space through dentin tubules, and then pulp inflammation is triggered [25]. Earlier research found that LPS has strong pathogenicity and biological toxicity, and it is a potent inducer of pulpitis [26]. LPS can directly damage local tissues by releasing inflammatory mediators and cytokines, such as tumor necrosis factors and interleukins [27, 28]. Previous studies found that different concentrations of LPS have different effects on the expression of genes in dental pulp cells [29, 30]. Therefore, we set a concentration gradient to detect the concentration of LPS which has the strongest promotive effect of AGGF1 in DPCs. During the experiment, it was found that the expression of AGGF1 increased significantly when LPS concentration was 1 μg/mL, so we used this concentration in the subsequent experiments. We also found that the expression of AGGF1 decreased when stimulated with 5 μg/mL, which might be contributed to the immunogenic effect of LPS. High concentrations of LPS triggered inflammatory cascade, caused the destruction of cellular ultrastructures, such as mitochondria, lysosomes, and Golgi apparatus, as well as reduced cellular energy metabolism, sugar utilization, and cellular protein synthesis [31]. IL-6 is a pleomorphic cytokine, which is involved in the response to trauma and infection and development of inflammation and tumors [32]. IL-8, as a major neutrophil chemokine, can be rapidly synthesized locally in the inflammation under the stimulation of LPS [33]. In a previous study in this field, Zehnder et al. measured the mRNA levels of IL-1α, IL-1β, IL-6, and IL-8 in DPCs and found that their expression levels were significantly higher than uninfected cells [34]. Similarly, our study found that after stimulating DPCs with LPS, the mRNA and protein levels of IL-6, IL-8, and CCL-2 were significantly unregulated.
Earlier studies found that the expression of AGGF1 in vascular endothelial cells was upregulated by the stimulation of TNF-α, and AGGF1 could regulate the expression of inflammatory factors in vascular endothelial cells [8]. In lymphoblasts, AGGF1 was enhanced after being stimulated with ionizing radiation, indicating AGGF1 was closely related to inflammation [35]. Studies also found that AGGF1 could inhibit vascular inflammation and improve endothelial function [36]. By using neutralizing antibody to inhibit the expression AGGF1 in myocardial ischemia/reperfusion injury, the expression of inflammation cytokines, such as p65, IL-1β, IL-6, and TNF-α, increased [37]. Since the role of AGGF1 in pulpitis has not been elucidated, we hypothesized that AGGF1 could inhibit the occurrence of the inflammation in dental pulp. In our study, we found that the expression of AGGF1 in inflamed dental pulp tissues was dramatically enhanced compared with healthy tissues, which suggested that AGGF1 was associated with pulp inflammation. Furthermore, when DPCs were stimulated with LPS, the expression of AGGF1 was upregulated. After AGGF1 knockdown with siRNA, in LPS-stimulated DPCs, the expression of inflammatory cytokines, such as IL-6, IL-8, and CCL-2, significantly increased compared with untransfected DPCs.
When stimulated by microbial, mechanical, chemical, and thermal factors, DPCs undergo an inflammatory immune response [38]. DPCs express TLR4, which can act as a receptor of LPS and recognize pathogen-associated molecular pattern molecules (PAMPs), thereby promoting the expression of inflammatory cytokines [39, 40]. NF-κB signaling pathway in DPCs mediated by TLR has important regulatory effects on the immune responses [41]. As an important transcriptional regulatory factor in cells, NF-κB usually binds to IκBα in the form of a p50/p65 heterodimer and is in a resting state [42]. Once NF-κB is activated by stimulations, such as LPS, IκBα is degraded after phosphorylation, which frees and transfers p65 from the cytoplasm to the nucleus [43], and promotes the expression of various inflammatory cytokines [44,45,46,47]. Thus, we speculated that the anti-inflammation mechanism of AGGF1 in DPCs might be relative to its suppressive effects on NF-κB activation. In this present study, we found that when LPS was applied to DPCs transfected with siAGGF1, the phosphorylation of NF-κB p65 was enhanced, and more p65 was translocated to the nucleus. After using NF-κB inhibitors, the expression of IL-6, IL-8, and CCL-2 was downregulated, which was consistent with earlier reports [47]. To sum up, our research clarified that AGGF1 knockdown upregulated the levels of inflammatory cytokines induced by LPS and explored the underneath mechanisms. The proposed mechanism illustration was presented in Fig. 7. However, further study should be performed in the future to clarify whether other signaling pathways are involved in this process.

