
Abstract
Platinum-based drugs are widely recognized efficient anti-tumor agents, but faced with multiple undesirable effects. Here, four dinuclear platinum(II) complexes, [{Pt(1,2-pn)Cl}2(μ-pydz)]Cl2 (C1), [{Pt(ibn)Cl}2(μ-pydz)]Cl2 (C2), [{Pt(1,3-pn)Cl}2(μ-pydz)]Cl2 (C3) and [{Pt(1,3-pnd)Cl}2(μ-pydz)]Cl2 (C4), were designed (pydz is pyridazine, 1,2-pn is ( ±)-1,2-propylenediamine, ibn is 1,2-diamino-2-methylpropane, 1,3-pn is 1,3-propylenediamine, and 1,3-pnd is 1,3-pentanediamine). Interactions and binding ability of C1–C4 complexes with calf thymus DNA (CT-DNA) has been monitored by viscosity measurements, UV–Vis, fluorescence emission spectroscopy and molecular docking. Binding affinities of C1–C4 complexes to the bovine serum albumin (BSA) has been monitored by fluorescence emission spectroscopy. The tested complexes exhibit variable cytotoxicity toward different mouse and human tumor cell lines. C2 shows the most potent cytotoxicity, especially against mouse (4T1) and human (MDA-MD468) breast cancer cells in the dose- and time-dependent manner. C2 induces 4T1 and MDA-MD468 cells apoptosis, further documented by the accumulation of cells at sub-G1 phase of cell cycle and increase of executive caspase 3 and caspase 9 levels in 4T1 cells. C2 exhibits anti-proliferative effect through the reduction of cyclin D3 and cyclin E expression and elevation of inhibitor p27 level. Also, C2 downregulates c-Myc and phosphorylated AKT, oncogenes involved in the control of tumor cell proliferation and death. In order to measure the amount of platinum(II) complexes taken up by the cells, the cellular platinum content were quantified. However, C2 failed to inhibit mouse breast cancer growth in vivo. Chemical modifications of tested platinum(II) complexes might be a valuable approach for the improvement of their anti-tumor activity, especially effects in vivo.
Graphical abstract
Dinuclear platinum(II) complex [{Pt(ibn)Cl}2(μ-pydz)]Cl2 shows anti-tumor activity, triggers the apoptosis and reduces the proliferation of mouse breast cancer cells in vitro. However, its inhibitory effect on tumor growth in vivo is absent.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Growing incidence and mortality of malignant diseases worldwide instigated the urgent need for the improvement of tumor therapy, including development of new therapeutics [1]. Platinum-based drugs, such as cisplatin, carboplatin, oxaliplatin and others, are widely recognized as the efficient anti-tumor agents included in therapy of various types of malignancies [2]. It is well known that complexes such as cisplatin, carboplatin and oxaliplatin bind to DNA by covalent interaction [3, 4]. There are also complexes that achieve non-covalent interactions with DNA such as groove binding, intercalative binding, and electrostatic binding [5]. Mononuclear platinum(II) complexes containing aromatic ligands mainly bind to DNA by intercalation as well as minor-groove binding [6, 7]. Highly-charged polynuclear platinum(II) complexes binding by backbone tracking or groove spanning [8, 9]. Such formed platinum-based complex/DNA adduct inhibits tumor cell growth [10]. Platinum-based compounds damage tumor cells DNA thus inhibiting its synthesis and repair, induce cell cycle arrest and trigger both intrinsic and extrinsic pathways of apoptosis [2, 10,11,12]. The investigation of the interactions of platinum(II) complexes with bovine serum albumin (BSA), which serves as a model for human serum albumin (HSA), contributes to the understanding of the transport of platinum-based drugs through the blood system [13,14,15,16].
However, as conventional therapeutics, platinum-based drugs have been accompanied with multiple undesirable effects such as systemic toxicity, low selectivity and intrinsic or acquired resistance development [11, 12, 17]. In order to overcome the limitations of their clinical usage, much effort was put into the designing of novel platinum-based compounds and testing their anti-tumor efficacy in the last decades. So far, various newly synthesized and modified platinum-based compounds have shown anti-tumor activity against different tumor cell lines, including substantial cytotoxicity, induction of cell cycle arrest and apoptosis, as well as anti-angiogenic effects [18,19,20,21,22,23,24].
The first polynuclear platinum complex which has entered phase II clinical trials was trinuclear bifunctional complex [{trans-PtCl(NH3)2}2-μ-trans-{Pt(NH3)2(H2N(CH2)6NH2)2}](NO3)4 (Triplatin, BBR3464) [25]. This complex contains three Pt centers connected by a bridging diamine ligand [26, 27] Platinum units interact with DNA through electrostatic and H-bonding interactions showing significant noncovalent binding affinity to the DNA [28]. Another polynuclear complex with significant antitumor activity is trinuclear platinum(II) complex [{trans-Pt(NH3)2(NH2(CH2)6(NH3+)}2-μ-trans-{Pt(NH3)2(NH2(CH2)6NH2)2}]8+, (TriplatinNC) containing three Pt(II) centers with possibilities for H-bonding to DNA adduct. Both complexes are linked by flexible hydrophobic molecules such as linear aliphatic molecules with different chain lengths [29]. These linkers enable them to form long-range cross-links with DNA [30, 31]. Some polynuclear complexes contain platinum centers connected through a rigid bridging molecule such as azine and azoles [32,33,34], exhibiting different DNA-binding mechanisms than clinically used mononuclear complexes [35] most likely by minimizing distortion of the DNA double helix in a cross-link [36]. Also, series of dinuclear platinum(II) complexes with six-membered heterocyclic diazines (pyridazine (pydz), pyrimidine (pm), and pyrazine (pz)) as bridging ligands, showed improved antitumor activity in vitro in cisplatin-resistant cell lines, as well as in several tumor cell lines [23, 32, 37].
In order to continue search for additional candidates for platinum-based tumor therapy, four dinuclear platinum(II) complexes have been synthesized and characterized including ([{Pt(1,2-pn)Cl}2(μ-pydz)]Cl2 (C1), [{Pt(ibn)Cl}2(μ-pydz)]Cl2 (C2), [{Pt(1,3-pn)Cl}2(μ-pydz)]Cl2 (C3) and [{Pt(1,3-pnd)Cl}2(μ-pydz)]Cl2 (C4) where pydz is pyridazine, 1,2-pn is (±)-1,2-propylenediamine, ibn is 1,2-diamino-2-methylpropane, 1,3-pn is 1,3-propylenediamine, and 1,3-pnd is 1,3-pentanediamine). Fluorescence emission measurements and molecular docking were used to investigate the binding affinity of C1–C4 complexes towards DNA and BSA. Their cytotoxic activities were evaluated using different tumor cell lines of mouse [melanoma (B16-F10), breast cancer (4T1) and Lewis lung carcinoma (LLC1)] and human origin [melanoma (A375), lung adenocarcinoma (A549), breast cancer (MDA-MB 468)] and human fibroblast (MRC-5) cell line. Further, their effects on the tumor cells death and proliferation were analyzed. Due to the highest cytotoxicity, especially against mouse and human breast cancer cells, and notable effects on mouse breast cancer cells death and probably proliferation, detected by in vitro assays, the activity of C2 on the mouse breast cancer growth was also tested in vivo.
Materials and methods
Materials
Chemicals and reagents deuterium oxide (D2O), dimethylsulfoxide-d6 (DMSO-d6), (±)-1,2-propylenediamine (1,2-pn), 1,2-diamino-2-methylpropane (isobutylenediamine, ibn), 1,3-propylenediamine (1,3-pn), 1,3-pentanediamine (1,3-pnd), pyridazine (1,2-diazine, pydz), deoxyribonucleic acid from calf thymus (CT-DNA), bovine serum albumin (BSA), 3,8-diamino-5-ethyl-6-phenylphenanthridiniumbromide (ethidium bromide, EtBr), phosphate-buffered saline (PBS, 10 mM, pH 7.4, containing 2.7 mM KCl and 137 mM NaCl) and potassium tetrachloroplatinate(II) (K2[PtCl4]) were obtained from the Sigma-Aldrich Chemical Co. Mononuclear Pt(II) complexes, [Pt(L)Cl2] (L is bidentately coordinated (±)-1,2-propylenediamine (1,2-pn), 1,2-diamino-2-methylpropane (isobutylenediamine, ibn), 1,3-propylenediamine (1,3-pn), 1,3-pentanediamine (1,3-pnd)), was synthesized according to a modified procedure previously described in the literature [38, 39]. The purity and structure of the complexes were verified by elemental microanalysis, 1H and 13C NMR spectroscopy. To record 1H and 13C NMR spectra of the [Pt(L)Cl2] complex, DMSO-d6 was used as a solvent. Other chemicals used in this work were commercial products of analytical grade purity and were purchased from a domestic manufacturer. Distilled water was used to prepare the solutions of the above-mentioned reagents.
Physical measurements
The 1H and 13C NMR spectra were recorded on a Varian Gemini 2000 spectrometer (1H at 200 MHz, 13C at 50 MHz) using 5 mm NMR tubes, at 25 °C in DMSO-d6 or D2O containing TSP (sodium 3-(trimethylsilyl)propionate) as the internal reference. Chemical shifts were reported in parts per million (ppm). The UV–Vis spectra were recorded on a Shimadzu double-beam spectrophotometer equipped with thermostated 1.00 cm quartz Suprasil cells, after dissolving the corresponding dinuclear Pt(II) complexes in water, over the wavelength range of 200–600 nm. The concentrations of the Pt(II) complexes were 5⋅10−5 mol/L. Fluorescence measurements were carried out on a RF-1501 PC spectrofluorometer (Shimadzu, Japan). The excitation and emission bandwidths were both 10 nm. Elemental microanalyses for carbon, hydrogen and nitrogen were performed by the Microanalytical Laboratory, Faculty of Chemistry, University of Belgrade.
Synthesis of dinuclear [{Pt(L)Cl}2(µ-pydz)]Cl2 complexes
Dinuclear complexes of platinum(II) were synthesized starting from the corresponding mononuclear [Pt(L)Cl2] complexes according to the procedures described in the literature [39,40,41].
Mononuclear [Pt(L)Cl2] complexes were converted into the corresponding monodimethylformamide complexes ([Pt(L)Cl(DMF)]+) by treatment with 0.98 equivalents of AgNO3. To a suspension containing 0.30 mmol of mononuclear complexes in 10 mL of dimethylformamide (DMF) a solution containing 0.29 mmol of AgNO3 (49.26 mg) in 5 mL of DMF was added. The reaction mixture was left on a stirrer overnight at room temperature in the dark. The AgCl precipitate was separated by filtration, and the filtrate containing [Pt(L)Cl(DMF)]+ complexes was used as a starting material for the synthesis of dinuclear [{Pt(L)Cl}2(µ-pydz)]Cl2 complexes.
