
Abstract
We previously reported that the bismuth(III) dithiocarbamate derivative, bismuth diethyldithiocarbamate (1) exhibited greater cytotoxicity while inducing apoptosis via the intrinsic pathway in MCF-7 cells. We further evaluated the other bismuth(III) dithiocarbamate derivatives, Bi[S2CNR]3, with R = (CH2CH2OH)(iPr), (CH2)4, and (CH2CH2OH)(CH3), denoted as 2, 3, and 4, respectively, in the same MCF-7 cell line. 2–4 were found to exhibit IC50 values of 10.33 ± 0.06 µM, 1.07 ± 0.01 µM and 25.37 ± 0.12 µM, respectively, compared to that of cisplatin at 30.53 ± 0.23 µM. Apoptotic promotion via the mitochondrial-dependent pathway was due to the elevation of intracellular reactive oxygen species (ROS), promotion of caspases, release of cytochrome c, fragmentation of DNA, and results of staining assay observed in all compound-treated cells. 2–4 are also capable of suppressing MCF-7 cell invasion and modulate Lys-48 also Lys-63 linked polyubiquitination, leading to proteasomal degradation. Analysis of gene expression via qRT-PCR revealed their modulation, which supported all activities conducted upon treatment with 2–4. Altogether, bismuth dithiocarbamate derivatives, with bismuth(III) as the metal center bound to ligands, isopropyl ethanol, pyrrolidine, and methyl ethanol dithiocarbamate, are potential anti-breast cancer agents that induce apoptosis and suppress metastasis. Further studies using other breast cancer cell lines and in vivo studies are recommended to clarify the anticancer effects of these compounds.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
GLOBOCAN (2020) reported for the first time that breast cancer (BC) has become the most commonly diagnosed cancer in women, with an estimated 2.3 million new cases representing 11.7% of all new cases, surpassing lung cancer (11.4%). This is particularly due to its high prevalence in low- and middle-income countries (LMICs). Nevertheless, the number of deaths caused by the disease still comes top by lung cancer with 18%, and breast cancer comes fifth (6.9%) after stomach cancer (7.7%), liver cancer (8.3%), and colorectal cancer (9.4%) [80].
The major recent advances in breast cancer treatment can be summarized as follows: (1) implementation of minimally invasive surgery, (2) optimization of systemic treatment under the guidance of molecular diagnosis, and (3) establishment of a multidisciplinary team for decision-making [47].
During the last few decades, several advances in biological targeted approaches for cancer therapy have occurred; however, despite recent progress, there remains a need for novel and effective anticancer agents for a variety of clinical cases. For invasive diseases, such as triple-negative BC (TNBC) and other metastatic BCs, wide-spectrum and effective chemotherapy agents are still required. Currently, this clinical area is covered only by platinum(II) coordination complexes, including the worldwide approved cisplatin (cDDP), oxaliplatin and carboplatin, together with the restricted nedaplatin and lobaplatin [13, 76].
Moreover, cancer chemoresistance to platinum-based complex treatments is an increasing clinical concern for all chemotherapy regimens [34]. This has fueled the search for novel advanced therapeutics based on alternative metals and their coordination complexes, showing possible cellular selectivity but retaining bioactivity versus malignancies as a function of unique mechanisms of action [77].
Cisplatin is an effective broad-spectrum anticancer drug that originates from metals, such as platinum. The antitumor toxicities of platinum compounds and their clinical application in the late 1970s were a milestone in the development of successful cancer chemotherapeutic agents. Patients usually have a good initial response to cisplatin-based chemotherapy, but later relapse, because the development of cisplatin resistance, either acquired or intrinsic, markedly reduces its clinical effectiveness. Furthermore, the drug’s toxicity profile, which is linked to various toxic side effects, including nausea, nephrotoxicity, cardiotoxicity, hepatotoxicity, and neurotoxicity [79], worsens the outcome of cisplatin in cancer treatment, including breast cancer.
Bismuth and bismuth-based materials have been utilized in pharmaceuticals, photocatalysts, and other solid-state materials because of their attractive properties, including non-toxicity, high thermodynamic stability, and low cost [2]. The medical use of bismuth can be traced back to the past 200 years, and it has been frequently used in various diseases before the discovery of antibiotics. The major applications of bismuth in medicine and health care are related to its high effectiveness in treating burns, intestinal disorders, and stomach ulcers as well as its potential activities against microorganisms, viruses, and malignant tumors [89].
Bismuth complexes, bismuth coordinated to the ligands, such as thiosemicarbazone/thiocarbohydrazone, hydrazine, dithiocarbamate and halide, have been widely found to exhibit potential antineoplastic activities against malignant tumor cell lines. Apart from its potential as an antiproliferative agent, bismuth has also been reported to exhibit antimicrobial, antiulcer, and antiviral properties. Bismuth can ameliorate the side effects of cisplatin.
As a biological target, it has a higher affinity for binding to various ligands, such as N, S, and O, leading to stable bismuth(III) complexes of carboxylates and thiolates [39, 85]. Various activities have been reported to be exerted by bismuth(III) complexes, including anticancer, antileishmanial, and antimicrobial activities [36, 39, 48]. As a ligand, dithiocarbamate can exert beneficial effects by inhibiting certain enzymes in biological systems [3]. Furthermore, their high affinity for binding metal ions, especially bismuth(III), through various modes of coordination, has a strong tendency to form metal complexes (reviewed by Ozturk et al. [61]). On the other hand, bismuth compounds have been shown to have great medicinal potentials [26], however, bismuth complexes formed with dithiocarbamate ligands attached to various functional groups have not yet been extensively studied.
To investigate the molecular diagnosis of breast cancer as an in vitro model in cancer research, it is important to select suitable cell lines that act as important tools. MCF-7 cell line is a potent candidate that can be used in studies to mimic the estrogen receptor (ER)-positive breast cancer and many sub-clones, which have been established for different classes of ER-positive tumors [19].
In this study, we first reported the antiproliferative and anti-invasive effects of various bismuth(III) dithiocarbamate complexes, Bi[S2CNR]3, R = (CH2CH2OH)[CH(CH3)2], (CH2)4, and (CH2CH2OH)(CH3), denoted as 2, 3, and 4, respectively, and the possible underlying apoptotic pathways in human breast adenocarcinoma MCF-7 cells. The findings were realized by measuring the cell membrane permeability and apoptotic rate, degree of fragmentation of DNA, amount of intracellular reactive oxygen species (ROS), release level of cytochrome c, magnitude of caspases, activities of invaded cells, and polyubiquitination of Lys63/Lys48 upon treatment with 2–4. In addition, various targeted human cancer gene expression analyses using polymerase chain reaction (PCR) were conducted to support the findings obtained. It is of utmost hope that the current findings will add to the list of metal-based drugs acting as chemotherapeutics, as well as adjuvants, for the readily available platinum-based drugs used in hospitals.
Materials and methods
Chemicals
Cisplatin was used as the positive control and was purchased from Sigma-Aldrich, (USA). Bismuth(III) compounds, labeled 2, 3, and 4, were synthesized as described below. Chemical solvents, such as absolute ethanol and dimethylsulfoxide (DMSO), were obtained from HmbG Chemicals (USA) and Merck (Germany), respectively. Other solvents used were of analytical grade and commercially available.
Synthesis of bismuth(III) dithiocarbamate derivatives
Preparation of compounds 2–4 was conducted following the protocol of [30]. The 1H and 13C spectra of the compounds were determined using nuclear magnetic resonance (NMR) spectroscopy (Bruker AVANCE 400, Germany) by dissolving the compounds in CDCl3 at 0 h and in DMSO-d6 at 25 °C for 24 and 36 h to mimic the solubility of the compounds in the aqueous environment of the biological system. The functional groups in the synthesized compounds were determined using a Fourier-Transform Infrared (FTIR) spectrophotometer (Perkin Elmer RX1, USA) with KBr pellets. Chloroform or DMSO was used to dissolve the compounds analyzed using a Cary-60 UV–Visible Spectrophotometer (Agilent, Santa Clara, USA) and a Cary Eclipse Fluorescence Spectrophotometer (Agilent, Santa Clara, USA).
A melting point meter (Krüss KSP1N, Germany) was used to determine the melting point of the compounds. An elemental analyzer (Perkin Elmer PE 2400 CHN, USA) was used to perform other elemental analyses.
The preparation of compounds 2–4 and their characterization are included in the Results section, under the Chemistry subsection.
Cell culture
Cell line, MCF-7 (catalog no. HTB-22) were purchased from ATCC (USA). Cells were thawed and cultured as previously described [15]. 2–4, and cisplatin were dissolved in dimethyl sulfoxide (DMSO) (1 mL) to a concentration of 10–2 M, according to Ishak et al. [31], and stored as stock solutions. The DMSO solution, which was less than 1%, did not affect the stability of the compounds used in biological assays. Stock solutions of the compounds (2–4, cisplatin) were diluted in DMEM (ScienCell Research Laboratories, USA) and approximately 100 µL was added to the wells to obtain a final concentration ranging from 0 to 80 µM for the cell proliferation assay.
Cell proliferation assay
Following the protocol described by Mossman [56], MCF-7 cells at > 90% confluence were incubated for 24 h under standardized conditions (37 °C, 5% CO2) in 96-well plates. Upon treatment with 2–4 and cisplatin at concentrations ranging from 0 to − 80 µM, cells were further incubated for 24 h, before adding 20 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, USA) solution (5 mg/mL in PBS) to each well. Incubation was continued for another 4 h under standardized conditions, and the old medium was discarded and replaced with 100 µL of DMSO. A microplate reader (BIOTEK, EL808) was used to measure the absorbance of each well and compare it with that of the untreated control cells at 570/630 nm. The percentage of cells that survived the treatment was calculated using the following formula:
From the curve plot of cell viability vs. log compound concentration (Origin v7.5), the IC50 values of the compounds (2–4, cisplatin) were obtained.
Determination of the cell death mode
Detection of the mode of cell death upon treatment with 2–4 and comparison with cisplatin were conducted via acridine orange/propidium iodide (AO/PI) double staining at the IC50 values of the compounds, following previously described protocols [15]. Overnight incubation of MCF-7 cells (1 × 106 cells/well) in a 6-well plate under standardized conditions (37 °C, 5% CO2 allowed cell attachment. Next, further incubation for 24 h was continued with untreated (negative control and treated cells, which were then harvested and centrifuged for 5 min at 1200 rpm. Continuous centrifugation was performed for another 5 min to remove the remaining medium, once the cells were washed (2× PBS, and the supernatant was discarded. An equal volume of fluorescent dye (AO, 1 mg/L; PI, 1 mg/mL was added to each well before incubation for 20 min in the dark. Freshly stained cell suspensions were aliquoted onto glass slides and covered with coverslips. Glass slides containing stained cell suspensions were viewed using a fluorescence microscope (Olympus BX-51, Japan), and their microscopic images were captured.
DNA fragmentation study
MCF-7 cells (3.0 \(\times \) 106 cells/well per 6-well plate) were incubated overnight under standardized conditions (37 °C, 5% CO2) prior to treatment for 24 h at their IC50. Untreated and treated cells were then incubated for 24 h. After trypsinization of cells with PBS (2×), they were centrifuged for another 24 h, before extracting the DNA from the cells using a G-spin™ Total DNA Extraction Kit (iNtRON Biotechnology, Korea) according to the manufacturer’s protocol. Cells were harvested, centrifuged, and washed with PBS (2 × ). Cell suspensions (1 mL in PBS) in a 1.5-mL centrifuge tube were centrifuged to discard the supernatant at 13,000 rpm for 1 min. The sample tube was added to lysis buffer, proteinase K, and RNase (Sigma-Aldrich, USA) in a volume of 200, 20, and 5 µL, respectively, and vigorously vortexed for an equal mixture before it was further incubated for 20 min at 56 °C in a water bath. Next, 200 µL of binding buffer was added to the lysate, followed by immediate incubation for 5 min at 70 °C in a water bath. Next, centrifugation at 13,000 rpm was conducted on the mixture and absolute ethanol (200 µL) was added for 1 min to discard the filtrate. Further centrifugation at 13,000 rpm was continued for 1 min after the addition of 700 µL of washing buffer to the spin column. The loading dye ((iNtRON Biotechnology, Korea) at a ratio of 6:1 (30 µL of DNA: 5 µL of loading dye) was then added (6\(\times \)) to the extracted DNA and mixed, before the samples were electrophoresed on a 1.5% agarose gel for 2 h at 66 V in TAE running buffer (Bioneer, USA). The gel image was captured under UV light via a transilluminator (Alpha lnnotech, USA) prior to its staining and destaining processes.