Putative mechanism for the role of AGGF1 in NF-κB signaling pathway. LPS accelerated the activation of NF-κB signaling pathway via TLR4/MyD88-dependent pattern to upregulate the expression of pro-inflammatory cytokines. Our study demonstrated that LPS promoted AGGF1 expression and AGGF1 inhibited the phosphorylation and nuclear translocation of NF-κB p65, which reduced the expression of LPS-upregulated inflammatory cytokines. LPS, lipopolysaccharide; TLR4, Toll-like receptor 4; MyD88, myeloid differentiation primary response protein 88; NF-κB, nuclear factor kappa B
As for angiogenesis, existing evidences indicated that vascular regeneration of dental pulp is a complex process, and it requires a series of cellular cascades of activities, which are strongly related to cytokines. An earlier study found that scaffold materials containing VEGF could promote the formation of blood vessels and connective tissues in the root canals [48], and the effect was achieved through Wnt/β-catenin signaling pathway [49]. Fibroblast growth factor 2 (FGF-2) could be secreted by dental pulp stem cells or dental fibroblasts [50], and it could promote the proliferation and migration of the cells during angiogenesis [51]. A previous study found that AGGF1 was initially identified as a vascular endothelium-derived protein and promoted angiogenesis as strongly as VEGF [9], while its effect was independent from VEGF [14]. A series of studies confirmed this conclusion. Tian et al. found that the occurrence of vascular disease Klippel-Trenaunay syndrome was related to the overexpression of AGGF1 [9, 36]. During the embryonic development of zebrafish, AGGF1 is necessary for promoting the differentiation of pluripotent hemangioblasts to blood cells [52]. Zhou et al. proved that AGGF1 had a strong ability to block vascular permeability and opened up possibilities for the treatment of coronary artery diseases and myocardial infarctions [53]. Furthermore, AGGF1 overexpression improved blood flow in hindlimb ischemia model of perivascular diseases in mouse [54]. More and more evidences showed that AGGF1 promoted angiogenesis by activating phosphoinositide-3-kinase (PI3K)/protein kinase B (PKB/AKT) signaling pathway and maintained vascular integrity by inhibiting VE-cadherin phosphorylation [52, 55]. Likewise, in our study, we found that when knocked down AGGF1 with siRNA in DPCs, the expression of VEGF mRNA was significantly downregulated and the generation of vascular-like structures were reduced as well, which indicated that AGGF1 promoted dental pulp angiogenesis, but the specific mechanism has not been elucidated, and further in vivo study on dental pulp regeneration will be carried out.
Conclusion
In conclusion, our research verified that more AGGF1–positive cells were found in dental inflamed pulp tissues than in healthy tissues. When DPCs were stimulated with LPS, the expression of AGGF1 was elevated in a dose-dependent manner. AGGF1 knockdown upregulated the expression of inflammatory cytokines induced by LPS through NF-κB signaling pathway and inhibited the formation of vascular-like structures and production of VEGF. These results indicated that AGGF1 plays a dual role in controlling inflammation and promoting angiogenesis in DPCs; therefore, our study provides a new strategy for the treatment of pulpitis and regeneration of dental pulp tissues. However, in vivo experiments should also be performed to evaluate the effect and safety for its clinical application.