Into a solution containing 0.01 g of pyridazine (0.15 mmol) in 5 mL of DMF, a solution of the previously obtained monodimethylformamide complexes was slowly added dropwise. The reaction mixture was left on the stirrer in the dark for 24 h at room temperature. Dimethylformamide was evaporated on a rotary vacuum evaporator, and the dry residue was washed with ether. After that, the dry residue was dissolved in an aqueous LiCl solution with a concentration of 0.5 mol/L. Light yellow crystals of dinuclear platinum(II) complexes were separated by filtration, then washed with ether and dry on air. Yield was 40–60%. The purity and structure of the complexes were checked using the results of elemental microanalysis, NMR (1H and 13C) and UV–Vis spectroscopy.
[{Pt(1,2-pn)Cl}2(µ-pydz)]Cl2 (C1). Anal. Calcd. for (C10H24N6Cl4Pt2: FW = 760.31): C, 15.80; H, 3.18; N, 11.05%. Found: C, 15.46; H, 3.08; N, 11.19%. 1H NMR (D2O, 200 MHz, δ, ppm) 1.46 (m, CH3 1,2-pn), 2.79 (m, CH2/CH 1,2-pn), 8,14 (m, H4/H5 pydz), 9,58 (m, H3/H6 pydz); 13C NMR (D2O, 50 MHz, δ, ppm): 18 (CH3 1,2-pn), 54, 60 (CH2/CH 1,2-pn), 137 (C4/C5 pydz), 164 (C3/C6 pydz). UV–Vis (H2O, λmax, nm): 301 (ε = 3.3⋅103 M−1 cm−1).
[{Pt(ibn)Cl}2(µ-pydz)]Cl2 (C2). Anal. Calcd. for (C12H28N6Cl4Pt2: FW = 788.36): C, 18.28; H, 3.58; N, 10.66%. Found: C, 18.41; H, 3.46; N, 10.72%. 1H NMR (D2O, 200 MHz, δ, ppm) 1.46 (m, CH3 ibn), 2.66 (s, CH2, ibn), 8.18 (m, H4/H5, pydz), 9.62 (m, H3/H6, pydz); 13C NMR (D2O, 50 MHz, δ, ppm): 26 (CH3 ibn), 64, 65 (CH2/C ibn), 135 (C4/C5 pydz), 162 (C3/C6 pydz). UV–Vis (H2O, λmax, nm): 301 (ε = 3.5⋅103 M−1 cm−1).
[{Pt(1,3-pn)Cl}2(µ-pydz)]Cl2 (C3). Anal. Calcd. for (C10H24N6Cl4Pt2: FW = 760.31): C, 15.80; H, 3.18; N, 11.05%. Found: C, 15.52; H, 3.02; N, 11.11%. 1H NMR (D2O, 200 MHz, δ, ppm): 1.89 (m, CH2 1,3-pn), 2.79 (m, CH2 1,3-pn), 8.14 (m, H4/H5 pydz), 9.57 (m, H3/H6 pydz); 13C NMR (D2O, 50 MHz, δ, ppm): 30, 45, 46 (CH2 1,3-pn), 139 (C4/C5 pydz), 166 (C3/C6 pydz).UV–Vis (H2O, λmax, nm): 282 (ε = 4.0⋅103 M−1 cm−1).
[{Pt(1,3-pnd)Cl}2(µ-pydz)]Cl2 (C4). Anal. Calcd. for (C14H32N6Cl4Pt2: FW = 816.41): C, 20.60; H, 3.95; N, 10.29%. Found: C, 20.41; H, 3.82; N, 10.16%. 1H NMR (D2O, 200 MHz, δ, ppm): 0.97 (m, CH3 1,3-pnd), 1,82 (m, CH2 1,3-pnd), 2.85 (m, CH2/CH 1,3-pnd), 8,15 (m, H4/H5 pydz), 9.63 (m, H3/H6 pydz); 13C NMR (D2O, 50 MHz, δ, ppm): 12 (CH3 1,3-pnd), 30, 33, 34, 43, 44, 56, 57 (CH2/CH 1,3-pnd), 135 (C4/C5 pydz), 163 (C3/C6 pydz). UV–Vis (H2O, λmax, nm): 292 (ε = 3.8⋅103 M−1 cm−1).
Structural and stability studies of C1–C4 complexes by UV–Vis spectroscopy and high resolution mass spectrometry
The stability of 5·10–5 M C1–C4 complexes in the phosphate-buffered saline (PBS, 0.01 M, pH = 7.4) was studied by observing the changes in their absorption spectra over 72 h at room temperature. The UV–Vis spectra in the range of 250–380 nm were recorded using a Shimadzu double-beam spectrophotometer equipped with thermostatted 1 cm quartz Suprasil cuvettes. In addition, the stability of C1–C4 complexes was investigated in D2O as solvent by 1H NMR spectroscopy.
High resolution mass spectra (HRMS) were recorded using the Quadrupole Time-of-Flight mass detector (6550 iFunnel Q-TOF MS, Agilent Technologies; Santa Clara, CA, USA) equipped with a dual spray Agilent Jet Stream (AJS) electrospray ion source. Mass detector operated in accurate TOF/MS scanning mode recording positive ions in the m/z range of 100–1500, under following conditions: capillary voltage, 3500 V, fragmentor voltage, 175 V, nozzle voltage, 1000 V, skimmer 1, 65 V, octupole RF peak, 750 V, desolvation gas (nitrogen) temperature, 150 °C, desolvation gas (nitrogen) flow, 14 L min−1, nebulizer, 35 psig, sheath gas (nitrogen) temperature, 300 °C, sheath gas (nitrogen) flow, 11 L min−1. Ions m/z 121.05087300 and 922.00979800 were used as a lock mass for accurate mass measurements. A personal computer system running Agilent MassHunter software (revisions B.06.01 and B.07.00) was used for data acquisition and processing, respectively.
Liquid chromatograph (1290 Infinity LC system; Agilent Technologies, Waldbronn, Germany) with a quaternary pump, a column oven, diode-array detector (DAD), and an autosampler, was used to introduce into the system in a mobile phase stream composed of a solvents A (0.1% formic acid in water) and B (ACN) (1:1, v/v) with a flow rate of 0.20 cm3 min−1. The injection volumes of samples dissolved in ACN:H2O (1:5), c = 1 mg cm−3 were 1 μL.
Interactions of [{Pt(L)Cl}2(µ-pydz)]Cl2 complexes with biomolecules
Interaction with CT-DNA
Uv–Vis spectroscopy: The binding of complexes C1–C4 towards CT-DNA was studies by UV–Vis spectroscopy to investigate the possible DNA-binding modes as well as to calculate the binding constants (Kb). The series of Pt(II)/CT-DNA solutions were prepared by mixing the fixed concentration of the complex solution (25.7 µM) with increments of DNA stock solution (0–25.7 μM). The buffer (0.01 M PBS, pH = 7.4 at 37 °C) was used for absorption measurements and absorbance were recorded at 37 °C after each successive addition of CT-DNA solution and equilibration (24 h). The value of intrinsic binding constant (Kb) was determined using the following equation [42]:
where [DNA] is the concentration of CT-DNA, εa is the extinction coefficient of the complex at a given CT-DNA concentration, εf and εb are the extinction coefficients of the complex in free solution and when it is fully bound to CT-DNA, respectively (εb corresponds to Aobsd/[complex] (Aobs = Ameasured − ADNA)). The plot of [DNA]/(εa − εf) versus [DNA] give a straight line with 1/(εb − εf) and 1/[Kb(εb − εf)] as slope and intercept, respectively. The value of Kb was calculated from the ratio of the slope of the intercept.
Emission fluorescence spectroscopy: The interactions of dinuclear [{Pt(L)Cl}2(µ-pydz)]Cl2 complexes with CT-DNA were examined in the presence of EtBr by using emission fluorescence spectroscopy. A platinum(II) complex solution was added to the solution obtained by mixing the EtBr and CT-DNA solution with concentrations of 8 μM in a 1:1 molar ratio, so that the concentration ratio of the platinum(II) complex and CT-DNA was in the range of 0.0 to 1.0. Phosphate buffer solution (0.01 M, pH = 7.4) was used to prepare the above mentioned solutions. In order to investigate the competitive reactions of dinuclear platinum(II) complexes and EtBr with CT-DNA, fluorescence spectra of EtBr/CT-DNA were recorded in the absence and presence of dinuclear Pt(II) complexes. Emission spectra were recorded in the wavelength range 550–750 nm, with extinction at 527 nm and fluorescence emission at 612 nm. The Stern–Volmer constant (Ksv) is determined based on the equation:
where I0 and I are the fluorescence intensities before and after the addition of the platinum(II) complex to the EtBr/CT-DNA solution while [Q] is the concentration of the Pt(II) complex. The obtained results are shown graphically as the dependence of I0/I on [Q]. The Stern–Volmer constant (Ksv) was determined from the slope of the obtained line. The stability constant (Ka), as well as the number of binding sites (n), was obtained based on the equation: log (I0 − I)/I = logKa + n∙log[Q] [43] where I0 and I are the fluorescence intensities before and after addition of the platinum(II) complex to the EtBr/CT-DNA solution while [Q] is the concentration of the Pt(II) complex. The obtained results are shown graphically as the dependence of log (I0–I)/I on log[Q]. The Ka value is obtained from the intersection of the line with the y axis, and the number of binding sites (n) from the slope of the line.
Viscosity: Viscosity measurements were performed using Anton Paar 4100 M rolling ball viscometer at 25.0 ± 0.1 °C. Relative viscosity of a CT-DNA solutions ηr = η/η0 (η is the CT-DNA viscosity in the presence of the complex, η0 is the viscosity of CT-DNA in PBS buffer) at constant concentration of CT-DNA and with different concentrations of C1–C4 complexes was measured. The data were presented as (η/η0)1/3 versus the ratio [complex]/[CT-DNA].
Interactions of the complex with BSA
The interactions of dinuclear platinum(II) complexes, [{Pt(L)Cl}2(µ-pydz)]Cl2 with BSA were investigated using emission fluorescence spectroscopy. Emission spectra were recorded in the range of 300–500 nm, with excitation at 295 nm [21]. The binding effect of the tested complexes to BSA was observed based on the decrease in the emission intensity of BSA at 360 nm. In the series of tested solutions, the concentration of BSA was constant (8 µM). BSA and [{Pt(L)Cl}2(µ-pydz)]Cl2 complexes were mixed in different molar ratios, so that the concentration of the complex was from 0 to 160 µM. All emission spectra were recorded under the same experimental conditions in 0.01 M PBS (pH = 7.4) and at a temperature of 25 °C. The fluorescence spectra of the [{Pt(L)Cl}2(µ-pydz)]Cl2 complex were recorded in a buffer solution under the same experimental conditions, and no fluorescence emission was detected. The Stern–Volmer constant (Ksv) is determined based on the equation:
where I0 is the initial fluorescence intensity of tryptophan, and I is the fluorescence intensity of tryptophan in BSA after the addition of the [{Pt(L)Cl}2(µ-pydz)]Cl2 complex to the protein solution, kq is the fluorescence quenching constant, τ0 is the average fluorescence time of albumin in the absence of the complex (τ0 = 10–8 s) and [Q] is the concentration of the complex. The Stern–Volmer constant is obtained graphically from the slope of the line when the values I0/I are plotted on the y axis and [Q] on the x axis.