Apoptotic assay
Annexin V/Fluorescein isothiocyanate (FITC) assay (Chemicon/Millipore, USA) was conducted to quantify the number of apoptotic cells upon treatment with 2–4 and cisplatin, respectively. Cultured MCF-7 cells (1 × 106 cells/mL) were incubated overnight to allow adherence and further incubated for another 24 h under standard conditions (37 °C, 5% CO2) before they were treated with 2–4 and cisplatin, respectively, at their corresponding IC50 values. Next, the cells were re-suspended in 500 µL ice-cold 1 × binding buffer, harvested, and rinsed with ice-cold PBS 2 × . Cell suspensions were added to 10 µL Annexin V-FITC and 8 µL PI (Sigma-Aldrich, USA), and a pipette was used to thoroughly mix the suspensions before they were incubated again for 20 min in the dark at room temperature and placed on ice. Flow cytometric analysis was performed using a band pass filter for FITC (530 nm) and PI (600 nm). To remove debris and dead cells during calibration, forward scatter (FSC) and side scatter (SSC) were slightly altered.
Caspases assay (caspase 3/7, -8, -9 and -10)
CaspaTagCaspase 3,7 CaspaTagCaspase 8, and CaspaTagCaspase 9 In Situ Assay Kits (Chemicon/Millipore, USA) were used for qualitative assessment of caspase 3/7, -8, -9, and -10 activation, respectively. For quantitative analysis, Fluorometric Assay Kits consist of caspases-3/7, -8, -9, and -10 (Merck Millipore, Germany) were used. Qualitative and quantitative analyses followed the protocols described by the manufacturers, and a luminometer (Perkin Elmer HTS 7000, France) was used according to the manufacturer’s protocol. Cultured MCF-7 cells (1 × 106 cells/mL/96-well plates) were treated with 2–4 and cisplatin in a final volume of 200 µL. Then, untreated and treated cells were incubated for 1 h at room temperature, immediately after adding 50 µL caspase reaction buffer, and measured.
Assessment of intracellular reactive oxygen species (ROS)
Prior to pretreatment with 2–4 and cisplatin at its respective IC50 values for 24 h, MCF-7 cells (1 \(\times \) 105/well/96-well black plate) were incubated overnight. Next, further incubation for 30 min was conducted on the cells with 10 µM 5-(And-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA, Sigma, USA) in the dark, before the cells were then centrifuged for 5 min at 1200 rpm to remove its supernatant, and replenishment of prewarmed PBS 1 \(\times \) (100 uL) in the wells. A fluorescence microplate reader (TECAN Infinite 200, Switzerland) was used to measure the wells at 488 nm (excitation and emission wavelengths of) and 525 nm (emission wavelength).
Detection of cytochrome c release
The FlowCellect™ cytochrome c kit (Millipore, Germany) was used to detect cytochrome c release upon treatment with 2–4 and cisplatin on MCF-7 cells. Cells (1 × 106 cells/T-75 cm2 flask) were treated with test samples at their respective IC50 values for 24 h before harvesting, permeabilization, and fixation. Anti-cytochrome c-FITC antibody (10 µL) was then added before 1 h of incubation in the dark at room temperature, and the staining process was continued. The cells were then washed with blocking buffer, and the supernatant was removed after centrifugation for 5 min at RCF < 280 g. The cell pellet was then re-suspended in 200 µL blocking buffer before analysis using a flow cytometer (FACSCalibur, BD Bioscience, US) with a band pass filter at 488 nm. The cell debris was removed by altering the FSC and SSC gates.
Lys48 and Lys63 polyubiquitination assay
Detection of Lys48 and Lys63 ubiquitination using Milli-Mark™ anti-ubiquitin, Lys-48-specific-FITC, clone Apu2, Milli-Mark™ anti-ubiquitin, Lys-63-specific-Alexa Fluor® 647, and clone Apu3 (Millipore, Billerica, MA, USA) was conducted on MCF-7 cells after treatment with 2–4 and cisplatin, respectively. Briefly, cells (1 × 106 cells/mL/6-well plate) were incubated overnight under standard conditions (37 °C, 5% CO2) to allow attachment before they were pretreated with test samples and incubated for another 24 h. The cells (untreated and treated) were harvested and centrifuged at 1200 rpm for 5 min to discard the supernatant. The cell pellet was then suspended in 1 mL of 1 × PBS and transferred to a 1.5-mL microcentrifuge tube for centrifugation before being re-suspended in 90 µL of 1 × PBS. Next, the cell suspensions were added to the working antibody solution, prepared by dilution of the primary antibody with PBS (1:5), in a volume of 10 µL, to obtain a total reaction volume of 100 µL. Further, 30 min incubation of the reaction mixtures took place at room temperature in the dark before they were analyzed by illumination at 495/647 nm.
Cell invasion assay
Invasion of MCF-7 cells was assessed using the BioCoat™ Matrigel™ Invasion Chamber (BD Biosciences, San Jose, CA) by following the manufacturer’s protocol. Cells (5 × 104 cells/well) were pretreated with 2–4 and cisplatin before they were incubated for 24 h. The non-invading cells were removed by gently scrubbing the inserts with cotton tips. Next, the cells were fixed with methanol for 10 min, and hematoxylin and eosin (H&E) staining of the inserts was performed before rinsing with PBS and air-drying. Microscopic images of the invaded cells were viewed using an inverted microscope at 200 × magnification by placing the insert on a glass slide. The number of invading cells in each field from ten random selected fields was counted according to the following formula:
RT2 profiler PCR microarray (human cancer drug target)
RT2 Profiler PCR microarray [Human Cancer Drug Target (PAHS-507ZR)] (Qiagen, Germany) was used by following the manufacturer’s protocol. MCF-7 cells (1 × 106 cells/mL/flask) in 25-cm2 culture flasks were incubated overnight to allow cell attachment. The RNeasy Mini Kit (Qiagen, USA) was used to extract total RNA from the cells, and a spectrophotometer (Eppendorf, BioPhotometer) was used to determine its purity and concentration by measuring its absorbance. The RT2 profiler PCR array consisted of 84 target genes related to human cancer pathogenesis.
Statistical analysis
Three independent experiments were conducted in triplicate, and the calculated mean ± standard deviation (SD) was statistically analyzed using GraphPad Prism version 7 software (GraphPad Software, La Jolla, California, USA). The software was also used for quantitative image acquisition. Pairwise multiple comparisons were performed using one-way analysis of variance (ANOVA), followed by a post hoc Tukey’s HSD test. Statistical significance was set at p < 0.05.
Results
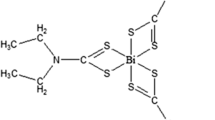
Scheme 1 illustrates the syntheses of compounds 2–4. Detailed methods are described in the chemistry section below.

Synthesis of bismuth(III) dithiocarbamate, Bi[S2CNR]3. a: stirred for 3 h; recrystallized with acetonitrile: chloroform (3:1) overnight
Chemistry
Preparation of tris(N,N′-isopropylethanoldithiocarbamato)bismuth(III), Bi[S2CN(CH2CH2OH)(CH3CHCH3]3 (2)
Sodium salt of isopropylethanol dithiocarbamate NaS2CN(CH3CHCH3)(CH2CH2OH) was priorly synthesized by mixing 2-(isopropylamino)ethanol (0.05 mol, 5.8 mL) in 52.0 mL acetone with carbon disulfide (0.05 mol, 3.0 mL) and sodium hydroxide, NaOH (50% concentration) in an ice-bath for 1–2 h. The white precipitates formed were collected and filtered before rinsing several times with distilled water. It was then left overnight to air dry to yield 4.87 g of the ligand. Next, 2.02 g of the synthesized ligand (10.0 mmol) was dissolved in distilled water (38.0 mL). The salt was slowly added to a suspension of bismuth(III) chloride (3.39 mmol, 1.07 g) in ethanol (28.0 mL). After stirring for 3 h, the pale yellow precipitate was filtered and recrystallized using an acetonitrile:chloroform (3:1) solution overnight to yield 1.93 g of Bi[S2CN (CH2CH2OH) (CH3CHCH3)]3 (2) as a pale yellow powder.
Percentage yield: 62.4%; M.P = 163.7 °C. Elemental analysis: Found C, 26.52; H, 5.63; N, 5.62. C18H36O3BiN3S6 requires: C, 26.50; H, 5.89; N, 5.15. IR (KBr disk, cm−1):1445 ν (C–N); 1037 v and 974 ν (C–S), 1239 v (C–O), 3329 v (OH). 1H NMR: δ 3.65 (t, CH2CH2, J = 5.18 Hz): δ 3.36 (m, CH2CH2, J = 5.25 Hz), δ 4.92 (t, OH, J = 5.20 Hz); δ 1.24 (d, CH3, J = 6.71 Hz): δ 5.21 (s, CH, J = 6.67 Hz) and 13C{1H} NMR: δ 202.5 (CS2);
54.9 (CH2CH2); 58.7 (CH2CH2); 20.1 (CH3); 49.8 (CH).
Preparation of (pyrrolidinedithiocarbamato)bismuth(III), Bi[S2CN(CH2)4]3 (3)
Ammonium salt of pyrrolidine dithiocarbamate, NH4[S2CN(CH2)4] (Sigma-Aldrich, USA) (10.0 mmol, 1.68 g) in 38.0 mL of distilled water was added to bismuth(III)chloride (3.39 mmol, 1.12 g) in 25.0 mL of ethanol for 3 h. The precipitate was filtered to obtain yellow crystals in the filtrate after an overnight incubation. The crystals were harvested and air-dried. They were then recrystallized with acetonitrile: chloroform (3:1) solution to yield 2.21 g of yellow crystalline material, Bi[S2CN(CH2)4]3 (3).
Percentage yield: 78.9%; M.P = 195 °C. Elemental analysis: found C, 27.94; H, 3.12; N, 6.27. C15H24BiN3S6 requires: C, 27.82; H, 3.74; N, 6.49. IR (KBr disk, cm−1):1433 ν (C–N); 1041 v and 940 ν (C–S). 1H NMR: δ 2.03 (tt, CH2CH2CH2, J = 3.48 Hz): δ 3.91 (t, NCH2CH2, J = 2.03 Hz) and 13C{1H} NMR: δ 199.0 (CS2); δ 54.0 (NCH2CH2); δ 26.0 (CH2CH2CH2).
Preparation of tris(N,N′-methylethanoldithiocarbamato)bismuth(III), Bi[S2CN(CH3)(CH2CH2OH)]3 (4)
The ligand potassium methylethanol dithiocarbamate, K[S2CN(CH3)(CH2CH2OH)], was first synthesized prior to the reaction with bismuth(III) chloride by mixing 2-(methylamino)ethanol (0.01 mmol, 0.8 mL), 50% potassium hydroxide, and carbon disulfide (0.01 mmol, 0.7 mL) in diethyl ether (1.0 mL). The mixture was stirred in an ice-bath for 1–2 h. Subsequently, bismuth(III) chloride (3.39 mmol, 0.77 g) in 38.0 mL distilled water was added to the mixture. The yellow precipitate was filtered and purified using an acetonitrile/chloroform (3:1) solvent system. Yellow–orange crystals were formed after a few days, harvested, and dried to yield 1.02 g of the crystalline material, Bi[S2CN(CH3)(CH2CH2OH)]3 (4).
Percentage yield: 61%; M.P. = 124 °C. Elemental analysis: found C, 22.0; H, 3.50; N, 6.36. C12H24O3BiN3S6 requires: C, 21.85; H, 3.67; N, 6.37. IR (KBr disk, cm−1):1433 ν (C–N); 1056 v and 943 ν (C–S), 1255 v (C–O), 3167 v (OH). 1H NMR: δ 3.84 (t, CH2CH2, J = 5.79 Hz): δ 3.70 dt, CH2CH2, J = 5.64 Hz), δ 3.70 (s, OH); δ 4.93 (t, CH3, J = 5.34 Hz) and 13C{1H} NMR: δ 202.4 (CS2); δ 58.4 (CH2CH2); δ 43.2 (CH2CH2); δ 59.0 (CH3).