References
Caplan DJ, Cai J, Yin G, White BA (2005) Root canal filled versus non-root canal filled teeth: a retrospective comparison of survival times. J Public Health Dent 65:90–96
Bergenholtz G (1981) Inflammatory response of the dental pulp to bacterial irritation. J Endod 7:100–104
Keller JF, Carrouel F, Staquet MJ, Kufer TA, Baudouin C, Msika P, Bleicher F, Farges JC (2011) Expression of NOD2 is increased in inflamed human dental pulps and lipoteichoic acid-stimulated odontoblast-like cells. Innate Immun 17:29–34
Staquet MJ, Carrouel F, Keller JF, Baudouin C, Msika P, Bleicher F, Kufer TA, Farges JC (2011) Pattern-recognition receptors in pulp defense. Adv Dent Res 23:296–301
Tecles O, Laurent P, Zygouritsas S, Burger AS, Camps J, Dejou J, About I (2005) Activation of human dental pulp progenitor/stem cells in response to odontoblast injury. Arch Oral Biol 50:103–108
Stashenko P (1990) Role of immune cytokines in the pathogenesis of periapical lesions. Endod Dent Traumatol 6:89–96
Baldwin AS Jr (1996) The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 14:649–683
Hu FY, Wu C, Li Y, Xu K, Wang WJ, Cao H, Tian XL (2013) AGGF1 is a novel anti-inflammatory factor associated with TNF-alpha-induced endothelial activation. Cell Signal 25:1645–1653
Tian XL, Kadaba R, You SA, Liu M, Timur AA, Yang L, Chen Q, Szafranski P, Rao S, Wu L, Housman DE, DiCorleto PE, Driscoll DJ, Borrow J, Wang Q (2004) Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature 427:640–645
Chen D, Li L, Tu X, Yin Z, Wang Q (2013) Functional characterization of Klippel-Trenaunay syndrome gene AGGF1 identifies a novel angiogenic signaling pathway for specification of vein differentiation and angiogenesis during embryogenesis. Hum Mol Genet 22:963–976
Zhou B, Zeng S, Li L, Fan Z, Tian W, Li M, Xu H, Wu X, Fang M, Xu Y (2016) Angiogenic factor with G patch and FHA domains 1 (Aggf1) regulates liver fibrosis by modulating TGF-beta signaling. Biochim Biophys Acta 1862:1203–1213
Fan C, Ouyang P, Timur AA, He P, You SA, Hu Y, Ke T, Driscoll DJ, Chen Q, Wang QK (2009) Novel roles of GATA1 in regulation of angiogenic factor AGGF1 and endothelial cell function. J Biol Chem 284:23331–23343
Xu W, Zeng S, Li M, Fan Z, Zhou B (2017) Aggf1 attenuates hepatic inflammation and activation of hepatic stellate cells by repressing Ccl2 transcription. J Biomed Res 31:428–436
Lu Q, Yao Y, Hu Z, Hu C, Song Q, Ye J, Xu C, Wang AZ, Chen Q, Wang QK (2016) Angiogenic factor AGGF1 activates autophagy with an essential role in therapeutic angiogenesis for heart disease. PLoS Biol 14:e1002529
Kim JY, Xin X, Moioli EK, Chung J, Lee CH, Chen M, Fu SY, Koch PD, Mao JJ (2010) Regeneration of dental-pulp-like tissue by chemotaxis-induced cell homing. Tissue Eng Part A 16:3023–3031
Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, DenBesten P, Robey PG, Shi S (2002) Stem cell properties of human dental pulp stem cells. J Dent Res 81:531–535
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S (2000) Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A 97:13625–13630
Huang GT, Gronthos S, Shi S (2009) Mesenchymal stem cells derived from dental tissues vs. those from other sources: their biology and role in regenerative medicine. J Dent Res 88:792–806
Tran-Hung L, Laurent P, Camps J, About I (2008) Quantification of angiogenic growth factors released by human dental cells after injury. Arch Oral Biol 53:9–13
Wang W, Li GY, Zhu JY, Huang DB, Zhou HC, Zhong W, Ji CS (2015) Overexpression of AGGF1 is correlated with angiogenesis and poor prognosis of hepatocellular carcinoma. Med Oncol 32:131
Doyon GE, Dumsha T, von Fraunhofer JA (2005) Fracture resistance of human root dentin exposed to intracanal calcium hydroxide. J Endod 31:895–897