The binding constant (Ka) as well as the number of binding sites (n) were obtained based on the logarithmic Scatchard equation:
The obtained results are shown graphically as the dependence of log(I0 − I)/I on log[Q]. The Ka value is obtained from the intersection of the line with the y axis, and the number of binding sites (n) from the slope of the line.
Molecular docking
The molecular docking study was performed on chloro dinuclear Pt(II) complexes (C1–C4) and their hydrolyzed derivatives (C1W–C4W). The structures of mentioned dinuclear Pt(II) complexes were optimized by B3LYP method, with 6–31 g** basis set for non-metal atoms and lanl2dz basis set for Pt. The Merz–Kollman atomic charges were calculated for all atoms, at same level of theory, according to the scheme via the RESP procedure [44].
The structure of DNA was extracted from Protein Data Bank (pdb code: 1BNA) [45] and used for docking study as a target for tested complexes. Selected structure represents synthetic double stranded dodecamer with two G≡C rich region and one A=T rich region, inserted in between, without (sequence: d(CpGpCpGpApApTpTpCpGpCpG)) DNA intercalation gap. Docking study was also performed at BSA and HSA, which structures were extracted from the PDB with 4F5S [46] pdb code for BSA and 4L8U [47] pdb code for HSA.
The preparation of DNA, BSA and HSA structures was done in AutoDock 4.2 program [48] that includes the removing of water and ligands from crystal structures of biomolecules. To generate grid and docking parameter files for the optimized structures of dinuclear Pt(II) complexes, DNA and both transport protein structures, the AutoDockTools program (64) was used. The structures of DNA and proteins were treated as a rigid one, while the single acyclic bonds of metal complexes were allowed to rotate freely. To accommodate the investigated complexes, the grid box containing the whole biomolecule was used. The Lamarckian genetic algorithm, with 100 runs for each docking screen, was used as the search method for virtual screening. The visualization and analysis of the docking results was performed in the Discovery Studio (BIOVIA Software product) [49].
Cell lines
The cytotoxic effects of C1–C4 complexes were evaluated using several tumor cell lines including mouse melanoma (B16–F10), breast cancer (4T1) and Lewis lung carcinoma (LLC1) cell lines, human melanoma (A375), lung adenocarcinoma (A549) and breast cancer (MDA-MB 468) cell lines, as well as human fibroblast (MRC-5) cell line, all purchased from the American Type Culture Collection (ATCC, VA, USA).
All cells were maintained in complete growth medium which consisted of high-glucose Dulbecco's Modified Eagle Medium (DMEM, Sigma Aldrich, Munich, Germany) supplemented with 10% fetal bovine serum (FBS, Sigma Aldrich), penicillin G (100 IU/mL, Sigma Aldrich) and streptomycin (100 μg/mL, Sigma Aldrich), and in the atmosphere containing 5% CO2 at 37 °C at absolute humidity. Cell number and viability were determined by trypan blue staining and only cell suspensions with ≥ 95% viable cells were used.
Assessment of cytotoxicity
The cytotoxicity of the platinum(II) complexes ([{Pt(1,2-pn)Cl}2(μ-pydz)]Cl2 (C1), [{Pt(ibn)Cl}2(μ-pydz)]Cl2 (C2), [{Pt(1,3-pn)Cl}2(μ-pydz)]Cl2 (C3) and [{Pt(1,3-pnd)Cl}2(μ-pydz)]Cl2 (C4)) was evaluated by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay and compared with cytotoxicity of cisplatin. Tumor cells were harvested from the culture flasks and 5 × 103 cells/well were seeded into 96-well cell culture plates. The cells were allowed to adhere and after 24 h the cells were treated with two-fold growing concentrations of C1–C4 complexes or cisplatin (7.8–500 μM). Control wells were prepared by the addition of complete cell culture medium alone. All cell lines were incubated at 37 °C in the atmosphere containing 5% CO2 at absolute humidity for 48 and 72 h. The complex-containing medium was removed from each well and MTT solution at final concentration of 0.5 mg/mL in PBS was added to each well and incubated for additional 4 h at 37 °C in the 5% CO2 incubator. The optical density of each well was determined at 595 nm using a Zenyth 3100 multimode microplate detector. Experiments were repeated three times in triplicates. Also, by using Microsoft Office Excel 2010, IC50 values were calculated. The percentage of viable cells was calculated using the formula: % viable cells = (E − B)/(S − B) × 100, where B is for background of medium alone, S is for total viability/spontaneous death of untreated target cells and E is for experimental well.
Platinum cellular uptake assay
In order to explore compound cellular uptake, 4T1 cells were seeded for 12 h at 37 °C and treated with C1–C4 or cisplatin (30 μM) for 12 h. After treatment, the cells were harvested and then washed with PBS, and pelleted for further analysis. The digestion of the cells was performed on Advanced Microwave Digestion System (Ethos 1, Milestone, Italy) using HPR-1000/10S high pressure segmented rotor. The cells were digested with of 4.5 mL HNO3 (65 wt.%, Suprapur®) and 0.5 mL H2O2 (30 wt.%, Suprapur®) (both of Merck KGaA, Darmstadt, Germany) by microwave-assisted digestion technique for ICP-OES measurement. The content of the Pt in solution samples was determined by inductively coupled plasma optical emission spectrometry, ICP-OES (iCAP 6500 Duo ICP, Thermo Fisher Scientific, Cambridge, United Kingdom). The calibration solutions were made from certified plasma standard solution: Platinum, plasma standard solution, Specpure®, Pt 1000 µg/mL (Alfa Aesar GmbH & Co KG, Germany). The quantification was performed on wavelength Pt II 214.423 nm. Quality control was carried out using blank samples, matrix-matched calibration solutions and triplicate analyses of each sample. The reliability of measurements was approved by a relative standard deviation (RSD) < 0.5%. The limit of detection (LOD) was 0.962 µg/L, and the limit of quantification (LOQ) was 3.206 µg/L in solutions of totally mineralized samples. The analytical process quality control (QC) performed by the use of standard addition of the platinum complex on untreated cells (control two) and by performing the entire analytical process. Recovery for platinum content was 99.7%. Concentrations of platinum of sample were expressed as ngPt/1 × 106 cells.
Detection of apoptosis
For apoptosis detection, 4T1 and MDA-MB 468 cells were exposed to the C2 or cisplatin at concentrations of 10 and 30 μM. After 12 h of the treatment with tested compound, the cells were harvested, washed two times in ice cold PBS, and double stained with fluorescein isothiocyanate (FITC) labeled Annexin V (BD Pharmingen, CA, USA) and Propidium Iodide (PI) (Sigma-Aldrich) in ice cold binding buffer (10 × binding buffer: 0.1 M Hepes/NaOH (pH 7.4), 1.4 M NaCl, 25 mM CaCl2) at the room temperature in the dark for 15 min. For determination of the percentages of dead cells, FACS Calibur flow cytometer (BD Biosciences, CA, USA) was used, and the data were analyzed using FlowJo software (Tree Star, Inc., OR, USA). The fluorescence distribution was displayed as a two-color dot plot analysis, and the percentage of fluorescent cells in each quadrant was determined. Four different populations of cells are distinguished: Annexin V−PI− are viable cells, Annexin V+PI− are cells in the early stage of apoptosis, Annexin V+PI+ are cells in the late stage of apoptosis, while Annexin V−PI+ are necrotic cells.
Cell cycle analysis
4T1 and MDA-MB 468 cells were exposed to C2 or cisplatin at the concentration of 30 μM for 12 h and cell cycle analysis was performed with Vybrant® DyeCycle™ Ruby stain (Thermo Fisher Scientific, MA, USA) according to manufacturer’s instructions. The results were analyzed by FACSCalibur flow Cytometer (BD Biosciences) and FlowJo (Tree Star) software was used for the cell cycle distribution analysis.
Flow cytometric analysis
4T1, grown in culture medium, were treated with C2 or cisplatin at the concentration of 30 μM for 12 h. The treated cells were fixed and permeabilized with permeabilization buffer (BD Bioscience) and incubated with anti-mouse cyclin E antibody (#MA5-14336, Thermo Fisher Scientific) and allophycocyanin (APC) conjugated anti-mouse protein kinase AKT phosphorylated on S473 (pAKT) antibody (#17-9715-42, eBioscience). For staining cyclin E cells were additionally incubated with secondary goat anti-mouse IgG FITC (ab6785, Abcam, Cambridge, UK). Flow cytometry was performed on FACSCalibur flow cytometer (BD Biosciences). The data were analyzed using FlowJo software (Tree Star).
Real-time PCR analysis
After the treatment with 30 μM of cisplatin or C2 for 12 h, total RNA from 4T1 cells was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Total RNA (2 μg) was reverse transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). qRT-PCR was performed using Power SYBR MasterMix (Applied Biosystems) and miRNA-specific primers for mouse caspase 3 (forward 5′-AAATTCAAGGGACGGGTCAT-3′ and reverse 5′-ATTGACACAATACACGGGATCTGT-3′), caspase 9 (forward 5′-GCTCTTCCTTTGTTCATCTCC-3′ and reverse 5′-CATCTGGCTCGGGGTTACTGC-3′), RIPK1 (forward 5′-AGAAGAAGGGAACTATTCGCTGG-3′ and reverse 5′-CATCTATCTGGGTCTTTAGCACG-3′), Cyclin D3 (forward 5′-CCGTGATTGCGCACGACTTC-3′ and reverse 5′-TCTGTGGGAGTGCTGGTCTG-3′), p27 (forward 5′-AGGAGAGCCAGGATGTCAGC-3′ and reverse 5′-CAGAGTTTGCCTGAGACCCAA-3′), c-Myc (forward 5′-CGGACACACAACGTCTTGGAA-3′ and reverse 5′-AGGATGTAGGCGGTGGCTTT-3′) and β-actin (forward 5′-AGCTGCGTTTTACACCCTTT-3′ and reverse 5′-AAGCCATGCCAATGTTGTCT-3′) as a housekeeping gene. qPCR reactions were initiated with a 10-min incubation time at 95 °C followed by 40 cycles of 95 °C for 15 s and 60 °C for 60 s in a Master cycler ep realplex (Eppendorf, Hamburg, Germany). Relative amount of mRNA was normalized to the β-actin content for each sample. Relative expression of genes was calculated according to the formula 2−(Ct−Ctactin), where Ct is the cycle threshold of the gene of interest and Ctactin is the cycle threshold value of the housekeeping gene (β-actin). Data points are represented by the expression ratio and mean ± SD fold of control in 4T1 cells.