All the synthesized complexes (2–4) showed a thioureide band (C–N) in the 1433–1445 cm−1 range and a (C–S) band at 940–1055 cm−1 in the IR spectra, proving the presence of dithiocarbamate groups in the compounds. Compounds 2 and 4 showed broad and intense peaks at 3329 and 3167 cm−1 respectively which indicating the presence of hydroxyl (OH) groups. These values are lower than the stretching of the free hydroxyl group at 3640 cm−1. This proves the possibility of intermolecular hydrogen bonding in the complex. The presence of metal–sulfur (M–S) bonds was observed at approximately 350–398 cm−1.
The 1H- and 13C-NMR spectral data of all complexes are shown in Tables S1 and S2, respectively. Deuterated DMSO-d6 and CDCl3 solvents were used for the analysis. In compound 2, the two methyl groups of protons (6 H) give a chemical shift at 1.24 ppm. By applying the n + 1 rule, the adjacent single CH splits the CH3 resonance into a 1:1 doublet (n + 1 = 2), which indicates the presence of a CH group in isopropyl alcohol, while the lone methine proton (CH) was de-shielded by the presence of adjacent electronegative nitrogen atom, thus shifting the NMR signal downfield to 5.21 ppm as a single peak. Protons on carbon adjacent to the alcohol oxygen appear in the region of 3.4–4.5 ppm. The electronegativity of the alcohol oxygen de-shields these protons, causing them to appear downfield compared to alkane protons. The position of the –OH peak varies dramatically depending on the conditions, for example, what solvent is used, the concentration, and the purity of the alcohol, especially on whether it is completely dry. Different sources quote totally different chemical shifts for the hydrogen atom in the -OH group in alcohols, often inconsistently. In this case, the splitting of OH indicates that the complex is not completely free of water, which may be related to the rinsing of the compound during purification.
In compound 3, the methylene proton on the pyrrolidine ring shifted the NMR signal to 2.03 ppm. The adjacent methylene protons on both sides split the CH2 resonance into (2 + 1) and (2 + 1); the triplet confirms its presence in the pyrrolidine ring. The methylene proton adjacent to the nitrogen atom was slightly deshielded, thus shifting its NMR signal to a lower downfield at 3.91 ppm as a triplet due to the splitting of neighboring methylene protons.
In compound 4, similar to the NMR signal in 2, the methylene protons in ethanol were observed in the region of 3.70–3.84 ppm. However, despite the multiplet displayed in 2, the CH2 protons were resonated by their adjacent protons into triplet and doublet triplets, respectively. Additionally, methyl protons adjacent to the nitrogen atom were deshielded and shifted to 4.93 ppm as triplet (due to three distinct CH3 groups) in the complex. The free OH group in 4 displayed a single peak at 3.70 ppm.
In the 13C spectra (Tables S3), the carbon in the RCH2OH group displayed single peaks in the region of 50–65 ppm. In compound 2, their signals are still within the mentioned region; 54.9 and 58.7 ppm, respectively. However, in 4, one carbon was slightly decreased to 43.2 ppm, whereas another carbon was maintained at 58.4 ppm.
In addition, in 2, the carbon in the methyl groups appeared at 20 ppm, which is slightly higher than the normal region of 10–15 ppm. This was probably due to the presence of a nearby nitrogen atom. Its electronegativity involves pulling electrons away from the methyl groups and, which tends to slightly increase the chemical shift. On the other hand, the C-N chemical shift was in the range of 50–70 ppm, which validates the presence of carbon adjacent to the nitrogen atom in this molecule at 49.8 ppm.
In 3, the carbon in the methylene group, R2CH2, is in the region of 15–25 ppm, which agrees with our data. The carbon atom in the methylene group adjacent to the N atom was slightly shifted to 54.0 ppm. Finally, in 4, apart from the carbon in the alcohol group, the carbon in the methyl group displayed a drastic increase in chemical shift to 59.0 ppm due to the higher influence of the neighboring nitrogen atom. All compounds also showed a signal in the 199–202.4 ppm that corresponded to the NCS2 group.
UV–visible and fluorescence spectroscopy were performed to obtain a molar absorptivity of 2–4, which was calculated using the Beer–Lambert law (Table 1). This value was used to determine the concentration of compounds 2–4 for biological appraisal.
The UV–visible spectra of the compounds were recorded between 259 and 264 nm range, corresponding to all compounds having similar distorted octahedral geometries. Based on the molar absorptivity value of more than 105 M−1 cm−1, 2–4 displayed strong spin–orbit coupling due to spin-allowed transitions (intra-ligand π → π* charge transfer) in their electronic arrangements, confirming the predicted distorted octahedral geometry of the compounds. The fluorescence wavelengths for 2–4 were measured to be between 359 and 407 nm range which can also be correlated to the similar electronic transition from the excited state to the ground state of the compounds [30].
Differential scanning calorimetry (DSC) analysis of the compounds revealed that the crystallization temperatures, Tc, of 2–4 were 180.7 °C, 178.7 °C and 130.1 °C, respectively, without any evidence of polymorphism.
The stability of complexes 2–4 in DMSO-d6 was assessed over 24 and 36 h at room temperature and 40 °C. After 24 and 36 h, the resulting 1H NMR spectra of 2–4 at both temperature conditions remained unchanged from the initial spectra (t = 0), indicating that the complexes were stable toward decomposition or hydrolysis in DMSO during the timeframe used for biological assays.
Biological assay
Compounds 2–4 exhibited antiproliferative activity against MCF-7 cell lines
We first evaluated the ability of compounds 2–4 to inhibit MCF-7 breast cancer cell proliferation, using cisplatin as a positive control. After 24 h, MCF-7 cell proliferation was significantly reduced in a concentration-dependent manner upon treatment with 2 starting from 1.25 µM till 10 µM. At higher concentrations (20–80 µM), the cells plateaued, with the maximum number of non-viable cells within 90% (Fig S1A).
Interestingly, 3 was shown to significantly reduce the number of cells to more than 50% at a lower concentration (1.25 µM). As the concentration increased, cells reached a plateau, with more than 90% of the cells already killed. (Fig S1B). However, 4 displayed a delayed response in reducing the cells independently. At 40 µM, compound 4 only started to significantly reduce the number of cells to 33%. As the concentration increased, the cells reached a plateau with less than 10% viable cells (Fig. S1C).
Cisplatin was observed to exhibit similar reduction pattern with 4 at higher concentration of 40 µM which straightly reached plateau state (Fig S1D) The IC50 of 2–4 and cisplatin, were then found to be at 10.33 ± 0.02 µM, 1.07 ± 0.01 µM, 25.37 ± 0.12 µM and 30.53 ± 0.23 µM, respectively (Table 2). Compared that of cisplatin, the IC50 of compounds 2–4 were 2.9-, 28.5-, and 1.2-fold lower which ranked the cytotoxicity of the compounds as follows; 3 > 2 > 4 > cisplatin.
Compound 2–4 promoted MCF-7 cells apoptosis
We then determined the mode of MCF-7 cell death by applying AO/PI double staining to cells treated with 2–4 and cisplatin, and examined them under a fluorescence microscope (Fig. 1). Apoptotic, necrotic, and viable MCF-7 cells were observed, manifested by different fluorescence colors; early apoptotic and viable cells are represented by bright green fluorescence color, late apoptotic cells by orange fluorescence color, and necrotic cells are shown by bright red fluorescence under 488 nm excitation [50].

Fluorescent micrographs of AO/PI double-stained MCF-7 cells i untreated and treated with ii cisplatin and iii 2 iv 3 v 4 at their respective IC50 values. Viable cells are indicated by purple arrows, membrane blebbing is indicated by blue arrows, fragmented nuclei and condensed chromatin are indicated by yellow arrows, and late apoptotic cells are indicated by red arrows. Magnification A 200 \(\times \) and B 400 \(\times \)
Upon treatment with 2–4, most cells turned into apoptotic cells manifested by a multiple-yellowish fluorescence color, blebbing of cell membranes, fragmented nuclei, and condensed chromatin within the cells (Fig. 1ii–v), indicating the common morphology of apoptosis observed. Control cells, however, exhibited bright green fluorescence, representing viable cells (Fig. 1i).
Compounds 2–4 induced DNA fragmentation in MCF-7 cells
To further confirm that the cells underwent apoptosis upon treatment with 2–4 and cisplatin, we conducted a chromosomal DNA fragmentation study, which is believed to be a relatively late event in the apoptotic process [29]. Upon treatment with 2–4 and cisplatin at their respective IC50 values, chromatin DNA is cleaved into multiples of approximately 180–200 base pairs (bp) of internucleosomal fragments, which can be detected by agarose gel electrophoresis as a typical DNA ladder pattern observed under ultraviolet (UV) light (Fig. 2L3–L6). Untreated cells showed no obvious DNA ladder (Fig. 2L2). The results demonstrated that 2–4 and cisplatin induced apoptosis in MCF-7 cells, as characterized by DNA fragmentation.

DNA gel electrophoresis of the internucleosomal DNA fragmentation pattern in a 1.5% agarose gel in MCF-7 cells after 24 h exposure to the negative control (untreated cells) (L2) and the corresponding IC50 of cisplatin (L3), 2 (L4), 3 (L5), and 4 (L6). Lane L1 represents 1-kb DNA laddering markers
Compound 2–4 exerted early apoptosis in MCF-7 cells
We further performed an Annexin V-FITC/PI binding assay to detect the different apoptotic stages of MCF-7 cells upon treatment with 2–4 and cisplatin. During apoptosis, membrane phospholipids, known as phosphatidylserine (PS), strongly bind to annexin V in the presence of Ca2+ ions and translocate from the inner side of the cell membrane to its outer side. PI can bind to DNA and can only enter necrotic or late apoptotic cells [41].
The qualitative assessment of the Annexin V-FITC/PI staining was conducted using flow cytometry to determine the cell distribution in four different quadrants: viable cells, early apoptosis, late apoptosis, and necrotic cells. The histogram illustrates the time course of Annexin V-FITC/PI binding to the phosphatidylserine (PS), in apoptotic cells (Fig. S2).
The untreated (negative control) group displayed more than 92% viable cells, indicating non-apoptotic characteristics throughout the incubation periods of 6, 12, and 24 h (Fig. S2a). The treated groups showed an increase in the percentage of cells undergoing early apoptosis (EA) in a time-dependent manner. The percentage of viable cells after treatment with 2–4 eventually decreased, ranging from 89.3% in 2, 83.1% in 3, and 88.3% in 4 at 6 h to 57.3%, 51.9%, and 65.9% at 24 h, respectively. Similarly, viable cells treated with cisplatin were also significantly altered from 90.7% at 6 h to 45.2% at 24 h. A noticeably higher rate of early apoptosis was observed in the 2–4-treated cells, which significantly increased from 7.56% to 36.43% (Fig. S2c), 12.44% to 43.61% (Fig. S2d), and 9.74% to 23.62% (Fig. S2e) within 24 h. Cisplatin significantly augmented early apoptosis from 6.84% to 44.92% within 24 h. Additionally, there was not much difference in the percentage of necrotic cells in the 2–4 and cisplatin-treated groups, ranging from 1.53% to − 3.82% to 2.37% to − 4.33%, within 24 h, respectively.
Compounds 2–4 activated caspases activities in MCF-7 cells
To study the involvement of caspases in 2–4-induced apoptosis, the activation of important key caspases (3/7, 8, 9, and 10) in apoptotic pathways was investigated upon treatment with 2–4 in MCF-7 cells and compared with cisplatin. Fluorescence images of caspase-3/7, caspase-8, and caspase-9 are shown in Figs. 3i, 4i, 5i. Quantification of caspases-3/7, -8, -9, and -10 is shown in Figs. 3ii, 4ii, 5ii and 6.