Murray PE, Garcia-Godoy F, Hargreaves KM (2007) Regenerative endodontics: a review of current status and a call for action. J Endod 33:377–390
Fouad AF (2011) The microbial challenge to pulp regeneration. Adv Dent Res 23:285–289
Goldberg M, Schmalz G (2011) Toward a strategic plan for pulp healing: from repair to regeneration. Clin Oral Investig 15:1–2
Love RM, Jenkinson HF (2002) Invasion of dentinal tubules by oral bacteria. Crit Rev Oral Biol Med 13:171–183
Hosoya S, Matsushima K (1997) Stimulation of interleukin-1 beta production of human dental pulp cells by Porphyromonas endodontalis lipopolysaccharide. J Endod 23:39–42
Graves DT, Oates T, Garlet GP (2011) Review of osteoimmunology and the host response in endodontic and periodontal lesions. J Oral Microbiol 3
Levin LG, Law AS, Holland GR, Abbott PV, Roda RS (2009) Identify and define all diagnostic terms for pulpal health and disease states. J Endod 35:1645–1657
Wu H, He M, Yang R, Zuo Y, Bian Z (2018) Astrocyte elevated gene-1 participates in the production of pro-inflammatory cytokines in dental pulp cells via NF-κB signalling pathway. Int Endod J 51:1130–1138
Qiao W, Huang Y, Bian Z, Sun X, Wang X, Gao Q, Peng Y, Meng L (2019) Lipopolysaccharide-induced DNA damage response activates nuclear factor κB signalling pathway via GATA4 in dental pulp cells. Int Endod J 52:1704–1715
Bucklin SE, Morrison DC (1995) Differences in therapeutic efficacy among cell wall-active antibiotics in a mouse model of gram-negative sepsis. J Infect Dis 172:1519–1527
Taga T, Kishimoto T (1997) Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol 15:797–819
Remick DG (2005) Interleukin-8. Crit Care Med 33:S466–S467
Zehnder M, Delaleu N, Du Y, Bickel M (2003) Cytokine gene expression—part of host defence in pulpitis. Cytokine 22:84–88
Long XH, Zhao ZQ, He XP, Wang HP, Xu QZ, An J, Bai B, Sui JL, Zhou PK (2007) Dose-dependent expression changes of early response genes to ionizing radiation in human lymphoblastoid cells. Int J Mol Med 19:607–615
Hu Y, Li L, Seidelmann SB, Timur AA, Shen PH, Driscoll DJ, Wang QK (2008) Identification of association of common AGGF1 variants with susceptibility for Klippel-Trenaunay syndrome using the structure association program. Ann Hum Genet 72:636–643
Liu Y, Yang H, Song L, Li N, Han QY, Tian C, Gao E, Du J, Xia YL, Li HH (2014) AGGF1 protects from myocardial ischemia/reperfusion injury by regulating myocardial apoptosis and angiogenesis. Apoptosis 19:1254–1268
Yu C, Abbott PV (2007) An overview of the dental pulp: its functions and responses to injury. Aust Dent J 52:S4–S16
Farges JC, Carrouel F, Keller JF, Baudouin C, Msika P, Bleicher F, Staquet MJ (2011) Cytokine production by human odontoblast-like cells upon Toll-like receptor-2 engagement. Immunobiology 216:513–517
Durand SH, Flacher V, Romeas A, Carrouel F, Colomb E, Vincent C, Magloire H, Couble ML, Bleicher F, Staquet MJ, Lebecque S, Farges JC (2006) Lipoteichoic acid increases TLR and functional chemokine expression while reducing dentin formation in in vitro differentiated human odontoblasts. J Immunol 176:2880–2887
Keller JF, Carrouel F, Colomb E, Durand SH, Baudouin C, Msika P, Bleicher F, Vincent C, Staquet MJ, Farges JC (2010) Toll-like receptor 2 activation by lipoteichoic acid induces differential production of pro-inflammatory cytokines in human odontoblasts, dental pulp fibroblasts and immature dendritic cells. Immunobiology 215:53–59
Reinders ME, Sho M, Izawa A, Wang P, Mukhopadhyay D, Koss KE, Geehan CS, Luster AD, Sayegh MH, Briscoe DM (2003) Proinflammatory functions of vascular endothelial growth factor in alloimmunity. J Clin Invest 112:1655–1665