Animals
Six to eight weeks old BALBc female mice were used for the experiments. Mice were equalized in weight and randomized in experimental and control groups. All mice were maintained in our animal facilities. Mice were housed in a temperature controlled environment with a 12 h light–dark cycle and were given standard laboratory chow and water ad libitum. All animals received humane care, and all experiments were approved by, and conducted in accordance with the Guidelines of the Animal Ethics Committee of the Faculty of Medical Sciences of the University of Kragujevac (number 01-14555).
Breast cancer model
Mice were inoculated with 3 × 104 4T1 tumor cells in 50 μl of PBS (Invitrogen), orthotopically into the fourth mammary fat-pad as described previously [50]. When tumors become palpable (7 days after the implantation of 4T1 cells), the mice received either C2 in two different doses (3 and 6 mg/kg), cisplatin (3 mg/kg) or vehicle (PBS) by intraperitoneal injection every 2–3 days, eight doses in total. Cisplatin was used as a conventional chemotherapeutic agent and its dose of 3 mg/kg was selected based on the previously published data [50, 51]. Primary 4T1 mammary tumors were measured every second day using caliper. Mice were sacrificed on 25th day of the experiment. Tumor volumes (mm3) were calculated as L × W2/2, where L represents the major axis of the tumor, while W represents minor axis.
Statistical analysis
Following the assessment of data normality distribution, the statistical analysis was conducted by two-tailed Student’s t-test or nonparametric Mann–Whitney Rank Sum test where appropriate, using SPSS 26.0 for Windows software (SPSS Inc., Chicago, IL, USA). For survival rate assessment, statistical testing between groups at each time point was determined using Fisher’s exact test. The data were shown as mean ± standard deviation (SD) or mean ± standard error (SE). Values of p < 0.05 were considered as statistically significant.
Results and discussion
Synthesis and structural characterization of the dinuclear platinum(II) complexes C1–C4
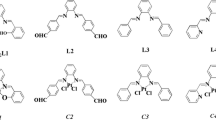
Dinuclear platinum(II) complexes, [{Pt(1,2-pn)Cl}2(μ-pydz)]Cl2 (C1), [{Pt(ibn)Cl}2(μ-pydz)]Cl2 (C2), [{Pt(1,3-pn)Cl}2(μ-pydz)]Cl2 (C3) and [{Pt(1,3-pnd)Cl}2(μ-pydz)]Cl2 (C4) (Fig. 1) were obtained from the corresponding mononuclear complexes according to a modified procedure previously described in the literature [32, 40, 41]. In the reaction of mononuclear [Pt(L)Cl2] complex (L is 1,2-pn, ibn, 1,3-pn, 1,3-pnd) with AgNO3 in a 1:0.98 molar ratio in DMF solution, substitution of one chlorido ligand with a DMF molecule occurs. In order to obtain dinuclear platinum(II) complexes, the corresponding mononuclear complex, [Pt(L)(Cl)DMF)]+, was mixed with pyridazine (pydz) in a 2:1 molar ratio in DMF and at room temperature. Dinuclear complexes crystallized from an aqueous solution of LiCl.

Structural formula of [{Pt(L)Cl}2(μ-pydz)]2+ complexes (C1–C4) employed in this study
1H and 13C NMR spectroscopy
As it is shown in Fig. 1, the synthesized dinuclear platinum(II) complexes have the same bridging pyridazine ligand (pydz) and different diamine ligands 1,2-pn, ibn, 1,3-pn, 1,3-pnd. 1H and 13C NMR spectrum of pyridazine as well as synthesized platinum(II) complexes were recorded in deuterated water (D2O) as solvent. Fig. S1 (Supplementary material) shows the corresponding 1H NMR spectra.
In the 1H NMR spectrum of free pyridazine, two multiplets appear at 7.83 and 9.20 ppm, which originate from the equivalent protons of the pyridazine ring (H4/H5 and H3/H6). After coordination of ligand to the platinum(II) ion, the signal shifts to a lower field, i.e. to a higher chemical shift by Δδ = 0.31 ppm (H4/H5), and by Δδ = 0.38 ppm (H3/H6) [41].
In the 13C NMR spectra, after the coordination of pyridazine to the Pt(II) ion, the signal shifts to a lower field (Fig. S1, Supplementary material).
After chelate coordination of 1,2-pn, ibn, 1,3-pn and 1,3-pnd ligands for Pt(II) ion in C1–C4 complexes, in the 1H NMR spectrum the signals originating from the free ligands shift to a lower field, i.e. to a higher chemical shift. The signals from the carbon atoms of the 1,2-pn, ibn, 1,3-pn and 1,3-pnd also shift to higher chemical shifts in regards to those for free ligands.
High resolution mass spectrometry
Results of LC–MS additionally confirm the structures of dinuclear Pt(II) complexes. Complexes represent bivalent cationic species, labeled in the mass spectrum with the letter M (Figs. S2–5, Supplementary material), because the C designation refers to a carbon atom. The first two complexes, Pt2C10N6H24Cl2 (M1 or [{Pt(1,2-pn)Cl}2(μ-pydz)]Cl2 complex) and Pt2C12N6H28Cl2 (M2 or [{Pt(ibn)Cl}2(μ-pydz)]Cl2 complex), form adducts with water molecule and acetonitrile ([M1 + H2O + CH3CN-H]+ and [M2 + H2O + CH3CN-H]+), corresponding to the highest m/z values in the spectra. In addition, [M-H]+ ion species were detected for both complexes. In the case of the third Pt2C10N6H24Cl2 complex (M3 or [{Pt(1,3-pn)Cl}2(μ-pydz)]Cl2 complex), the largest m/z value originates from [M3 + H2O + 2CH3CN + 2Cl + H]+ ion adduct, while the [M3-H]+ ion species was not detected. Fragmentation of the complex is most pronounced in MS of the fourth complex, Pt2C14N6H32Cl2 (M4 or [{Pt(1,3-pdn)Cl}2(μ-pydz)]Cl2 complex). In addition to the dinuclear complex M4, two mononuclear complexes, PtC9N4H18Cl (M4′) and PtC5N2H14Cl (M4"), were also detected (Fig. S5, Supplementary material). Ligand environment of Pt(II) is completely the same in M4 and M4′ complexes, except that pyridazine is a monodentate ligand in the M4′ complex. The structure of M4" complex can be obtained from M4′ by removing the pyridazine ligand. The peak with the highest m/z value originates from the [M4 + M4′ + 2H2O-2H]+ ion adduct, while the highest intensity peak corresponds to the [M4" + CH3CN]+ ion adduct.
Taking into account the results of the mentioned techniques, the structures of investigated complexes were drawn and optimized. Optimized structures are depicted in Fig. S6 (Supplementary material). The chloro ligands are located on different sides of the plane of the complex, which passes through the pyridazine bridging ligand. The mean planes of the chelate rings are normal to this plane, as a consequence of intramolecular hydrogen bonds between the Pt–N-H group and the chloro ligands.
UV–Vis spectrophotometry
The UV–Vis spectra of synthesized dinuclear complexes, C1–C4 are given in Fig. S7 (Supplementary material). Absorption maxima are located at 282 nm for C3, 292 nm for C4 and at 301 nm for C1 and C2 complex. The absorption maxima of the synthesized complexes have a bathochromic shift in relation to the absorption maxima of the ligands, due to π….π electronic transitions in the heterocyclic pyridazine ligand.
Stability of C1–C4 complexes
The stability of dinuclear C1–C4 complexes was investigated by UV–Vis spectroscopy during 72 h. Solutions of all complexes were prepared PBS buffer (0.01 M, pH = 7.4). The samples were stored at room temperature between measurements. After 72 h, only very slight changes in the intensity of the absorption maxima were observed (Fig. S8a, Supplementary material).
In addition, 1H NMR spectroscopy was used for investigation of stability of the C1–C4 complexes at different time intervals (t = 30 min, 24 and 72 h) (Fig. S8b for complex C3, Supplementary material). New very weak resonances in the aromatic region, arising from free pyridazine protons, appear in the spectrum after 72 h. These resonances correspond to the free bridging ligands, as result of very slow decomposition of dinuclear complexes. It could be concluded that during the experimental time the investigated complexes are stable.
Interactions C1–C4 complexes with biomolecules
Interactions of C1–C4 complexes with CT-DNA
The interactions of dinuclear platinum(II) complexes with the CT-DNA molecule were investigated using UV–Vis spectroscopy, emission fluorescence spectroscopy and viscosity mesuremants.
Since the interaction of complexes with DNA leads to perturbations in their electronic absorption spectra, UV–Vis spectroscopy is an tool to study these interactions [52]. The absorption spectra of C1–C4 with CT-DNA were recorded for a constant complex concentration with different complex/DNA ratios (r) and are shown in Fig. S9 (Supplementary material). The intensity of the absorption bands at λ = 260 nm is gradually increasing. The intrinsic equilibrium binding constants (Kb) were calculated to compare the binding affinity of the complexes with CT-DNA. The calculated Kb values for the C1–C4 complexes were obtained from the plot of [DNA]/(εa − εf) versus [DNA] (Fig. S10, Supplementary material, Table 1) indicating that mode of binding of these complexes to CT-DNA helix is not intercalative. Also, considering that there is no bathochromic shift of the absorption maximum [53], it can be concluded that the investigated C1–C4 complexes do not form covalent DNA adducts.
EtBr was used as a typical intercalator for these tests. Changes in the EtBr/CT-DNA emission spectra after addition of the complex solution are used to study the interactions of the complex with DNA. In the emission spectra after the addition of the complex, decrease or increase in fluorescence emission occurs, which indicates that the complex replaces EtBr, with formation of new adduct CT-DNA/Pt(II) complex [54]. The addition of the platinum(II) complex solution (the concentration of the complex increases) leads to a decrease in the emission intensity at 612 nm, which indicates that there is competition between EtBr and the examined platinum(II) complexes in relation to CT-DNA. However, the decrease in fluorescence intensity is very small, confirming that our complexes are weak intercalators. The addition of complexes caused the fluorescence emission of EtBr to drop up to 20% of initial maximal value for all dinuclear platinum(II) complexes (Fig. 2). The obtained values for the Stern–Volmer constant (Ksv), as well as for the stability constant (Ka), (Table 1) indicate that our examined complexes are not good intercalators and that they achieve very weak interactions with CT-DNA (Table 1). All complexes achieve approximately the same number of binding sites (n ≈ 1), which confirms that C1–C4 bind in the same way. However, the Ka constant for the C2 complex have the lowest value compared to the other investigated complex, which can be attributed to the steric effect of the two methyl groups in bidentately coordinated diamine ligands. A small decrease in the maximum of the emission band, as well as the obtained values for Ksv indicate that the dinuclear C1–C4 complexes do not displace ethidium bromide from the EtBr/CT-DNA adduct.