Detection of caspase-3/7 in cultured MCF-7 cells for 24 h i in situ in a untreated b cisplatin, c 2, d 3, and e 4 at the respective IC50 concentrations. The MCF-7 cell morphology was viewed under (L) phase contrast and (R) fluorescent caspase-3/7 substrate (FAM-DEVD-FMK). a Low caspase-3/7 levels were observed in the control MCF-7 cells. b–e High levels of caspase-3/7 were detected in cells after treatment. ii Quantitative analysis using ELISA. The negative control was normalized to 100%. Significant differences (p < 0.05) between experimental groups are indicated by different letters

Detection of caspase-8 in cultured MCF-7 cells for 24 h via i in situ in a untreated b cisplatin and c 2 d 3 e 4, at their respective IC50 values. MCF-7 cell morphology viewed under (L) phase-contrast and (R) fluorescent caspase-8 substrate (FAM-LETD-FMK). Notes: a moderate levels of caspase-8 were observed in the control MCF-7 cells. b–e High levels of caspase-8 were observed in treated cells. ii Quantitative analysis using ELISA: the negative control was normalized to 100%. Significant differences (p < 0.05) are indicated by different letters between the experimental groups

Detection of caspase-9 in cultured MCF-7 cells for 24 h via i in situ in a untreated b cisplatin and c 2 d 3 e 4, at their respective IC50 values. The MCF-7 cell morphology was viewed under (L) phase contrast and (R) fluorescent caspase-9 substrate (FAM-LEHD-FMK). Notes: a small amounts of caspase-9 were detected in control MCF-7 cells. b–e High levels of caspase-9 were detected in treated cells. ii Quantitative analysis using ELISA. The negative control was normalized to 100%. Significant differences (p < 0.05) between experimental groups are indicated by different letters

Quantitative analysis of caspase-10 using ELISA. The negative control was normalized to 100%. Significant differences (p < 0.05) between experimental groups are indicated by different letters
Caspase-controlled apoptosis involves a characteristic enzyme cascade that involves multiple caspases at different stages and pathways [96]. Caspases-8, -9, and -10 (initiator caspases) initiate the propagation of apoptotic signals, whereas caspases-3, -6, and -7 (effector caspases) execute apoptosis by cleaving numerous cellular proteins [7]. Furthermore, the activation of effector caspase-3 or -7 sets off the explosive feedback amplification of upstream apoptotic events, which is a key feature of apoptotic signaling essential for efficient apoptotic cell death [52]. In this study, significant increases in caspase-3/7, caspase-8, caspase-9, and caspase-10 levels were observed in cells treated with 2–4 compared to the negative control. As shown (Fig. 3ii), the expression of caspase-3/7 in the treatment groups was significantly elevated by threefold compared with that in the negative control group. Cisplatin was shown to be significantly different (p < 0.05) from 2 and 4, but not from 3. In addition, there was only a significant difference between 2 and 3.
All treated groups, including cisplatin, exhibited significant elevation, by threefold in caspase-8 and 2.5-fold in caspase-9 expression, compared to the negative (untreated) group. Nevertheless, no significant difference was found between any of the treated groups (Figs. 4ii and 5ii). In contrast, a significant comparison was ubiquitously observed in all treatment groups, including cisplatin, with compound 3 displaying the most elevated caspase-10 expression (Fig. 6).
Compounds 2–4 raised intracellular reactive oxygen species (ROS) production in MCF-7 cells
Activation of the mitochondrial (intrinsic) pathway is triggered by the elevation of intracellular ROS upon treatment with anticancer agents. ROS concentration in cancer cells is critical for anticancer therapy. Elevated ROS levels target malignant cells by augmenting ROS generation through exogenous agents or inhibitors of the antioxidant system [4]. Thus, the production of cellular ROS (mainly H2O2) was analyzed using DCFH-DA, a fluorogenic freely permeable tracer. Overall, compounds 2–4 demonstrated a significant elevation in ROS levels twofold with that the negative control (untreated cells) (Fig S3). In comparison to cisplatin, which exerted the lowest level of ROS, 2–4 significantly demonstrated higher ROS levels. Compound 4 had the highest ROS level compared to the other treatment groups, whereas compounds 2 and 3 generated comparable ROS production in MCF-7 cells (Fig. S3).
Compounds 2–4 promoted mitochondrial cytochrome c release into cytoplasm of MCF-7 cells
We further investigated the type of apoptotic pathway upon treatment with 2–4 by quantifying the level of cytochrome c released from the mitochondria at 6, 12, and 24 h in MCF-7 cells. The release of key mitochondrial proteins such as cytochrome c is an important hallmark of apoptosis and is considered a point of no return in the apoptosis process. The release of cytochrome c into the cytosol of apoptotic cells can be used to characterize the mitochondria-dependent pathway of cell death. Fluorescent signals were emitted upon binding of anti-cytochrome c (with secondary binding to FITC) to cytochrome c. Fig. S4 shows the shift in the fluorescence population from the right (M2) to the left (M1) in MCF-7 cells.
In untreated MCF-7 cells, cytochrome c was mostly retained in the mitochondria and not released into the cytoplasm, as indicated by the location of the peak in the graph at M2 (Fig. S4a). The peak of the graph shifted to M1, indicating the induction of apoptosis due to the release of cytochrome c from the mitochondria into the cytoplasm, which was bound by anti-cytochrome c-FITC to form a fluorescence signal. Cytochrome c release was observed as early as 6 h after treatment with 2–4, with a percentage ranging from 95.78 to 97.30%. Nevertheless, prolonged incubation only slightly increased cytochrome c release in cells treated with 2–4, indicating the time-independent effects of the compounds (Fig. S4c–e). In contrast, cisplatin elevated cytochrome c levels in the cells in a time-dependent manner, as evidenced by its release from 42.80% at 6 h to 87.66% at 24 h (Fig. S4b). From these results, we therefore concluded that compounds 2–4 were able to induce cytochrome c release into the cytosol, which decreased the mitochondrial membrane potential (MMP), leading to mitochondrial permeability, thus suggesting the initiation of the mitochondrial apoptotic pathway.
Compounds 2–4 modulated both Lys48- and Lys63-linked polyubiquitination
The ubiquitin–proteasome system (UPS) is essential for maintaining protein turnover, which can either trigger or inhibit apoptosis [1]. Lys48-linked chains are the predominant linkage type in cells and are primarily involved in proteasomal degradation [59]. In contrast, cellular Lys-63 chains have less proteasomal accessibility and proteasome-bound Lys-63 chains are more rapidly deubiquitinated, which could cause inefficient degradation of Lys-63 conjugates [32]. Compounds 2–4 were able to induce Lys-48 release in MCF-7 cells as early as 12 h after treatment, with the percentage of Lys-48 ranging from 54.71% in 2, 50.20% in 3, and 43.74% in 4. On the other hand, cisplatin had already shown more than 50% expression (59.46%) at the earlier hour, that is, at 6 h, but there was no significant increase in expression as the time was prolonged to 12 h (67.18%) and 24 h (67.38%) (Fig. S5b). Compounds 2–4 demonstrated their ability to increase ubiquitinated Lys-48 expression in a time-dependent manner, in which, at 24 h, higher expression was observed by 87.76%, 81.02%, and 92.33%, respectively (Fig. S5c–e).
Notwithstanding, highly consistent ubiquitinated Lys-63 expression (66.37%) was observed in untreated MCF-7 cells throughout the 24 h incubation period (Fig. 7a). Compounds 2–4 and cisplatin significantly arrested ubiquitinated Lys-63 expression, from 62.76%, 37.68%, 49.63%, and 64.02% at 6 h to 44.65%, 34.08%, 32.98%, and 48.52% at 12 h, and eventually to 11.23%, 26.48%, 8.57%, and 15.07% at 24 h (Fig. 7b–e). All treatment groups, including cisplatin, were shown to significantly suppress Lys-63 ubiquitin activity in a time-dependent manner, except for 3.

Flow cytometric analysis of ubiquitinated-FITC of Lys63 in MCF-7 cells a negative control, treated with b cisplatin c 2 d 3 and e 4 at its respective IC50 at the incubation period of 6 h, 12 h and 24 h, respectively. b–e Display the shift in fluorescence levels from M1 to M2 caused by binding of the anti-ubiquitin antibody
Compounds 2–4 inhibited MCF-7 cells invasion
Metastasis, the multiple process by which an original primary tumor develops into a distal secondary tumor, represents a hallmark of cancer and leads to treatment failure, resulting in the death of many patients. Metastasis is highly complex and involves multiple cellular mechanisms, including division from the primary tumor, invasion, evasion of immune surveillance, and regulation of the tissue microenvironment [64]. Metastasis and invasion are the most important biological characteristics of tumor cells. Thus, inhibition of metastasis and invasion is the most direct and effective method for tumor treatment. Compounds 2–4 exhibited significant inhibition of MCF-7 cell invasion (Fig. 8c–e) by 72.1%, 71.1%, and 71.7%, respectively, compared to cisplatin (Fig. 8b) by 58.2%, as shown in Table 3. Nevertheless, the anti-invasive effects of 2–4 displayed comparable efficacy (p > 0.05).

Qualitative analysis of MCF-7 cells in a the negative control and treated with b cisplatin and c 2 d 3 e 4 at their respective IC50 values. Magnification 100 \(\times \)
Compounds 2–4 regulated several human cancer-related genes in MCF-7 cells
Finally, to support our earlier findings, we further explored the effects of 2–4 in modulating the targeted genes involved in breast cancer. Exposure of 2–4 on MCF-7 cells was found to significantly downregulate a number of important cancer-related oncogenes responsible for apoptosis, survival and growth, metastasis, DNA damaged and repair and protein degradation in varying degrees i.e., ABCC1 (-10.63, -41.93, 1.28- fold), AKT1 (-2.5, -54.19, -5.13-fold), BIRC5 (-22.47, -834180.46, -122294.5 -fold), ERBB2 (-225067.78, -62866.33, -4.82-fold), ERBB3 (-9.51, -189258.69, -51063.33-fold), ESR1 (-10.48, -182.28, -9.19-fold), FIGF (-6.23, -6746.86, -7.57-fold), FLT1 (-4.47, -4.53, -47643.77-fold), FLT4(-8.69, -64, -6.96-fold), HDAC1 (-11.79, -3169.41, -37.27-fold), HDAC3 (-8.88, -430.54, -8.94-fold), HDAC4 (-2.89, -17682.08, -7.41-fold), HDAC6 (-4.08, -122294.5, 8.22-fold), HSP90B1 (-4.35, -88.03, 1.29-fold), IGF2 (-5.46, -855.13, -6.11-fold), KIT (-19483.97, -2.13, -4182.07-fold) MTOR (-16.56, -53.82, -41.93-fold), NFKB1 (-5.54, -1.54, -5.74-fold), PARP2 (-56.49, -4.96, -8.17-fold), PARP4 (-4.82, -584.07, -78.79-fold), PDGFRB (-15.56, -230.72, -803.41-fold), PRKCA (-120.26, -8, -9.19-fold), PRKCD (-7826848.02, -5690010.96, -3.43-fold), PRKCE (-28808675.6, -74.03, -3.36-fold), RHOB (-13.55, -7383.04, -13.27-fold), TERT (-32.67, -1016.93, -4.99-fold), TNKS (-4.89, -2310862.52, -2.95-fold) TOP2A (-8.46, -91.14, -182811.84-fold), TXN (-11.71, -225067.78, -2.11-fold) and TXRND1 (-4.06, -2.89, -1.36-fold).
While 3 and 4 significantly downregulated CTSB (-36865.92, -21321.18-fold), ERBB4 (-152.22, -199.47-fold), HDAC2 (-18875349.2, -155.42-fold), HIF1A (-1287.18,-2592.27-fold), PARP1 (-7.26, -3492.39-fold), and 2 significantly downregulated HDAC7 (-186653.11-fold), HDAC11 (-76331.98-fold).
In most of the downregulated genes, compound 3 was shown to exert its action more prominently than 2 and 4, indicating its higher potency as an antiproliferative agent.
Discussion
The sustained interest in dithiocarbamate complexes of bismuth could be attributed to several reasons. These include their structural diversity, which ranges from monomeric to polymeric supermolecular assemblies, their unique application as biological agents/catalysts, and their utilization in materials and surface chemistry. In addition, straightforward synthesis has contributed to their increased attention [3].
Previously, we reported the antiproliferative activity of bismuth(III) diethyldithiocarbamate, labeled 1, against MCF-7 cells [15]. Diethyldithiocarbamate is a good free radical scavenger because of its ability to trap free radicals, in some cases, forming complexes or new radicals [88]. Ajiboye et al. [5] stated that dithiocarbamate ligands and complexes are useful for several applications. However, when both ligands and complexes were found to be relevant in similar applications, the complexes appeared to be more potent than the ligands, as evidenced by several studies [8, 9, 62].
Although the DTC ligand alone has been reported to exhibit greater cytotoxicity, the functional groups linked to the ligand may either lower or augment the cytotoxicity of the metal complexes. As shown in Table 2, compound 3 exhibited the highest cytotoxicity, followed by compounds 2 and 4, respectively. These findings are in agreement with those of Lopez-Cardoso et al. [49] on the antiproliferative effects of new bismuth(III) complexes of pyrrolidinedithiocarbamate of dithia-bismolane and bismane, and oxodithia- and trithia-bismocane, respectively, against breast cancer cell lines in vitro. Pyrrolidine dithiocarbamate (PDTC) exerts higher cytotoxicity against small-cell lung cancer cells (SCLCs) [81]. In another study by Zhang et al. [95], a copper–PDTC complex was reported to suppress the proliferation of BE(2)C cells, a human neuroblastoma cell line, with an IC50 of 8.0 μmol/L, which was more potent than that of cisplatin (IC50 of 80 μmol/L). In addition to copper, Li et al. [45] reported potent cytotoxicity of 3 against various cancer cell lines (ranging from 0.009 to 0.131 µM), specifically with an IC50 of 0.006 µM against MCF-7. These findings demonstrate the potency of bismuth(III) bound to PDTC as its ligand.