Schonthaler HB, Huggenberger R, Wculek SK, Detmar M, Wagner EF (2009) Systemic anti-VEGF treatment strongly reduces skin inflammation in a mouse model of psoriasis. Proc Natl Acad Sci U S A 106:21264–21269
Park C, Lee SY, Kim HJ, Park K, Kim JS, Lee SJ (2010) Synergy of TLR2 and H1R on Cox-2 Activation in Pulpal Cells. J Dent Res 89:180–185
Horst OV, Tompkins KA, Coats SR, Braham PH, Darveau RP, Dale BA (2009) TGF-beta1 Inhibits TLR-mediated odontoblast responses to oral bacteria. J Dent Res 88:333–338
Farges JC, Keller JF, Carrouel F, Durand SH, Romeas A, Bleicher F, Lebecque S, Staquet MJ (2009) Odontoblasts in the dental pulp immune response. J Exp Zool B Mol Dev Evol 312b:425–436
Kang W, Shang L, Wang T, Liu H, Ge S (2018) Rho-kinase inhibitor Y-27632 downregulates LPS-induced IL-6 and IL-8 production via blocking p38 MAPK and NF-kappaB pathways in human gingival fibroblasts. J Periodontol 89:883–893
Yadlapati M, Biguetti C, Cavalla F, Nieves F, Bessey C, Bohluli P, Garlet GP, Letra A, Fakhouri WD, Silva RM (2017) Characterization of a vascular endothelial growth factor-loaded bioresorbable delivery system for pulp regeneration. J Endod 43:77–83
Zhang Z, Nor F, Oh M, Cucco C, Shi S, Nor JE (2016) Wnt/beta-catenin signaling determines the vasculogenic fate of postnatal mesenchymal stem cells. Stem Cells 34:1576–1587
Tran-Hung L, Mathieu S, About I (2006) Role of human pulp fibroblasts in angiogenesis. J Dent Res 85:819–823
Takeuchi N, Hayashi Y, Murakami M, Alvarez FJ, Horibe H, Iohara K, Nakata K, Nakamura H, Nakashima M (2015) Similar in vitro effects and pulp regeneration in ectopic tooth transplantation by basic fibroblast growth factor and granulocyte-colony stimulating factor. Oral Dis 21:113–122
Li L, Chen D, Li J, Wang X, Wang N, Xu C, Wang QK (2014) Aggf1 acts at the top of the genetic regulatory hierarchy in specification of hemangioblasts in zebrafish. Blood 123:501–508
Zhou B, Ma R, Si W, Li S, Xu Y, Tu X, Wang Q (2013) MicroRNA-503 targets FGF2 and VEGFA and inhibits tumor angiogenesis and growth. Cancer Lett 333:159–169
Lu Q, Yao Y, Yao Y, Liu S, Huang Y, Lu S, Bai Y, Zhou B, Xu Y, Li L, Wang N, Wang L, Zhang J, Cheng X, Qin G, Ma W, Xu C, Tu X, Wang Q (2012) Angiogenic factor AGGF1 promotes therapeutic angiogenesis in a mouse limb ischemia model. PLoS One 7:e46998
Zhang T, Yao Y, Wang J, Li Y, He P, Pasupuleti V, Hu Z, Jia X, Song Q, Tian XL, Hu C, Chen Q, Wang QK (2016) Haploinsufficiency of Klippel-Trenaunay syndrome gene Aggf1 inhibits developmental and pathological angiogenesis by inactivating PI3K and AKT and disrupts vascular integrity by activating VE-cadherin. Hum Mol Genet 25:5094–5110
Funding
This research was supported by the National Natural Science Foundation of China (No. 81670993 and 81873716) and The Construction Engineering Special Fund of “Taishan Scholars” of Shandong Province (No. ts20190975). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors declare that no financial or other potential competing interests exist with regard to this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Ethics approval was gained from the Medical Ethical Committee of School of Stomatology, Shandong University (Protocol Number: GR201801).
Informed consent
The signed information consent forms were acquired from all individuals who were informed of the research proposal.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shen, S., Shang, L., Liu, H. et al. AGGF1 inhibits the expression of inflammatory mediators and promotes angiogenesis in dental pulp cells. Clin Oral Invest 25, 581–592 (2021). https://doi.org/10.1007/s00784-020-03498-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00784-020-03498-9