The intensity of the fluorescence emission of EtBr at λem = 612 nm (λex = 527 nm) versus r (r = [Pt(II) complex]/[CT-DNA]) for C1–C4 complexes ([EtBr] = [DNA] = 8 μM; [complex] = 0–8 μM)
Viscosity measurements were used to further clarify the interaction mode of these four platinum(II) complexes (C1–C4) with CT-DNA. The addition of increasing amounts (up to r = 1.0) of each complex to a CT-DNA solution (20 μM) caused a slight decrease in the viscosity of CT-DNA (Fig. S10, Supplementary material) indicating the non-intercalative interaction of complexes with CT-DNA.
The molecular docking to DNA was performed for prediction of drug binding mode of investigated Pt(II) complexes (C1–C4). For this purpose, aqua complexes (C1W–C4W) were used for the calculations, because the investigated complexes (C1–C4) penetrate into the cell nucleus, where DNA is located, only as aqua derivatives. Binding energies of the most stable adducts are in the range from − 6.4 to − 7.3 kcal/mol (Fig. 3). The results of docking study have shown that the groove spanning (adducts with complexes C1 and C2) and backbone tracking binding mode (adduct with complex C3) are the most stable binding modes (Fig. 3), except for the complex C4, where the major groove bonding occurs, in A-T rich region of DNA. A slightly higher binding energies for binding to the phosphate backbone is result of the hydrogen bonds between (Pt)–N-H and (Pt)–O-H groups from the metal complex and backbone oxygens of DNA (Fig. S11, Supplementary material), that are electrostatically reinforced. It is obvious that the six-membered chelate ring of C4 complex, substituted with an additional ethyl group, sterically hinders the approach of coordinated water and ligating NH2 groups to form the electrostatically strengthened hydrogen bonds with the phosphate backbone. It is known that dinuclear complexes, due to their binding to the phosphate backbone, cause the displacement of intercalated EtBr from double helix of DNA [9], as a consequence of the bending of DNA structure. The bending of DNA structure that results in the condensation of DNA, is the reason for a slight decrease in the viscosity of CT-DNA. One can conclude that reduction of viscosity and the displacement of intercalated EtBr can be also caused by binding in the major groove, as in the case of complex C4.

The structures of the most stable adducts of dinuclear Pt(II) complexes and DNA, as assessed by molecular docking. The complexes represent aqua derivatives (C1W–C4W) of the corresponding chloride Pt(II) complexes (C1–C4)
Interactions of platinum(II) complexes with BSA
Serum albumin, one of the most studied blood plasma proteins, plays an important role in the transport of metal ions and their complexes through the blood system to cells and tissues [55, 56]. Examining the interactions of serum albumin proteins and transition metal ion complexes, which are used as drugs, is of great importance, because it can provide answers to very important questions, both about biological activity and drug transport in the body. BSA is the most studied serum albumin due to its high structural similarity to HSA. Namely, human serum albumin contains one tryptophan at position Trp-214, while BSA contains two tryptophan residues, Trp-134 and Trp-214. BSA solution shows intense fluorescence emission at λem,max = 365 nm, and when excited at 295 nm [57, 58].
After the addition of C1–C4 complexes to the BSA solution, a decrease in fluorescence intensity is observed at λ = 365 nm (Fig. S12, Supplementary material), based on which it can be concluded that the tested complexes interact with this biomolecule. The decrease in fluorescence intensity can be attributed to changes in the protein structure, which are caused by a change in the environment of tryptophan in BSA, due to binding of proteins to complexes [57, 58].
The value of the dynamic Stern–Volmer constant (Ksv) and the fluorescence quenching constant (kq) for the interactions of the complex with BSA were determined using the Stern–Volmer equation and are shown in Table 2. In addition, Table 2 shows the values of the binding constants (Ka) as and the number of binding sites (n) of albumin for the investigated platinum(II) complexes, which were obtained based on the Scatchard equation. The obtained results are shown graphically as the dependence of log(I0 − I)/I on log[Q]. The Ka value is obtained from the intersection of the line with the y axis, and the number of binding sites (n) from the slope of the line.
Fluorescence quenching constants (kq) (> 1012 M−1 s−1) are higher in relation to different biopolymers that lead to fluorescence quenching, indicating the existence of a statistical interaction mechanism [58].
The values of the constants obtained based on the interaction of the investigated complexes with BSA can indicate the possibility of distribution of the potential drug (platinum(II) complex) in the blood plasma via BSA. Based on the Ka value, it can be concluded that C1–C4 complexes can bind to BSA and thus transport to the cell. Since the strongest known noncovalent interactions of avidin with diverse ligands (1015 M−1), from Ka values obtained for C1–C4 it could be concluded that these complexes can be released from BSA even before arrival to the target cells [59, 60]. Ka value for C2 complex is lower than Ka value for other investigated Pt(II) complexes indicating that C2 complex binds weaker to BSA (Table 2). The values of n are less than 1, which suggests that there is only one potential site in the BSA molecule where the corresponding complexes can bind.
Docking studies on BSA and HSA were performed with chloro complexes, because these two transport proteins are components of the extracellular matrix, where the complexes occur in the chloro form (C1–C4). BSA and HSA contains three domains (I, II and III), each consisting of two subdomains (A and B), while HSA has nine drug binding sites, marked as a DBS in Fig. 4. Molecular docking results have shown that C1–C4 complexes are bound to BSA in the IIA subdomain, in a binding site that is not considered transportable (Fig. 4). Binding energies are in range from − 4.22 to − 5.07 kcal/mol, indicating a moderate binding to the surface of the transport protein. Docking studies on BSA confirm the previous assumptions derived from the Ka values, which suggest that complexes can be released from BSA even before arrival to the target cells.

The most stable binding site of dinuclear Pt(II) complexes (C1–C4) to BSA and HSA, as assessed by molecular docking
All four complexes have a unique binding site on HSA, but this time between the IA and IIA subdomains. The assessed binding site is not a drug binding site (DBS). In Fig. 4 DBS are highlighted, as well as the compounds (painted black) that crystallized together with the selected HSA. Binding energies (from − 4.26 to − 4.44 kcal/mol) also indicate a moderate binding of C1–C4 complexes to HSA, as in the case of BSA. Electrostatically strengthened hydrogen bonds between the Pt–N-H group of positively charged complexes and negatively charged amino acids (Asp307 and Glu332) are mainly responsible for binding of investigated complexes in the most stable binding site (Fig. S13, Supplementary material). One can also conclude that in the binding site of the complex for BSA there are no amino acids to which the complex could be covalently bound. As the complexes are bound on the surface of the transport protein, at non-transportable site, it can be assumed that the complexes will be easily released during transport through the bloodstream.
Evaluation of C1–C4 complexes cytotoxicity
The effects of C1–C4 complexes on the viability of several tumor cell lines (4T1, MDA-MB 468, LLC1, A549, B16-F10, A375), as well as non-tumor MRC-5 cells, were determined by MTT assay. The cells were exposed to two-fold growing concentrations of C1–C4 complexes or cisplatin, as a comparative compound, for 48 h (Fig. 5) and 72 h (Fig. 6). IC50 values of tested complexes and cisplatin after 48 h and 72 h treatment were shown in Table 3 and 4, respectively.

The dose-dependent cytotoxicity of C1–C4 complexes toward mouse and human tumor cell lines after 48 h treatment. Tumor cell lines including mouse (4T1) and human (MDA-MD 468) breast cancer cells (a), mouse (LLC1) and human (A549) lung carcinoma cells (b), mouse (B16-F10) and human (A357) melanoma cells (c), as well as human fibroblasts (MRC-5) (d) were treated with two-fold growing concentrations of C1–C4 complexes or cisplatin (CDDP) for 48 h. The cell viability was determined by MTT assay. The data are presented as the mean from three independent experiments performed in triplicates

The dose-dependent cytotoxicity of C1–C4 complexes toward mouse and human tumor cell lines after 72 h treatment. Tumor cell lines including mouse (4T1) and human (MDA-MD 468) breast cancer cells (a), mouse (LLC1) and human (A549) lung carcinoma cells (b), mouse (B16-F10) and human (A357) melanoma cells (c), as well as human fibroblasts (MRC-5) (d) were treated with two-fold growing concentrations of C1–C4 complexes or cisplatin (CDDP) for 72 h. The cell viability was determined by MTT assay. The data are presented as the mean from three independent experiments performed in triplicates
The obtained data revealed that, following 48 h exposure, C2 showed dose-dependent cytotoxic effect against mouse breast cancer cells (4T1) (IC50 = 146.48 ± 75 μM), while C1, C3 and C4 were cytotoxic for these cells only at highest concentrations (250 and 500 μM) (Fig. 5a; Table 3). Interestingly, both C1 (IC50 = 16.3 ± 18.8 μM) and C2 (IC50 = 15.9 ± 0.8 μM) exhibited marked cytotoxic potential toward human breast cancer cells (MDA-MB 468) and their effects were similar to cisplatin (IC50 = 12.0 ± 1.5 μM) (Fig. 5a; Table 3). The cytotoxic potential of C3 and C4 toward MDA-MB 468 was observed only at highest concentrations (Fig. 5a). The dose-dependent cytotoxic activity of C1 and particularly C2 (IC50 = 56.3 ± 10.8 μM), was also recorded toward the mouse lung carcinoma cell line (LLC1), while C3 and C4 were cytotoxic for these cells only at highest concentrations after 48 h treatment (Fig. 5b; Table 3). However, it appears that the viability of human lung carcinoma cells (A549) was not significantly affected by tested complexes (Fig. 5b). Also, in contrast to the cisplatin treatment, the tested complexes exhibited weak cytotoxic potential for both mouse (B16-F10) and human (A375) melanoma cell lines observed only at highest concentrations (Fig. 5c). However, the obtained data have indicated that the tested complexes exhibit weak cytotoxic selectivity between tumor and non-tumor MRC-5 cell lines, although they show less toxicity against normal cells in comparison with cisplatin (Fig. 5d; Table 3).