Compound 3 showed a 30 times higher potency than cisplatin in terms of cytotoxicity, and compound 2 showed a threefold increase in cytotoxicity compared to the IC50 of cisplatin. In contrast, compound 4 did not show any significant changes compared with cisplatin (Table 2). Compound 2, with isopropyl ethanol as its functional group, increased the IC50 to 3.5-fold that of compound 3. This finding agrees with that of Li et al. [45], who reported a higher IC50 value for the isopropyl functional group bound to the DTC ligand in bismuth(III), with an IC50 value of 8.489 µM in MCF-7 cells.
In terms of the substitution pattern in bismuth(III) dithiocarbamate complexes, Li et al. [45] ranked Me < Et > n-Pr > n-Bu for straight-chain alkyl potency and n-Pr > i-Pr, with smaller rings for N(CH2)n being more potent. Surprisingly, our current results for IC50 values of 2 and 4 contradict the ranking generated by Li et al. [45]. This might be due to the –OH group attached to both the methyl and isopropyl chains in 2 and 4, respectively, which inevitably influences the cytotoxicity of the DTC ligands attached to bismuth(III). Increasing the polarity of the hydroxyl group to the central metal atom eventually leads to reduced biological activity of the metal-based complex [78], which is in agreement with our recent findings on altered cytotoxicities manifested by bismuth(III)-DTC complexes bound to the –OH group in its ligands, 2 and 4. In another study by Turk et al. [87] on bismuth(III) halide complexes against MCF-7, higher cytotoxicity was observed for complexes containing thiophene-2-carbaldehyde thiosemicarbazone than for their ligands alone. Nevertheless, similar complexes containing ethanol in their ligands exhibited lower cytotoxicity, which is in agreement with our findings. In fact, our findings showed that not only the metal ion but also the ligand type and geometry of the complex significantly affect the antiproliferative activity of the complex. In vivo, inclusion of the –OH group in the DTC moieties increases the solubility of the bismuth(III)-DTC complex, which in turn may increase the bioavailability of bismuth(III) complexes as antitumor agents.
Additionally, Yarar et al. [92] highlighted that the type of ligand coordinated to the bismuth(III) ion played a crucial role in the activity of the compounds. The choice of dithiocarbamates compared to other related compounds is attributed to their poor solubility in water, ease of preparation under laboratory conditions, and formation of more stable compounds than several complexes made from other common analytical ligands [38]. Ozturk et al. [61] reported on higher cytotoxicity by bismuth(III) dithiocarbamate complexes, {[BiCl(Me2DTC)2]n} (2) and {[Bi(Et2DTC)3]2} (3) against MCF-7 and HeLa cells, respectively.
Induction of early apoptosis in MCF-7 cells by 2–4 was evident from light microscopic observations of changes in cell morphology, minor loss of cell membrane integrity, shrinkage of cells, appearance of blebs, and the absence of apoptotic bodies (Fig. 1) [72]. The indication of early apoptosis manifested by 2–4 treatment in MCF-7 cells is shown in Fig. 1, revealing the pro-apoptotic activities of 2–4 in a time-dependent manner. In our study, the nuclear morphological and cell surface externalization staining results clearly revealed the relationship between the cytotoxic properties of a substance and the early and late events in apoptosis. Although both 2 and 4 exhibited higher IC50 values than 3, all compounds induced apoptosis in proliferating cancer cells. Shakibaei et al. [75] reported that bismuth nanoparticles induced late apoptosis and necrosis in HT29 cells at the IC50 concentration, without affecting caspase-3 activity. A study comparing the activities of bismuth(III) and diorganotin(IV) complexes of bis(2-acetylpyridine) thiocarbohydrazone in HepG2 cells showed that the bismuth(III) complex induced apoptosis via the mitochondrial pathway [23]. To date, only a few studies of bismuth(III) complexes have thoroughly discussed the mode of cancer cell death.
Early apoptosis signaling focuses on the activation of signaling molecules downstream of death receptors and/or activation of pro-apoptotic members of the B cell lymphoma 2 (Bcl-2) pathway. Detecting early events in apoptosis allows for the identification of the pathway (extrinsic or intrinsic) by which apoptosis is induced.
Our recent findings demonstrated the activation of caspases-8, -9, and -10 by 2–4 (Figs. 3, 4, 5 and 6), indicating that all compounds, 2–4, may induce apoptosis via both intrinsic and extrinsic pathways. To clarify this, we reported an increase in ROS levels by 2–4 (Fig. S2), indicating the compounds’ activation of oxidative stress, which applies to death signals originating from cellular stress, in turn activating the intrinsic apoptotic pathway mediated largely by the mitochondria.
DTC has also been reported to alter ROS concentrations and to exhibit antioxidant activity. The ROS-modulating ability of DTCs is associated with their metal-binding activity (reviewed in [88]. Metals and DTC–metal complexes can produce reactive oxygen species using the Fenton and Haber–Weiss reactions, ultimately leading to the regeneration of metals and highly toxic hydroxyl species are consequently produced [40]. Many studies revealed the importance of ROS playing a part of the mechanism in inducing cancer cell death after treatment with DTCs, with or without metal.
Interestingly, a variety of dithiocarbamic derivatives have been studied in cell culture models, demonstrating their effectiveness in the redox regulation of apoptosis as either prooxidants or antioxidants, depending on the environment in which the compound is administered. In our study, 2–4 initially acted as a pro-oxidant by promoting ROS production to initiate apoptosis.
The increase in cytochrome c by 2–4 (Fig. S3) is manifested by its release via mitochondrial outer membrane permeabilization (MOMP), which induces the formation of a multiprotein complex called the apoptosome. Apoptosomes include cytochrome c, procaspase-9, and the adaptor protein Apaf-1 [43], which support the activation of caspase-9 through enforced multimerization, which in turn cleaves and activates the effector caspase-3/7, resulting in the subsequent degradation of cellular death substrates. As caspase-3 is not detected in MCF-7 cells owing to the deletion of a certain gene in these cells [33], the activation of caspase-9 activates effector caspase-7. Figure 3 shows the activation of caspase-7 by compounds 2–4.
The cleavage of chromosomal DNA into oligonucleosomal size fragments is an integral part of apoptosis [94]. Tang and Kidd [83] reported that activation of caspase-3 is essential for the initiation of DNA fragmentation. Nevertheless, in caspase-3 deficient cells, MCF-7 and caspase-7 have been shown to activate DNA fragmentation, as shown in Fig. 2, which suggests that caspase-7 may mediate this apoptotic hallmark. This also suggests that caspase-3 is not necessarily essential for DNA fragmentation associated with apoptosis [53]. Our study demonstrated that the effects of 2–4 on activating DNA fragmentation were mediated by the induction of caspase-7. Furthermore, in addition to caspase-7, non-caspase proteases, such as cathepsins or endonuclease G, may also participate in the apoptosis induced by these bismuth(III) complexes.
Ultimately, 2–4 were shown to induce intrinsic apoptosis of MCF-7 cells in this study, as evidenced by our findings on the augmentation of ROS, release of cytochrome c, and DNA fragmentation by the compounds. Nevertheless, extrinsic apoptosis mediated by the compounds could not be clarified, as further investigation of other markers involved in extrinsic apoptosis was not conducted. Moreover, an important apoptotic marker, p53, encoded by TP53, was not upregulated by 2–4 (Table S3), indicating that it was not influenced by the compounds that induced extrinsic apoptosis. p53 promotes apoptosis mainly by inducing transcriptional upregulation of pro-apoptotic proteins, such as death receptor 5 (DR5), TNFR1, and Fas, which results in the activation of caspase-8 [58].
Breast cancer metastasis and invasion are complex processes that include tumor cell proliferation, extracellular matrix and basement membrane degradation, tumor cell migration, and microvascular formation [99]. Numerous deaths resulting from breast cancer are caused by cancer cell invasion into the surrounding tissues and metastasis to distal tissues. Breast cancer metastasis is a complex process that involves many genes that interact with and affect each other. Therefore, we further assessed the inhibitory effects of 2–4 on cell invasion, as shown in Table 3 and Fig. 8. Compounds 2–4 inhibited metastasis at equal percentages. These findings are in agreement with studies reporting that PDTC exerts antiproliferative effects in metastatic renal cancer cell lines [55]. Yang et al. [91] also reported the synergistic effect of PDTC when combined with paclitaxel in suppressing MCF-7 cell invasion. In addition, Ouyang et al. [60] reported the ability of bismuth(III)-based complexes to suppress colony formation and migration and significantly induce apoptosis in the human lung cancer cells A549 and H460, attributed to its metal, not the parent ligand, thus demonstrating the potency of the bismuth(III) metal itself in exhibiting therapeutic activities.
The roles of Lys48- and Lys63-linked polyubiquitins in protein degradation and cellular signaling are well characterized. The K63-linked ubiquitin chain participates in various intracellular signal transduction pathways, such as DNA damage and immune responses, rather than degradation [18]. With the recognition of bortezomib, a proteasome inhibitor, in 2003, numerous efforts to search for new and structurally diverse antiproteasome agents, unrelated to bortezomib, have focused on new potential metal-based complexes as proteasome inhibitors with reduced toxicity and improved clinical effectiveness. The DTC moieties of metal-complexing compounds have become an emerging class of anticancer proteasome inhibitors with great potential to overcome the limitations of bortezomib [17, 25]. As dithiocarbamates have been found to inhibit proteasomes and NF-κB [20], we believe that bismuth(III) complexes bound to DTCs as ligands also act as proteasome and NF-κB inhibitors. This is the first study to report DTCs bound to bismuth(III) as proteasome inhibitors. Previously, we reported bismuth(III) diethyldithiocarbamate as a proteasome inhibitor [15], and diethyldithiocarbamate was previously reported to inhibit proteasomes [51]. Diethyldithiocarbamate, as well as pyrrolidine dithiocarbamate (PDTC), a synthetic antioxidant and inhibitor of NF-κB, have been reported to inhibit proteasomes when bound to other metals, such as copper [27], other endogenous metals, such as Mn, Zn [73], gold(I), and gold (III) [16], but none has ever reported it on bismuth(III) yet. Therefore, compound 3, with its ligand PDTC, is ubiquitously potentiated as a proteasome inhibitor by specifically inhibiting NF-κB and acting as an antioxidant agent; however, compounds 2 and 4 may also act similarly, as evidenced by the downregulation of the NFKB1 gene by all compounds (Table S3). Thus far, apart from DTC, diethyldithiocarbamate, PDTC, isopropylethanol DTC, and methylethanol DTC as ligands bound to bismuth(III) have not yet been reported as proteasome inhibitors.
To elucidate the potential mediators/pathways involved in the antiproliferative and antimetastatic effects, apoptotic promotion, and DNA fragmentation of compounds 2–4, we performed human cancer gene expression analysis upon treatment with the compounds. ER-α, encoded by ESR1, plays a crucial role in breast epithelial cell proliferation and survival as well as in mammary tumorigenesis. Interestingly, a high correlation between ER-α, ErbB2, and ABCC11 expression was reported by Honorat et al. [28], which is in agreement with the downregulation of these oncogenes by 2–4 (Table S3). Pertaining to ER-α expression in breast cancer cells, MCF-7, RhoB is critical for the cells, suggesting their role as positive regulators in this model [54]. Compounds 2–4 also downregulated RHOB gene expression (Table S3). Insulin-like growth factor (IGF)-1 and IGF-2 stimulate the growth, development, and proliferation of human cancer cells, including breast cancer cells. IGF-2 has been reported to promote cyclin D (CCND1) and CDK4 to induce MCF-7 cell proliferation [84], which is in line with our finding that 2–4 downregulate these oncogenes (Table S3).