The similar pattern of C1–C4 complexes cytotoxicity toward tested cell lines was determined following 72 h exposure (Fig. 6). 4T1 cells viability was markedly dose-dependently reduced when treated with C2 (IC50 = 51.1 ± 22.4 μM), but not other tested complexes (Fig. 6a; Table 4). Furthermore, both C1 (IC50 = 3.0 ± 1.3 μM) and C2 (IC50 = 4.0 ± 2.1 μM) were highly cytotoxic for MDA-MB 468 cells, similarly to cisplatin, while C3 and C4 exhibited weak cytotoxic potential toward these cells after 72 h treatment (Fig. 6a; Table 4). Also, the viability of LLC1 cells was dose-dependently reduced when treated with C1 (IC50 = 53.2 ± 2.42 μM) and C2 (IC50 = 30.2 ± 21.6 μM), but not C3 and C4 (Fig. 6b; Table 4). Moreover, lower IC50 values after 72 h when compared to 48 h exposure implicates time-dependent effect of complexes, especially C1 and C2. After 72 h exposure, the tested complexes exhibited low cytotoxic potential toward A549 (Fig. 6b), as well as B16-F10 and A375 cells (Fig. 6c), only at highest concentrations.
In addition, lower cytotoxicity of tested complexes for non-tumor MRC-5 cell line in comparison with cisplatin was determined following 72 h treatment (Fig. 6d; Table 4). The selectivity of C1–C4 and cisplatin for tumor cells was analyzed using selectivity index, calculated as the ratio of IC50 value for non-tumor cells and IC50 value for cancer cells [61]. After 72 h exposure, the selectivity indexes of cisplatin for all tested tumor cell lines were less than 2, which indicated general toxicity [61]. On the other hand, among tested complexes, C1 and C2 exhibited highest toxicity and also selectivity for human breast cancer cells MDA-MB 468 after 72 h treatment. In addition, both C1 and C2 showed selectivity for mouse LLC1 cells, while C2 also exhibited selectivity for mouse 4T1 cells.
In order to evaluate the uptake of tested complexes by tumor cells, the platinum content was quantified in 4T1 cells incubated with 30 μM of C1–C4 or cisplatin for 12 h. The platinum accumulation level was lower in tumor cells exposed to C1 and C2, while the highest cellular uptake was detected in cells treated with C4 (Fig. S14, Supplementary material). To explain the cellular uptake values, lipophilicity (determined by logP) and water solubility were predicted by SwissADME [62], a free web tool. A negative logP values for complexes (Table S1, Supplementary material) denotes a higher affinity of complexes for the aqueous than for the lipid phase. However, there is no relation between the values of logP and the measured values of the cellular uptake. On the other hand, the cellular uptake is inversely proportional to the water solubility of the Pt(II) complexes. It is obvious that less soluble complexes (like the C4 complex) penetrate the cell more easily.
Overall, among all tested complexes, C2 exhibits the strongest cytotoxic activity against tumor cells in vitro, in particular breast cancer cells, in both dose- and time-dependent manner.
C2 induces apoptosis of breast cancer cells
It has been well established that the dysregulation of apoptosis is one of the critical events in tumor development and also the important target for anti-tumor therapy [63]. Given the notable cytotoxic effect of C2 toward breast cancer cell lines, the next aim of the study was to evaluate the potential mechanisms involved in C2-induced tumor cell death. Therefore, the breast cancer cells of mouse (4T1) and human (MDA-MB 468) origin were treated with 10 and 30 μM of cisplatin or C2 for 12 h for further Annexin V and PI staining. The obtained data have shown that both cisplatin (at concentrations of 10 and 30 μM) and C2 (only at the higher concentration of 30 μM) trigger the apoptosis of 4T1 cells as demonstrated by significantly increased percentage of early apoptotic (Annexin V+PI−), but not late apoptotic (Annexin V+PI+) cells, when compared to untreated cells (Fig. 7a). Furthermore, this effect seems to be dose-dependent which has been implicated by the increased percentage of early apoptotic 4T1 cells treated with higher concentration of cisplatin or C2 in comparison with the cells treated with lower concentration of tested compounds (Fig. 7a). Also, there was significantly increased percentage of late apoptotic (Annexin V+PI+) 4T1 cells treated with higher when compared to lower concentration of C2 (Fig. 7a). The small percentage of 4T1 cells treated with cisplatin at both tested concentrations or C2 at higher concentration have undergone necrosis (Fig. 7a). It appears that cisplatin, at lower dose of 10 μM, was more efficient in the induction of both apoptosis and necrosis of 4T1 cells as shown by the increased percentage of early apoptotic and necrotic cells in comparison with the cells exposed to the same concentration of C2 (Fig. 7a). However, when applied at the higher dose, C2 and cisplatin exhibited similar effects on the induction of both 4T1 cells apoptosis and necrosis (Fig. 7a).

The effect of C2 on the apoptosis of 4T1 and MDA-MB 468 breast cancer cells. 4T1 (a) and MDA-MD 468 (b) cells were exposed to 10 and 30 μM of cisplatin (CDDP) or C2 for 12 h and stained with FITC-labeled Annexin V and Propidium iodide (PI) for further flow cytometric analysis. The data are presented as mean ± SD. *p < 0.05 vs. untreated cells; §p < 0.05 higher (30 µM) vs. lower (10 µM) concentration of cisplatin or C2; ¶p < 0.05 cisplatin vs. C2 at the same concentration of 10 or 30 µM. The representative dot plots denote the populations of viable, early apoptotic, late apoptotic and necrotic 4T1 and MDA-MD 468 cells following cisplatin or C2 treatment
Further, there was marked dose-dependent increase in the percentage of both early (Annexin V+PI−) and late (Annexin V+PI+) apoptotic MDA-MB 468 cells after either cisplatin or C2 treatment at both tested concentrations (Fig. 7b). Although cisplatin exhibited better effect on the induction of MDA-MB 468 cell apoptosis than C2 as shown by the increased percentages of early apoptotic cells (at the dose of 10 μM) and late apoptotic cells (at the dose of 30 μM) (Fig. 7b), the obtained results are in line with the high sensitivity of MDA-MB 468 cells to C2 treatment determined by cytotoxicity assays. Apart from 4T1 cells, it seems that neither cisplatin nor C2 affected the necrosis of MDA-MB 468 cells (Fig. 7b).
C2 promotes breast cancer cell accumulation at sub-G1 phase of cell cycle and triggers the intrinsic pathway of apoptosis
Next, the cell cycle profile of 4T1 and MDA-MB 468 breast cancer cells has been investigated after exposure to C2 or cisplatin (30 μM) for 12 h. As shown in Fig. 8a, upper panel, it appears that C2, similar to cisplatin, trend toward increased 4T1 cells accumulation at sub-G1 phase, while consequently decreased the percentages of cells at G0/G1 phase of cell cycle. The effect of cisplatin, but not C2, on the accumulation of cells at sub-G1 phase was accompanied with decreased percentage of cells in S phase compared to C2-treated cells and the cells in G2/M phase compared to untreated cells (Fig. 8a, upper panel). Also, the percentages of MDA-MB 468 cells at sub-G1 phase were significantly increased following 12 h treatment with cisplatin or C2 when compared to untreated cells (Fig. 8a, lower panel). The percentages of MDA-MB 468 cells at G0/G1 phase were accordingly decreased by cisplatin or C2 treatment (Fig. 8a, lower panel). Hence, it seems that C2 induces 4T1, and in particular MDA-MB 468 breast cancer cell accumulation at sub-G1 phase after 12 h treatment. This result is in line with findings that cisplatin promotes accumulation of human promyelocytic leukemia cells in sub-G1 phase of cell cycle, which indicate the cells entering into apoptosis or necrosis [64].

The effects of C2 on breast cancer cell cycle progression and death. a 4T1 and MDA-MD 468 cells were treated with 30 μM of cisplatin (CDDP) or C2 for 12 h. The cells cycle was analyzed by flow cytometry and the percentage of cells in different phases of the cell cycle was presented. The effects of cisplatin (30 µM) and C2 (30 µM) on the expression level of mRNA of caspase 3 (b), caspase 9 (c) and RIPK1 (d) by 4T1 cells were analyzed using RT-PCR after 12 h exposure. β-actin mRNA was used as an internal control. All data are presented as mean ± SD. *p < 0.05 vs. untreated cells; ¶p < 0.05 C2 vs. cisplatin
Among different cell death modalities, anti-cancer agents act mainly by the induction of tumor cell apoptosis. The essential participants in apoptotic machinery are proteases named caspases. Caspase 3 is the main executive caspase that cleaves cellular components leading to cell death [65]. The effect of C2 on the induction of 4T1 cells apoptosis was further documented by significantly higher expression of effector caspase 3 mRNA following cisplatin or C2 treatment (Fig. 8b). Various signals may activate apoptotic programs via intrinsic and extrinsic pathways. The DNA damage is one of the triggers of intrinsic (mitochondrial) apoptotic signaling conducting via releasing of cytochrome c from mitochondria, formation of apoptosome and cleavage of initiator caspase 9 which further activates caspase 3 [66]. The trend toward increases in the expression of caspase 9 mRNA by 4T1 cells following cisplatin treatment (Fig. 8c) is in line with the finding that cisplatin activates intrinsic pathway of apoptotic program [67]. Accordingly, the significant increase in caspase 9 mRNA expression by 4T1 cells following C2 exposure in comparison with untreated cells, indicates that C2 induces mouse breast cancer cells death by triggering the intrinsic apoptotic pathway (Fig. 8c).
Apart from necrosis and apoptosis, one of the alternative ways of cell death is necroptosis which represents programmed necrosis, mediated by the activation of the receptor-interacting protein kinase (RIPK) 1 and RIPK3 [68]. Cisplatin induces necroptosis through the formation of necrosome containing RIP1, RIP3 and mixed lineage kinase domain like (MLKL) protein involved in the membrane damage and subsequent cell death [69]. Here, both cisplatin and C2 increased the expression of RIPK1 mRNA expression thus indicating that C2 might additionally induce 4T1 cells necroptosis (Fig. 8d).
C2 reduces mouse breast cancer cells proliferation
In addition to the induction of apoptosis, the next aim of the study was to examine the effect of C2 on cancer cells proliferation by the analysis of the expression of cell cycle regulators. Cyclins are the critical regulators of different phases of cell cycle progression, strongly implicated in the proliferation and differentiation of tumor cells [70, 71]. Cyclin D3 and cyclin E are mainly expressed during G1 phase of cell cycle and drive the G1/S phase transition [70]. Also, cyclin D3 and cyclin E are important prognostic markers in various malignancies, including breast cancer [72,73,74,75]. Following 12 h exposure to cisplatin or C2, there was significantly decreased expression of cyclin D3 mRNA and the percentage of cyclin E positive 4T1 cells when compared to untreated cells (Fig. 9a, b). Further, the effect of C2 on the gene expression of cyclin dependent kinase (Cdk) inhibitor, p27, has been analyzed. p27 is ubiquitously expressed in both normal and tumor cells that regulates G1 phase of cell cycle by binding cyclin E-Cdk2 complex to inhibit kinase activity [76, 77]. Both cisplatin and C2 markedly elevated p27 mRNA level, thus indicating the important inhibitory effect on 4T1 cells proliferation (Fig. 9c). In addition, it is known that overexpression of p27 activates tumor cells apoptosis, including breast cancer cells [77], which is in line with the effect of C2 on the induction of 4T1 cells death.