Tables S3 shows the downregulation of ERBB2, 3, and 4 oncogenes to varying degrees by 2–4, which agrees with a study by Ross and Fletcher [68], who concluded that alterations in ErbB2 alone are insufficient for breast tumor progression from a relatively benign to a more malignant phenotype. Paradoxically, a higher degree of ErbB2 overexpression has been reported in early forms of breast cancer than in more advanced invasive carcinomas. As the activation of ErbB2 may require the presence of erbB3 protein, or alternatively, is stabilized by erbB3, downregulation of ErbB2 and erbB3 by 2–4 was shown to be in equilibrium, as both oncogenes are dependent on each other [46].
Dysregulation and mutation of several HDACs, including HDAC1, HDAC2, HDAC3, HDAC4, HDAC6, HDAC7, and HDAC11, has been reported in human breast cancer [44]. Overexpression of HDAC2 and HDAC6 is also associated with ErbB2 [57, 74], and 2–4 significantly downregulated all aforementioned oncogenes to varying degrees (Table S3), indicating that these compounds have antineoplastic effects. Interestingly, downregulation of HDACs may also correlate with downregulation of HSP90B [93].
In addition to cytochrome c, other mitochondrial proteins released during apoptosis include the second mitochondria-derived activator of caspase/direct inhibitor of apoptosis protein (IAP) binding protein with a low pI (Smac/DIABLO), endonuclease G (Endo G), apoptosis-inducing factor (AIF), HtrA2/Omi, and heat shock proteins (Hsps) reviewed in [70].
It has also been reported that HSP90B1 helps cells escape apoptosis and influences the functions of various proto-oncogenes essential for breast cancer growth [82]. Inhibition of HSP90 binding to AKT results in loss of AKT activity and increased sensitivity to apoptosis [71]. Interestingly, this is in agreement with our current findings, in which 2–4 downregulated HSP90B1 while simultaneously downregulating AKT1 (Table S3), indicating that these compounds can suppress MCF-7 cell growth by inducing apoptosis.
HSP90 also plays an important role in the mitochondrial apoptotic pathway. Pandey et al. [63] reported that Hsp90 binds to Apaf-1 and blocks activation of procaspase-3. The results also showed that Hsp90 inhibited cytochrome c-mediated oligomerization of Apaf-1, thereby activating procaspase-9. As shown in Fig. 52–4 activated caspase-9, indicating that Hsp90 downregulation enabled cytochrome c-mediated oligomerization of Apaf-1 in the presence of dATP to activate procaspase-9 to form apoptosomes. The CARD domain of Apaf-1 in the apoptosome interacts with the prodomain (also a CARD domain) of procaspase-9, resulting in the recruitment and subsequent activation of procaspase-9 zymogen, thereby forming a complex with caspase-9 [14]. Activated caspase-9 cleaves downstream caspases, such as caspase-3, -6, and -7, initiating the caspase cascade.
In most cases, activation of caspase-9 leads to activation of procaspase-3, thus activating caspase-3. This leads to the cleavage of PKCδ encoded by the PRKCD gene. However, as MCF-7 cells do not express caspase-3, the expression of PKCδ is altered, which is manifested by the downregulation of PRKCD by 2–4 (Table S3).
Additionally, hypoxia-inducible factor 1 alpha (HIF1α) is overexpressed in MCF-7 cell proliferation and invasion, confirming its role in cancer cell growth and metastasis [21, 97]. This is in line with the ability of compounds 3 and 4 to downregulate HIF1A in this study, thus supporting the use of these compounds as antimetastatic agents.
The receptor tyrosine kinase (RTKs) family, which comprises growth factors, c-Kit, platelet-derived growth factor receptor (PDGFR), insulin growth factor receptor (IGFR), and epidermal growth factor receptor (EGFR), controls the main cellular events, such as cell migration, differentiation, apoptosis, and proliferation [37]. c-Kit signals the main cellular functions, such as cell chemotaxis, survival, differentiation, proliferation, and adhesion. Moreover, it promotes apoptosis and improves invasive potential by stimulating several signal transduction pathways, such as STAT3, the MAP kinase cascade, PLC, and PI3-K. Abnormalities in c-Kit expression have also been reported in breast cancer [69]. Our results showed that 2–4 downregulated c-KIT and PDGFRβ, encoded by the KIT and PDGFRB genes (Table S3), respectively.
Overexpression of thioredoxin (TXN) and its enzyme, thioredoxin reductase (TXNRD), results in increased VEGF production and excessive tumor angiogenesis [90]. Compounds 2–4 also downregulated both TXN and TXRND1 (Table S3), demonstrating the role of these mediators in the intrinsic apoptosis of MCF-7 cells induced by these compounds.
Furthermore, 2–4 mediated the downregulation of DNA repair gene expression, PARP1 and PARP2, and DNA replication gene, TOP2A, in MCF-7 cells, in which all genes play pivotal roles in DNA damage repair, chromatin structure modulation, and cause the breakage of double-stranded DNA [24, 66].
Interestingly, proteasome inhibitors have also been associated with metastasis by suppressing NF-κB, as reported by Jones et al. [35] and Baritaki et al. [11]. Additionally, a part of the mechanism of action of bortezomib, the first proteasome inhibitor, is a decrease in angiogenic cytokines [65], in which metastasis plays a pivotal role in angiogenesis and tumor growth. Activation of NF-κB is attributed to its oncogenic state in several ways: by driving proliferation, enhancing cell survival, or promoting angiogenesis or metastasis [98].
Modulation of several genes by 2–4, which are responsible for protein degradation and anti-angiogenic/antimetastatic activity (Table S3), confirmed the mechanistic pathway of these compounds as potential anticancer agents in vitro. Based on the variation in their active sites, cathepsins play a prominent role in cancer metastasis, and high expression of cathepsins is found in breast cancer patients. Interestingly, CTSB was highly downregulated in 3 and 4 but not in 2. Related metastasis-associated oncogenes, such as VEGF-D, encoded by its FIGF gene; VEGF receptor genes, FLT1 and FLT4 [6],and PARP4 [12], are downregulated by 2–4 to varying degrees, indicating their roles as potential antimetastatic agents.
Owing to its multitude and target proteins, tankyrases denoted by its gene, TNKS has been implicated in various types of cancer, including breast cancer [10]. A strong correlation was observed between protein expression responsible for both processes, that is, TNKS and TERT. Downregulation of both oncogenes by 2–4 was found to varying degrees, in which 3 downregulated both genes by multiple times-fold, indicating that 3 was a strong proteasome inhibitor in this study, followed by 2 and 4.
Since the ubiquitin proteasome system (UPS) plays a major role in regulating key apoptotic proteins, and 2–4 are found to act as proteasome inhibitors by degrading the target proteins, further studies should be conducted on compounds with ubiquitinating abilities. Advantages reported on the compounds with protein degradation include their ability in reducing systemic drug concentrations and, hence, possible cytotoxic side effects, which may improve the success of cancer therapy [1].
Interestingly, most of the therapeutic activities were potentiated by the metal complexes, not just by the ligand DTC alone. Both DTC ligands and complexes are useful for several applications. However, when both ligands and complexes were found to be relevant in similar applications, the complexes appeared to be more potent than the ligands [5]. For instance, bismuth(III)–DTC complexes are more active against microbes than ligands from which they are formed [22]. This is in line with a report that MCF-7 cell growth was not suppressed by PDTC alone, as reported by Tu and Yao [86], which also agrees with our findings in clarifying the higher potency of bismuth(III)–DTC complexes than its ligands alone.
In vivo, Bi3+ is a borderline metal ion; thus, the thiophilic nature of Bi3+ is a clear manifestation that thiolate-containing biological molecules (e.g., cysteine residues) are likely to be their potential targets. According to Rosario et al. [67], thiol chelation generally improves the solubility and lipophilicity of bismuth-based compounds, thus resulting in the potentiation of antitumor activity, as observed for antimicrobial activity. The greater thermodynamic stability of Bi–S compounds compared to Bi–O compounds and the relatively low lability of Bi–S give thiobismuth compounds with broad biological applicability. Because thiobismuth compounds can promptly exchange with free thiols owing to a kinetically labile Bi–S bond, Bi3+ has high mobility inside cells, making it more suitable for reaching the desired biological targets. In a low-pH environment, chelation of metals by biologically compatible thiols or appropriate chelating ligands may represent a promising way to enhance the solubility and permeability of a given cationic metal. In general, properties, such as solubility in organic and non-organic solvents, and the thermal decomposition pattern of dithiocarbamate complexes are influenced by the nature of the dithiocarbamate ligand attached to the metal [5]. Regardless of the size of the dithiocarbamate-bound R groups, intermolecular Bi … S interactions dominate the crystal packing in Bi(S2CNR2)3 [42].
Conclusion
The bismuth(III) dithiocarbamate derivatives, Bi[S2CNR]3, with R = (CH2CH2OH)(iPr) (2), (CH2)4 (3), and (CH2CH2OH)(CH3) (4), demonstrated higher cytotoxicity against MCF-7 breast cancer cells with potent antiproliferative activity to that of cisplatin, which was used as the positive control. Compounds 2–4 also promoted apoptosis via a mitochondrial-dependent pathway and suppressed invasion, which may potentiate their use as antimetastatic agents. Their ability to modulate Lys48-linked and Lys63-linked polyubiquitination results in proteasome degradation, suggesting their roles as proteasome inhibitors. In addition to their role as NF-κB inhibitors, these compounds might also serve as mediators of the reactive oxygen scavenging system and have the potential to act as prooxidants and antioxidants, depending on the redox state of the biological environment. Modulation of cancer genes associated with cell growth and survival, apoptosis, DNA fragmentation, and invasion, including ABC111, CTSB, HSP90B1, HDAC2, ESR1, FIGF, IGF2, FLT4, NFKB1, PARP4, PRKCD, and TXN, further support these findings.
Data availability
The data are available from the corresponding author upon reasonable request.
References
Abbas R, Larisch S (2021) Killing by degradation: regulation of apoptosis by the ubiquitin-proteasome-system. Cells 10(12):3465. https://doi.org/10.3390/cells10123465
Adcock AK, Ayscue RL, Breuer LM, Verwiel CP, Marwitz AC, Bertke JA, Vallet V, Réal F, Knope KE (2020) Synthesis and photoluminescence of three bismuth(III)organic compounds bearing heterocyclic n-donor ligands. Dalton Trans 49:11756–11771. https://doi.org/10.1039/D0DT02360D
Adeyemi JO, Onwudiwe DC (2020) The mechanisms of action involving dithiocarbamate complexes in biological systems. Inorg Chim Acta 511:119809. https://doi.org/10.1016/j.ica.2020.119809
Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, Varol M, Jain A, Khan MA, Sethi G (2019) Role of reactive oxygen species in cancer progression: molecular mechanisms and recent advancements. Biomolecules 9(11):735. https://doi.org/10.3390/biom9110735
Ajiboye TO, Ajiboye TT, Marzouki R, Onwudiwe DC (2022) The versatility of dithiocarbamates. Int J Mol Sci 23(3):1317. https://doi.org/10.3390/ijms23031317
Akahane M, Akahane T, Shah A, Okajima E, Thorgeirsson UP (2005) A potential role for vascular endothelial growth factor-D as an autocrine growth factor for human breast carcinoma cells. Anticancer Res 25(2A):701–707
Algeciras-Schimnich A, Barnhart BC, Peter ME (2013) Apoptosis-dependent and independent functions of caspases. In: Madame Curie Bioscience Database [Internet]. Landes Bioscience, Austin (TX), 2000–2013. https://www.ncbi.nlm.nih.gov/books/NBK6198/. Accessed 30 June 2023
Al-Janabi ASM, Saleh AM, Hatshan MR (2021) Cytotoxicity, antimicrobial studies of M(II)-dithiocarbamate complexes, and molecular docking study against SARS COV2 RNA-dependent RNA polymerase. J Chin Chem Soc 68(6):1104–1115. https://doi.org/10.1002/jccs.202000504
Andrew FP, Ajibade PA (2018) Metal complexes of alkyl-aryl dithiocarbamates: structural studies, anticancer potentials and applications as precursors for semiconductor nanocrystals. J Mol Str 1155:843–855. https://doi.org/10.1016/j.molstruc.2017.10.106
Bao R, Christova T, Song S, Angers S, Yan X, Attisano L (2012) Inhibition of tankyrases induces Axin stabilization and blocks Wnt signaling in breast cancer cells. PLoS ONE 7(11):e48670. https://doi.org/10.1371/journal.pone.0048670
Baritaki S, Chapman A, Yeung K, Spandidos DA, Palladino M, Bonavida B (2009) Inhibition of epithelial to mesenchymal transition in metastatic prostate cancer cells by the novel proteasome inhibitor, NPI-0052: pivotal roles of Snail repression and RKIP induction. Oncogene 28(40):3573–3585. https://doi.org/10.1038/onc.2009.214
Baskaran Nair HB, Krishnegowda N, Nair B, Ganapathy N, Olga S et al (2010) Novel role of PARP4 in hormone-dependent breast cancer progression and metastasis. Cancer Res. https://doi.org/10.1158/1538-7445.AM10-4607
Biersack B, Schobert R (2019) Current state of platinum complexes for the treatment of advanced and drug-resistant breast cancers. Adv Exp Med Biol 1152:253–270. https://doi.org/10.1007/978-3-030-20301-6_13
Bratton SB, Salvesen GS (2010) Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Sci 123(Pt 19):3209–3214. https://doi.org/10.1242/jcs.073643
Chan PF, Ang KP, Hamid RA (2021) A bismuth diethyldithiocarbamate compound induced apoptosis via mitochondria-dependent pathway and suppressed invasion in MCF-7 breast cancer cells. Biometals 34:365–391. https://doi.org/10.1007/s10534-021-00286-0
Chen X, Yang Q, Xiao L, Tang D, Dou QP, Liu J (2017) Metal-based proteasomal deubiquitinase inhibitors as potential anticancer agents. Cancer Metastasis Rev 36(4):655–668. https://doi.org/10.1007/s10555-017-9701-1
Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP (2011) Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets 11(3):239–253. https://doi.org/10.2174/156800911794519752
Cho J, Park J, Kim EE, Song EJ (2020) Assay systems for profiling deubiquitination activity. Int J Mol Sci 21(16):5638. https://doi.org/10.3390/ijms21165638
Comşa Ş, Cîmpean AM, Raica M (2015) The story of MCF-7 breast cancer cell line: 40 years of experience in research. Anticancer Res 35(6):3147–3154
Cvek B, Dvorak Z (2007) Targeting of nuclear factor-kappa B and proteasome by dithiocarbamate complexes with metals. Curr Pharm Des 13(30):3155–3167. https://doi.org/10.2174/138161207782110390
Ebright RY, Zachariah MA, Micalizzi DS, Wittner BS, Niederhoffer KL, Nieman LT, Chirn B, Wiley DF, Wesley B, Shaw B, Nieblas-Bedolla E, Atlas L, Szabolcs A, Iafrate AJ, Toner M, Ting DT, Brastianos PK, Haber DA, Maheswaran S (2020) HIF1A signaling selectively supports proliferation of breast cancer in the brain. Nat Commun 11(1):6311. https://doi.org/10.1038/s41467-020-20144-w
Ejelonu BC, Olagboye SA, Oyeneyin OE, Ebiesuwa OA, Bada OE (2018) Synthesis, characterization and antimicrobial activities of sulfadiazine Schiff base and phenyl dithiocarbamate mixed ligand metal complexes. Open J Appl Sci 8:346. https://doi.org/10.4236/ojapps.2018.88026
Fang Y, Wang Y, Zhao M, Lu Y, Li M, Zhang Y (2018) Bismuth(III) and diorganotin(IV) complexes of bis(2-acetylpyridine) thiocarbonohydrazone: synthesis, characterization, and apoptosis mechanism of action in vitro. Polyhedron 155:254–260. https://doi.org/10.1016/j.poly.2018.08.049
Fasching PA, Weihbrecht S, Haeberle L, Gasparyan A, Villalobos IE, Ma Y, Ekici AB, Wachter DL, Hartmann A, Beckmann MW, Slamon DJ, Press MF (2014) HER2 and TOP2A amplification in a hospital-based cohort of breast cancer patients: associations with patient and tumor characteristics. Breast Cancer Res Treat 145(1):193–203. https://doi.org/10.1007/s10549-014-2922-x
Frezza M, Schmitt S, Dou QP (2011) Targeting the ubiquitin-proteasome pathway: an emerging concept in cancer therapy. Curr Top Med Chem 11(23):2888–2905. https://doi.org/10.2174/156802611798281311
Gowda V, Sarma B, Laitinen RS, Larsson AC, Ivanov AV et al (2017) Structural insights into the polymorphism of bismuth(III) di-n-butyldithiocarbamate by X-ray diffraction, solid-state (13C/15N) CP-MAS NMR and DFT calculations. Polyhedron 129:123–132. https://doi.org/10.1016/j.poly.2017.03.018
Han J, Liu L, Yue X, Chang J, Shi W, Hua Y (2013) A binuclear complex constituted by diethyldithiocarbamate and copper(I) functions as a proteasome activity inhibitor in pancreatic cancer cultures and xenografts. Toxicol Appl Pharmacol 273(3):477–483. https://doi.org/10.1016/j.taap.2013.09.009
Honorat M, Mesnier A, Vendrell J, Guitton J, Bieche I, Lidereau R, Kruh GD, Dumontet C, Cohen P, Payen L (2008) ABCC11 expression is regulated by estrogen in MCF7 cells, correlated with estrogen receptor alpha expression in postmenopausal breast tumors and overexpressed in tamoxifen-resistant breast cancer cells. Endocr Relat Cancer 15(1):125–138. https://doi.org/10.1677/ERC-07-0189
Huang XC, Wang M, Wang HS et al (2014) Synthesis and antitumor activity of novel dipeptide derivatives derived from dehydroabietic acid. Bioorg Med Chem Lett 24(6):1511–1518. https://doi.org/10.1016/j.bmcl.2014.02.001
Ishak DHA (2017) Synthesis, characterization and biological activity of bismuth and antimony 1,1-dithiolates. MSc thesis. Universiti Malaya, Kuala Lumpur, Malaysia, 2017
Ishak DH, Ooi KK, Ang KP, Akim AM, Cheah YK, Nordin N, Halim SN, Seng HL, Tiekink ER (2014) A bismuth diethyldithiocarbamate compound promotes apoptosis in HepG2 carcinoma, cell cycle arrest and inhibits cell invasion through modulation of the NF-κB activation pathway. J Inorg Biochem 130:38–51. https://doi.org/10.1016/j.jinorgbio.2013.09.018
Jacobson AD, Zhang NY, Xu P, Han KJ, Noone S, Peng J, Liu CW (2009) The lysine 48 and lysine 63 ubiquitin conjugates are processed differently by the 26 s proteasome. J Biol Chem 284(51):35485–35494. https://doi.org/10.1074/jbc.M109.052928
Jänicke RU (2009) MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res Treat 117(1):219–221. https://doi.org/10.1007/s10549-008-0217-9
Jin J, Zhang W, Ji W, Yang F, Guan X (2017) Predictive biomarkers for triple negative breast cancer treated with platinum-based chemotherapy. Cancer Biol Ther 18(6):369–378. https://doi.org/10.1080/15384047.2017.1323582
Jones MD, Liu JC, Barthel TK, Hussain S, Lovria E, Cheng D, Schoonmaker JA, Mulay S, Ayers DC, Bouxsein ML, Stein GS, Mukherjee S, Lian JB (2010) A proteasome inhibitor, bortezomib, inhibits breast cancer growth and reduces osteolysis by downregulating metastatic genes. Clin Cancer Res 16(20):4978–4989. https://doi.org/10.1158/1078-0432.CCR-09-3293
Joshi S, Chauhan H, Carpenter N (2017) Preparation, spectroscopic characterization, and antimicrobial activities of mixed-metal (Sb and Bi) bridged derivatives with mixed sulfur donor ligands. J Mol Str 1128:221–229. https://doi.org/10.1016/j.molstruc.2016.08.063
Kadivar A, Kamalidehghan B, Akbari Javar H, Karimi B, Sedghi R, Noordin MI (2017) Antiproliferation effect of imatinib mesylate on MCF7, T-47D tumorigenic and MCF 10A nontumorigenic breast cell lines via PDGFR-β, PDGF-BB, c-Kit and SCF genes. Drug Des Devel Ther 11:469–481. https://doi.org/10.2147/DDDT.S124102
Kanchi S, Singh P, Bisetty K (2014) Dithiocarbamates as hazardous remediation agent: a critical review on progress in environmental chemistry for inorganic species studies of 20th century. Arab J Chem 7:11–25. https://doi.org/10.1016/j.arabjc.2013.04.026
Keogan DM, Griffith DM (2014) Current and potential applications of bismuth-based drugs. Molecules 19(9):15258–15297. https://doi.org/10.3390/molecules190915258
Kehrer JP (2000) The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 149(1):43–50. https://doi.org/10.1016/s0300-483x(00)00231-6
Kumar R, Saneja A, Panda AK (2021) An annexin V-FITC—propidium iodide-based method for detecting apoptosis in a non-small cell lung cancer cell line. In: Santiago-Cardona PG (ed) Lung cancer: methods in molecular biology, vol 2279. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1278-1_17
Lai C, Tiekink E (2007) Prevalence of intermolecular Bi S interactions in bismuth dithiocarbamate compounds: Bi(S2CNR2)3. Z Kristallogr 222(10):532–538. https://doi.org/10.1524/zkri.2007.222.10.532
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91(4):479–489. https://doi.org/10.1016/s00928674(00)80434-1
Li Y, Seto E (2016) HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med 6(10):a026831. https://doi.org/10.1101/cshperspect.a026831
Li H, Lai CS, Wu J, Ho PC, de Vos D, Tiekink ER (2007) Cytotoxicity, qualitative structure-activity relationship (QSAR), and antitumor activity of bismuth dithiocarbamate complexes. J Inorg Biochem 101(5):809–816. https://doi.org/10.1016/j.jinorgbio.2007.01.010
Liu B, Ordonez-Ercan D, Fan Z, Edgerton SM, Yang X, Thor AD (2008) Downregulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int J Cancer 120(9):1874–1882. https://doi.org/10.1002/ijc.22423
Liu L, Kawashima M, Toi M (2020) Breast cancer in global health: beyond diversity and inequality. Int J Surg: Global Health 3(6):e32. https://doi.org/10.1097/GH9.0000000000000032
Lizarazo-Jaimes EH, Monte-Neto RL, Reis PG, Fernandes NG, Speziali NL, Melo MN, Frézard F, Demicheli C (2012) Improved antileishmanial activity of Dppz through complexation with antimony(III) and bismuth(III): investigation of the role of the metal. Molecules 17(11):12622–12635. https://doi.org/10.3390/molecules171112622
López-Cardoso M, Tlahuext H, Pérez-Salgado M, Vargas-Pineda DG, Román-Bravo P et al (2020) Synthesis, crystal structure, antibacterial, antiproliferative, and QSAR studies of new bismuth(III) complexes of pyrrolidineditiocarbamates of dithia-bismolane and bismane, oxodithia-, and trithia-bismocane. J Mol Str 1217:128456. https://doi.org/10.1016/j.molstruc.2020.128456
Makeen HA, Mohan S, Al-Kasim MA, Sultan MH, Albarraq AA, Ahmed RA, Alhazmi HA, Alam MI (2021) Preparation, characterization, and anticancer activity of nanostructured lipid carriers containing imatinib. Pharmaceutics 13(7):1086. https://doi.org/10.3390/pharmaceutics13071086