The effects of C2 on 4T1 cells proliferation. The level of cyclin D3 mRNA (a), the percentage of cyclin E positive cells (b), the level of p27 mRNA (c), the level of c-Myc mRNA (d) and the percentage of pAKT positive cells (e) were detected in 4T1 cells after 12 h exposure to cisplatin (CDDP) or C2 at the concentration of 30 μM by RT-PCR or flow cytometry. For RT-PCR analyses β-actin mRNA was used as an internal control. The data are presented as mean ± SD. *p < 0.05
c-Myc oncogene is a transcription factor involved in control of cell growth, differentiation, and apoptosis [78]. It is recognized as an important therapeutic target as its aberrant expression is related to the development of many tumors [79]. c-Myc stimulates cell cycle via upregulation of cyclins and Cdks, as well as downregulation of the Cdk inhibitors, including phosphorylation and subsequent degradation of p27 [80]. C2 significantly reduced c-Myc mRNA level in 4T1 cells when compared to cisplatin treated cells (Fig. 9d) which might be related to the elevated p27 mRNA expression. Also, C2-induced repression of c-Myc, as well as cyclin D3 and cyclin E, correspond with the known effect of c-Myc on the stimulation of cell cycle by activation of cyclin/Cdk complexes, but also the antagonizing the activity of p21 and p27 cell cycle inhibitors [81].
Phosphorylated and activated form of protein kinase AKT (pAKT), is a major downstream effector of the phosphatidylinositol 3-kinase (PI3K) pathway, involved in further activation of mTOR (the Mammalian Target Of Rapamycin) signaling and strongly implicated in the dysregulation of cell proliferation, apoptosis and motility in the development and progression of many tumors, including breast cancer [82, 83]. Therefore, overexpression of pAKT is a considerable therapeutic target in different malignancies [82]. Both C2 and cisplatin significantly reduced the percentage of pAKT positive 4T1 cells in comparison with untreated cells (Fig. 9e). However, the effect of C2 on the decrease of pAKT expression was weaker than the effect of cisplatin (Fig. 9e). It has been reported that PI3K/AKT signaling affects cell proliferation via the upregulation of c-Myc, as well as targeting cyclins, Cdks and Cdk inhibitors [84, 85]. C2-reduced percentage of pAKT positive 4T1 cells implicates the inhibition of AKT signaling which corresponds with further decrease of c-Myc, cyclin D3 and cyclin E expression, as well as upregulation of p27 inhibitor. Also, PI3K/AKT signaling plays the critical role in the induction of cell survival by blocking apoptosis through different targets, including inhibition of caspase 9 and caspase 3 [86, 87]. Therefore, the lower pAKT expression in 4T1 cells following C2 exposure may be responsible for its pro-apoptotic activity documented by increased expression of caspase 9 and caspase 3 mRNA (Figs. 7, 8).
Taken together, it appears that the cytotoxic effect of C2 toward breast cancer cells relies mainly on the induction of apoptosis.
Analysis of C2 effects on the mouse breast cancer growth
Given the notable cytotoxic activity toward 4T1 cells in vitro, the effects of C2 have been further tested on mouse breast cancer growth in vivo. Following 4T1 cells implantation, the effects of C2 (3 mg/kg and 6 mg/kg), cisplatin (3 mg/kg) or vehicle treatment on the breast cancer growth and mice survival were monitored until 25 day of the experiment. The efficacy of cisplatin treatment was demonstrated by trend toward decrease in tumor volume compared to untreated mice, but the difference did not reach statistical significance probably due to the increased mortality in the group of untreated tumor-bearing mice (Fig. 10a). Accordingly, the analysis of the tumor-bearing mice mortality rate revealed significantly increased survival of cisplatin treated compared to untreated mice (Fig. 10b). Surprisingly, C2 treatment at both tested concentrations did not inhibit tumor growth which was in contrast with the notable cytotoxic effect demonstrated in vitro. The mice injected with the lower dose of C2 (3 mg/kg) had significantly higher tumor volumes in comparison with cisplatin treated mice started from 21 day of the experiment (Fig. 10a). Moreover, it appears that C2 enhanced tumor growth at lower tested dose (Fig. 10a). At 25 day, the average tumor volume was also increased in mice treated with higher dose of C2 (6 mg/kg) compared to cisplatin injected mice (Fig. 10a). However, the mice survival exhibited a slightly increased rate following C2 treatment, as denoted by significant difference reached at 18 day between mice treated with lower dose of C2 and untreated mice (Fig. 10b). On the other hand, the survival rate was decreased in mice treated with higher dose (6 mg/kg) of C2 in comparison with cisplatin 25 days after tumor cells implantation (Fig. 10b).

The effects of C2 on mouse breast cancer growth and survival. a Following the appearance of palpable tumor, started from day 7 after 4T1 cells implantation, the mice (n = 10 per group) were treated with C2 (3 mg/kg and 6 mg/kg), cisplatin (CDDP) (3 mg/kg) or vehicle, every 2–3 days, eight doses in total. The tumor growth was monitored for 25 days. Data are presented as mean ± SE. *p < 0.05 vs. cisplatin treated mice. b The mortality rate of tumor-bearing mice treated with C2 (3 mg/kg and 6 mg/kg), cisplatin (3 mg/kg) or vehicle. *p < 0.05 cisplatin treated vs. untreated mice; §p < 0.05 C2 (3 mg/kg) treated vs. untreated mice; ¶p < 0.05 C2 (6 mg/kg) vs. cisplatin treated mice
Overall, the results obtained in vitro show a good cytotoxic effect of C2 on mouse 4T1 cells. However, anti-breast cancer activity of C2 in vivo is limited. The several hypothesis may explain the discrepancy between in vitro and in vivo anti-tumor efficacy of tested dinuclear platinum(II) complex. It appears that C2 binds weaker to the surface of transport protein, thus possibly the lower amount of the complex reaches the tumor cells. Other studies indicate that the failure of metallotherapeutic anti-tumor activity in vivo may be related to its interactions with serum albumin proteins [88]. In addition, there is evidence that inactivation of platinum(II) complexes in vivo may be the consequence of their degradation into biologically inactive compounds by glutathione or other unknown metabolic pathways [89, 90].
Conclusions
The dinuclear platinum(II) complexes, ([{Pt(1,2-pn)Cl}2(μ-pydz)]Cl2 (C1), [{Pt(ibn)Cl}2(μ-pydz)]Cl2 (C2), [{Pt(1,3-pn)Cl}2(μ-pydz)]Cl2 (C3) and [{Pt(1,3-pnd)Cl}2(μ-pydz)]Cl2 (C4)) were synthesized and characterized by elemental microanalyses, NMR (1H and 13C), and UV–Vis spectroscopy. The fluorescence emission spectroscopy was used for the evaluation of DNA/BSA-binding affinity to C1–C4 complexes. Small hypochromism and decrease of the emission intensity indicates that the mode of binding of these complexes to CT-DNA helix is not intercalation and that these complexes are not so strong as intercalators. Ka value obtained in the reaction with BSA indicates that C2 binds weaker to BSA than other investigated Pt(II) complexes. The results of the docking study have shown that electrostatic attraction is responsible for binding of the complexes to target biomolecules. Namely, positively charged investigated complexes were mainly bound to the negatively charged phosphate backbone of DNA, or on the surface of BSA and HSA, electrostatically attracted by negatively charged amino acid residues.
The in vitro analyses of anti-tumor activity of C1–C4 platinum-based compounds have shown that C2 exhibits the best cytotoxic activity, specially toward breast cancer cells in the dose- and time-dependent manner, and possibly less toxicity for normal cells than cisplatin as a conventional therapeutic. C2 induces breast cancer cells death via triggering intrinsic pathway of apoptosis and possibly necroptosis. Also, C2 might reduce mouse breast cancer cells proliferation in vitro, as evaluated by the alterations in the expression of cell cycle regulators cyclin D3, cyclin E and Cdk inhibitor p27, as well as downregulation of c-Myc and pAKT oncogenes involved in the control of cell proliferation and death. Examination of interactions with BSA has shown that the binding of the C2 complex to a non-transport site, with the formation of weak interactions, can be considered as a potential reason for the lower in vivo activity of this complex in the mouse breast cancer model compared to its activity in vitro.