Matsuno T, Kariya R, Yano S, Morino-Koga S, Taura M, Suico MA, Shimauchi Y, Matsuyama S, Okamoto Y, Shuto T, Kai H, Okada S (2011) Diethyldithiocarbamate induces apoptosis in HHV-8-infected primary effusion lymphoma cells via inhibition of the NF-κB pathway. Int J Oncol 40(4):1071–1078. https://doi.org/10.3892/ijo.2011.1313
McComb S et al (2019) Efficient apoptosis requires feedback amplification of upstream apoptotic signals by effector caspase-3 or -7. Sci Adv. https://doi.org/10.1126/sciadv.aau9433
Mc Gee MM, Hyland E, Campiani G, Ramunno A, Nacci V, Zisterer DM (2002) Caspase-3 is not essential for DNA fragmentation in MCF-7 cells during apoptosis induced by the pyrrolo-1,5-benzoxazepine, PBOX-6. FEBS Lett 515(1–3):66–70. https://doi.org/10.1016/s0014-5793(02)02440-7
Médale-Giamarchi C, Lajoie-Mazenc I, Malissein E, Meunier E, Couderc B, Bergé Y, Filleron T, Keller L, Marty C, Lacroix-Triki M, Dalenc F, Doisneau-Sixou SF, Favre G (2013) RhoB modifies estrogen responses in breast cancer cells by influencing expression of the estrogen receptor. Breast Cancer Res 15(1):R6. https://doi.org/10.1186/bcr3377
Morais C, Pat B, Gobe G, Johnson DW, Healy H (2006) Pyrrolidine dithiocarbamate exerts antiproliferative and pro-apoptotic effects in renal cell carcinoma cell lines. Nephrol Dial Transplant 21(12):3377–3388. https://doi.org/10.1093/ndt/gfl543
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65(1–2):55–63. https://doi.org/10.1016/0022-1759(83)90303-4
Müller BM, Jana L, Kasajima A, Lehmann A, Prinzler J, Budczies J, Winzer J, Dietel M, Weichert W, Denkert C (2013) Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer - overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 13:215. https://doi.org/10.1186/1471-2407-13-215
O’Connor L, Harris AW, Strasser A (2000) CD95 (Fas/APO-1) and p53 signal apoptosis independently in diverse cell types. Cancer Res 60(5):1217–1220
Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K (2016) The K48–K63 branched ubiquitin chain regulates NF-kappaB signaling. Mol Cell 64:251–266. https://doi.org/10.1016/j.molcel.2016.09.014
Ouyang R, Yang Y, Tong X, Feng K, Yang Y, Tao H, Zhang X, Zong T, Cao P, Xiong F, Guo N, Li Y, Miao Y, Zhou S (2017) Potent anticancer activity of a new bismuth(III) complex against human lung cancer cells. J Inorg Biochem 168:18–26. https://doi.org/10.1016/j.jinorgbio.2016.12.006
Ozturk II, Banti CN, Kourkoumelis N, Manos MJ, Tasiopoulos AJ et al (2014) Synthesis, characterization and biological activity of antimony(III) or bismuth(III) chloride complexes with dithiocarbamate ligands derived from thiuram degradation. Polyhedron 67:89–103. https://doi.org/10.1016/j.poly.2013.08.052
Paca AM, Ajibade PA, Andrew FP, Nundkumar N, Singh M (2021) Synthesis, X-ray crystal structures and anticancer studies of four Pd(II) dithiocarbamate complexes. Arab J Chem 14(9):103326. https://doi.org/10.1016/j.arabjc.2021.103326
Pandey P, Saleh A, Nakazawa A, Kumar S, Srinivasula SM, Kumar V, Weichselbaum R, Nalin C, Alnemri ES, Kufe D, Kharbanda S (2000) Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J 19:4310–4322. https://doi.org/10.1093/emboj/19.16.4310
Park M, Kim D, Ko S, Kim A, Mo K, Yoon H (2022) Breast cancer metastasis: mechanisms and therapeutic implications. Int J Mol Sci 23(12):6806. https://doi.org/10.3390/ijms23126806
Rajkumar SV, Richardson PG, Hideshima T, Anderson KC (2005) Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol 23(3):630–639. https://doi.org/10.1200/JCO.2005.11.030
Richard IA, Burgess JT, O’Byrne KJ, Bolderson E (2022) Beyond PARP1: the potential of other members of the poly (ADP-Ribose) polymerase family in DNA repair and cancer therapeutics. Front Cell Dev Biol 9:801200. https://doi.org/10.3389/fcell.2021.801200
Rosário JDS, Moreira FH, Rosa LHF, Guerra W, Silva-Caldeira PP (2023) Biological activities of bismuth compounds: an overview of the new findings and the old challenges not yet overcome. Molecules 28(15):5921. https://doi.org/10.3390/molecules28155921
Ross JS, Fletcher JA (1998) The HER-2/neu oncogene in breast cancer: prognostic factor, predictive factor, and target for therapy. Stem Cells 16(6):413–428. https://doi.org/10.1002/stem.160413
Roussidis AE, Theocharis AD, Tzanakakis GN, Karamanos NK (2007) The importance of c-Kit and PDGF receptors as potential targets for molecular therapy in breast cancer. Curr Med Chem 14(7):735–743. https://doi.org/10.2174/092986707780090963
Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S (1999) Presence of a pre-apoptotic complex of procaspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J 18(8):2040–2048. https://doi.org/10.1093/emboj/18.8.2040
Sato S, Fujita N, Tsuruo T (2000) Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA 97(20):10832–10837. https://doi.org/10.1073/pnas.170276797
Saraste A, Pulkki K (2000) Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res 45(3):528–537. https://doi.org/10.1016/s0008-6363(99)00384-3
Sedlacek J, Martins LDMRS, Danek P, Pombeiro AJL, Cvek B (2014) Diethyldithiocarbamate complexes with metals used as food supplements show different effects in cancer cells. J Appl Biomed 12:301–304. https://doi.org/10.1016/j.jab.2014.04.002
Seo J, Min SK, Park R, Kim DH, Kwon MJ, Kim LS, Ju S (2014) Expression of HDAC1, HDAC2, HDAC3, and HDAC6 in invasive ductal carcinomas of the breast. J Breast Cancer 17(4):323–331. https://doi.org/10.4048/jbc.2014.17.4.323
Shakibaie M, Forootanfar H, Ameri A, Jafari M, Rahimi HR (2018) Cytotoxicity of biologically synthesised bismuth nanoparticles against HT-29 cell line. IET Nanobiotechnol 12(5):653–657. https://doi.org/10.1049/iet-nbt.2017.0295
Shamseddine AI, Farhat FS (2011) Platinum-based compounds for metastatic breast cancer treatment. Chemotherapy 57(6):468–487. https://doi.org/10.1159/000334093
Simpson P, Desai NM, Casari I, Massi M, Falasca M (2019) Metal-based antitumor compounds: beyond cisplatin. Future Med Chem 11(2):119–135. https://doi.org/10.4155/fmc-2018-0248
Singh S, Bharti N, Mohapatra PP (2009) Chemistry and biology of synthetic and naturally occurring antiamoebic agents. Chem Rev 109(5):1900–1947. https://doi.org/10.1021/cr068217k
Singh L, Aldosary S, Saeedan AS, Ansari MN, Kaithwas G (2018) Prolyl hydroxylase 2: a promising target to inhibit hypoxia-induced cellular metabolism in cancer cells. Drug Discov Today 23(11):1873–1882. https://doi.org/10.1016/j.drudis.2018.05.016
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71:209–249. https://doi.org/10.3322/caac.21660
Tahata S, Yuan B, Kikuchi H, Takagi N, Hirano T, Toyoda H (2014) Cytotoxic effects of pyrrolidine dithiocarbamate in small-cell lung cancer cells, alone and in combination with cisplatin. Int J Oncol 45(4):1749–1759. https://doi.org/10.3892/ijo.2014.2564
Takayama S, Reed JC, Homma S (2003) Heat-shock proteins as regulators of apoptosis. Oncogene 22(56):9041–9047. https://doi.org/10.1038/sj.onc.1207114
Tang D, Kidd VJ (1998) Cleavage of DFF-45/ICAD by multiple caspases is essential for its function during apoptosis. J Biol Chem 273(44):28549–28552. https://doi.org/10.1074/jbc.273.44.28549
Tomblin JK, Salisbury TB (2014) Insulin-like growth factor 2 regulation of aryl hydrocarbon receptor in MCF-7 breast cancer cells. Biochem Biophys Res Comm 443(3):1092–1096. https://doi.org/10.1016/j.bbrc.2013.12.112
Tiekink ERT (2002) Antimony and bismuth compounds in oncology. Crit Rev Oncol Hemat 42(3):217–224. https://doi.org/10.1016/s1040-8428(01)00217-7
Tu G, Yao ZX (2004) Pyrrolidine dithiocarbamate inhibits NF-κB activation and enhances TNF-induced apoptosis in human breast cancer cells. Chin J Cancer Res 16:99–104. https://doi.org/10.1007/s11670-004-0005-y
Turk K, Grześkiewicz AM, Banti CN, Hadjikakou SK, Kubicki M, Ozturk II (2022) Synthesis, characterization, and biological properties of mono-, di- and poly-nuclear bismuth (III) halide complexes containing thiophene-2-carbaldehyde thiosemicarbazones. J Inorg Biochem 237:111987. https://doi.org/10.1016/j.jinorgbio.2022.111987
Viola-Rhenals M, Patel KR, Jaimes-Santamaria L, Wu G, Liu J, Dou QP (2018) Recent advances in antabuse (disulfiram): the importance of its metal-binding ability to its anticancer activity. Curr Med Chem 25(4):506–524. https://doi.org/10.2174/0929867324666171023161121
Wang R, Li H, Sun H (2019) Bismuth: environmental pollution and health effects. Encyclopedia of environmental health, 2nd edn. Elsevier, Amsterdam, pp 415–423. https://doi.org/10.1016/B978-0-12-409548-9.11870-6
Welsh SJ, Bellamy WT, Briehl MM, Powis G (2002) The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1 protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res 62(17):5089–5095
Yang C, Zhang H, Huang W, Lin Q, Wei H (2010) Effect of combined use of PDTC and paclitaxel on proliferation and invasion of human breast cancer cell line MCF-7. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 27(5):1105
Yarar S, Ozturk I, Banti C, Panagiotou N, Papatriantafyllopoulou C, Manoli M, Manos M, Tasiopoulos A, Hadjikakou S (2018) Synthesis, characterization and cytotoxic properties of bismuth(III) chloride complexes with heterocyclic thioamides. Inorg Chim Acta 471:23–33. https://doi.org/10.1016/j.ica.2017.10.026
Yu S, Cai X, Wu C, Liu Y, Zhang J, Gong X, Wang X, Wu X, Zhu T, Mo L, Gu J, Yu Z, Chen J, Thiery JP, Chai R, Chen L (2017) Targeting HSP90-HDAC6 regulating network implicates precision treatment of breast cancer. Int J Biol Sci 13(4):505–517. https://doi.org/10.7150/ijbs.18834
Zhang J, Xu M (2000) DNA fragmentation in apoptosis. Cell Res 10:205–211. https://doi.org/10.1038/sj.cr.7290049
Zhang H, Wu J-S, Peng F (2008) Potent anticancer activity of pyrrolidine dithiocarbamate–copper complexes against cisplatin-resistant neuroblastoma cells. Anticancer Drugs 19(2):125–132. https://doi.org/10.1097/CAD.0b013e3282f2bdff
Zhang L, Lei J, Liu J, Ma F, Ju H (2015) In situ activation and monitoring of the evolution of the intracellular caspase family. Chem Sci 6(6):3365–3372. https://doi.org/10.1039/c5sc00471c
Zhang Y, Yan J, Wang L, Dai H, Li N, Hu W, Cai H (2017) HIF-1α promotes breast cancer cell MCF-7 proliferation and invasion through regulating miR-210. Cancer Biother Radiopharm 32(8):297–301. https://doi.org/10.1089/cbr.2017.2270
Zhang T, Ma C, Zhang Z, Zhang H, Hu H (2021) NF-κB signaling in inflammation and cancer. MedComm 2(4):618–653. https://doi.org/10.1002/mco2.104
Zhu X, Wang K, Zhang K, Xu F, Yin Y, Zhu L, Zhou F (2016) Galectin-1 knockdown in carcinoma-associated fibroblasts inhibits migration and invasion of human MDA-MB-231 breast cancer cells by modulating MMP-9 expression. Acta Biochim Biophys Sin (Shanghai) 48(5):462–467. https://doi.org/10.1093/abbs/gmw019
Acknowledgements
The authors thanked Prof Edward R.T. Tiekink for providing the sample and Universiti Malaya for providing the grants under High Impact Research Schemes and collaborated with us by providing the compounds and the funding. The authors also thanked Prof Dr Noor Saadah Abd Rahman, the Deputy Vice Chancellor (Research and Innovation) at the University of Malaya for giving us the consent to publish the data
Funding
This research was funded by the Ministry of Higher Education, Malaysia (grant numbers UM.C/HIR-MOHE/SC/03 and UM.C/HIR-MOHE/SC/12).
Author information
Authors and Affiliations
Contributions
Conceptualization, RAH; methodology, RAH; validation, CPF and RAH; formal analysis, CPF and AKP; investigation, CPF and AKP; data curation, CPF and AKP; writing—original draft preparation, CPF; writing—review and editing, RAH; visualization, CPF and AKP; supervision, RAH; project administration, RAH All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chan, P.F., Ang, K.P. & Hamid, R.A. Cytotoxicity of bismuth(III) dithiocarbamate derivatives by promoting a mitochondrial-dependent apoptotic pathway and suppressing MCF-7 breast adenocarcinoma cell invasion. J Biol Inorg Chem 29, 217–241 (2024). https://doi.org/10.1007/s00775-023-02041-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-023-02041-x