Data availability
The data are available from the corresponding author upon reasonable request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249. https://doi.org/10.3322/caac.21660
Dilruba S, Kalayda GV (2016) Platinum-based drugs: past, present and future. Cancer Chemother Pharmacol 77(6):1103–1124. https://doi.org/10.1007/s00280-016-2976-z
Jamieson ER, Lippard SJ (1999) Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev 99:2467–2498. https://doi.org/10.1021/cr980421n
Todd RC, Lippard SJ (2009) Inhibition of transcription by platinum antitumor compounds. Metallomics 1:280–291. https://doi.org/10.1039/B907567D
Lazarević T, Rilak A, Bugarčić Ž (2017) Platinum, palladium, gold and ruthenium complexes as anticancer agents: current clinical uses, cytotoxicity studies and future perspectives. Eur J Med Chem 142:8–31. https://doi.org/10.1016/j.ejmech.2017.04.007
Neidle S, Nunn CM (1998) Crystal structures of nucleic acids and their drug complexes. Nat Prod Rep 15:1–15. https://doi.org/10.1039/A815001
Han X, Gao X (2001) Sequence specific recognition of ligand-DNA complexes studied by NMR. Curr Med Chem 8(5):551–581. https://doi.org/10.2174/0929867003373337
Komeda SI, Qu Y, Mangrum JB, Hegmans A, Williams LD, Farrell NP (2016) The phosphate clamp as recognition motif in platinum–DNA interactions. Inorg Chim Acta 452:25–33. https://doi.org/10.1016/j.ica.2016.04.052
Konovalov B, Živković MD, Milovanović J, Djordjević D, Arsenijević A, Vasić I, Janjić G, Franich A, Manojlović D, Skrivanj S, Milovanović M, Djuran MI, Rajković S (2018) Synthesis, cytotoxic activity and DNA interaction studies of new dinuclear platinum(II) complexes with an aromatic 1,5-naphthyridine bridging ligand: DNA binding mode of polynuclear platinum(II) complexes in relation to the complex structure. Dalton Trans 47(42):15091–15102. https://doi.org/10.1039/C8DT01946K
Fantoni NZ, Brown T, Kellett A (2021) DNA-targeted metallodrugs: an untapped source of artificial gene editing technology. ChemBioChem 22(13):2184–2205. https://doi.org/10.1002/cbic.202000838
Dasari S, Tchounwou PB (2014) Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740:364–378. https://doi.org/10.1016/j.ejphar.2014.07.025
Florea AM, Büsselberg D (2011) Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel) 3(1):1351–1371. https://doi.org/10.3390/cancers3011351
Dömötör O, Hartinger CG, Bytzek AK, Kiss T, Keppler BK, Enyedy EA (2013) Characterization of the binding sites of the anticancer ruthenium(III) complexes KP1019 and KP1339 on human serum albumin via competition studies. J Biol Inorg Chem 18(1):9–17. https://doi.org/10.1007/s00775-012-0944-6
Timerbaev AR, Hartinger CG, Aleksenko SS, Keppler BK (2006) Interactions of antitumor metallodrugs with serum proteins: advances in characterization using modern analytical methodology. Chem Rev 106(6):2224–2248. https://doi.org/10.1021/cr040704h
Tian J, Liu J, Tian X, Hu Z, Chen X (2004) Study of the interaction of kaempferol with bovine serum albumin. J Mol Struct 691:197–202. https://doi.org/10.1016/j.molstruc.2003.12.019
He XM, Carter DC (1992) Atomic structure and chemistry of human serum albumin. Nature 358(6383):209–215. https://doi.org/10.1038/358209a0
Huang D, Savage SR, Calinawan AP, Lin C, Zhang B, Wang P, Starr TK, Birrer MJ, Paulovich AG (2021) A highly annotated database of genes associated with platinum resistance in cancer. Oncogene 40(46):6395–6405. https://doi.org/10.1038/s41388-021-02055-2
Arsenijevic M, Milovanovic M, Jovanovic S, Arsenijevic N, Markovic BS, Gazdic M, Volarevic V (2017) In vitro and in vivo anti-tumor effects of selected platinum(IV) and dinuclear platinum(II) complexes against lung cancer cells. J Biol Inorg Chem 22(6):807–817. https://doi.org/10.1007/s00775-017-1459-y
Živković MD, Kljun J, Ilić-Tomić T, Pavić A, Veselinović A, Manojlović DD, Nikodinović-Runić J, Turel I (2018) A new class of platinum(II) complexes with the phosphine ligand pta which show potent anticancer activity. Inorg Chem Front 5:39–53. https://doi.org/10.1039/C7QI00299H
Marković N, Zarić M, Živković MD, Rajković S, Jovanović I, Arsenijević N, Čanović P, Ninković S (2019) Novel platinum(II) complexes selectively induced apoptosis and cell cycle arrest of breast cancer cells in vitro. Chem Select 4(44):12971–12977. https://doi.org/10.1002/slct.201903290
Franich AA, Živković MD, Ćoćić D, Petrović B, Milovanović M, Arsenijević A, Milovanović J, Arsenijević D, Stojanović B, Djuran MI, Rajković S (2019) New dinuclear palladium(II) complexes with benzodiazines as bridging ligands: interactions with CT-DNA and BSA, and cytotoxic activity. J Biol Inorg Chem 24(7):1009–1022. https://doi.org/10.1007/s00775-019-01695-w
Franich AA, Živković MD, Ilić-Tomić T, Đorđević IS, Nikodinović-Runić J, Pavić A, Janjić GV, Rajković S (2020) New minor groove covering DNA binding mode of dinuclear Pt(II) complexes with various pyridine-linked bridging ligands and dual anticancer-antiangiogenic activities. J Biol Inorg Chem 25(3):395–409. https://doi.org/10.1007/s00775-020-01770-7
Vasić I, Rajković S, Arsenijević A, Milovanović M, Arsenijević N, Milovanović J, Živković MD (2021) In vitro and in vivoactivity of series of cationic dinuclearPt(II) complexes. J Inorg Biochem 225:111619. https://doi.org/10.1016/j.jinorgbio.2021.111619
Konovalov B, Franich AA, Jovanović M, Jurisević M, Gajović N, Jovanović M, Arsenijević N, Maric V, Jovanović I, Živković MD, Rajković S (2020) Synthesis, DNA-/bovine serum albumin-binding affinity, and cytotoxicity of dinuclear platinum(II) complexes with 1, 6-naphthyridine-bridging ligand. Appl Organomet Chem. https://doi.org/10.1002/aoc.6112
Rosa NMP, Ferreira FHDC, Farrell NP, Costa LAS (2019) TriplatinNC and biomolecules: building models based on non-covalent interactions. Front Chem 7:307. https://doi.org/10.3389/fchem.2019.00307
Farrell N (2003) Metal complexes as drugs and chemotherapeutic agents. Compr Coord Chem II 9:809–840. https://doi.org/10.1016/B0-08-043748-6/09021-6
Zhang J, Wang L, Xing Z, Liu D, Sun J, Li X, Zhang Y (2010) Status of bi- and multi-nuclear platinum anticancer drug development. Anticancer Agents Med Chem 10(4):272–282. https://doi.org/10.2174/187152010791162270
Harris AL, Yang X, Hegmans A, Povirk L, Ryan JJ, Kelland L, Farrell NP (2005) Synthesis, characterization, and cytotoxicity of a novel highly charged trinuclear platinum compound. Enhancement of cellular uptake with charge. Inorg Chem 44(26):9598–9600. https://doi.org/10.1021/ic051390z
Qu Y, Rauter H, Soares Fontes AP, Bandarage R, Kelland LR, Farrell N (2000) Synthesis, characterization, and cytotoxicity of trifunctional dinuclear platinum complexes: comparison of effects of geometry and polyfunctionality on biological activity. J Med Chem 43(16):3189–3192. https://doi.org/10.1021/jm990536z
Brabec V, Kaspárková J, Vrána O, Nováková O, Cox JW, Qu Y, Farrell N (1999) DNA modifications by a novel bifunctional trinuclear platinum phase I anticancer agent. Biochemistry 38(21):6781–6790. https://doi.org/10.1021/bi990124s
Farrell N, Appleton TG, Qu Y, Roberts JD, Fontes AP, Skov KA, Wu P, Zou Y (1995) Effects of geometric isomerism and ligand substitution in bifunctional dinuclear platinum complexes on binding properties and conformational changes in DNA. Biochemistry 34(47):15480–15486. https://doi.org/10.1021/bi00047a013
Komeda S, Kalayda GV, Lutz M, Spek AL, Yamanaka Y, Sato T, Chikuma M, Reedijk J (2003) New isomeric azine-bridged dinuclear platinum(II) complexes circumvent cross-resistance to cisplatin. J Med Chem 46(7):1210–1219. https://doi.org/10.1021/jm020004%2B
Komeda S, Lutz M, Spek AL, Chikuma M, Reedijk J (2000) New antitumor-active azole-bridged dinuclear platinum(II) complexes: synthesis, characterization, crystal structures, and cytotoxic studies. Inorg Chem 39(19):4230–4236. https://doi.org/10.1021/ic000273v
Reedijk J (2003) New clues for platinum antitumor chemistry: kinetically controlled metal binding to DNA. Proc Natl Acad Sci USA 100(7):3611–3616. https://doi.org/10.1073/pnas.0737293100
Manzotti C, Pratesi G, Menta E, Di Domenico R, Cavalletti E, Fiebig HH, Kelland LR, Farrell N, Polizzi D, Supino R, Pezzoni G, Zunino F (2000) BBR 3464: a novel triplatinum complex, exhibiting a preclinical profile of antitumor efficacy different from cisplatin. Clin Cancer Res 6(7):2626–2634
Komeda S, Ohishi H, Yamane H, Harikawa M, Sakaguchi K, Chikuma M (1999) An NMR study and crystal structure of [{cis-Pt(NH3)2(9EtG-κN7)}2(μ-pz)][NO3]3 (9EtG=9-ethylguanine) as a model compound for the 1,2-intrastrand GG crosslink. Dalton Trans. https://doi.org/10.1039/A903081F
Senerovic L, Zivkovic MD, Veselinovic A, Pavic A, Djuran MI, Rajkovic S, Nikodinovic-Runic J (2015) Synthesis and evaluation of series of diazine-bridged dinuclear platinum(II) complexes through in vitro toxicity and molecular modeling: correlation between structure and activity of Pt(II) complexes. J Med Chem 58(3):1442–1451. https://doi.org/10.1021/jm5017686
Hohmann H, van Eldik R (1900) Rate and equilibrium data for substitution reactions of diaqua(ethylenediamine)palladium(II) with chloride in aqueous solution. Inorg Chim Acta 174(1):87–92. https://doi.org/10.1016/S0020-1693(00)80282-0
Živković MD, Ašanin DP, Rajković S, Djuran MI (2011) Hydrolysis of the amide bond in N-acetylated l-methionylglycine catalyzed by various platinum(II) complexes under physiologically relevant conditions. Polyhedron 30(6):947–952. https://doi.org/10.1016/j.poly.2010.12.039
Ašanin DP, Živković MD, Rajković S, Warźajtis B, Rychlewska U, Djuran MI (2013) Crystallographic evidence of anion⋯π interactions in the pyrazine bridged {[Pt(en)Cl]2(μ-pz)}Cl2 complex and a comparative study of the catalytic ability of mononuclear and binuclear platinum(II) complexes in the hydrolysis of N-acetylated l-methionylglycine. Polyhedron 5:255–262. https://doi.org/10.1016/j.poly.2012.12.037
Rajković S, Rychlewska U, Warżajtis B, Ašanin DP, Živković MD, Djuran MI (2014) Disparate behavior of pyrazine and pyridazine platinum(II) dimers in the hydrolysis of histidine—and methionine-containing peptides and unique crystal structure of {[Pt(en)Cl]2(μ-pydz)}Cl2 with a pair of NH...Cl–...HN hydrogen bonds supporting the pyridazine bridge. Polyhedron 67:279–285. https://doi.org/10.1016/j.poly.2013.09.008
Hertzberg RP, Dervan PB (1982) Cleavage of double helical DNA by methidium-propyl-EDTA-iron(II). J Am Chem Soc 104:313–315. https://doi.org/10.1021/ja00365a069
Bi S, Pang B, Zhao T, Wang T, Wang Y, Yan L (2013) Binding characteristics of salbutamol with DNA by spectral methods. Spectrochim Acta A Mol Biomol Spectrosc 111:182–187. https://doi.org/10.1016/j.saa.2013.03.067
Bayly CI, Cieplak P, Cornell WD, Kollman PA (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 97(40):10269–10280. https://doi.org/10.1021/j100142a004
Drew HR, Wing RM, Takano T, Broka C, Tanaka S, Itakura K, Dickerson RE (1981) Structure of a B-DNA dodecamer: conformation and dynamics. Proc Natl Acad Sci USA 78(4):2179–2183. https://doi.org/10.1073/pnas.78.4.2179
Bujacz A (2012) Structures of bovine, equine and leporine serum albumin. Acta Crystallogr D Biol Crystallogr 68(Pt 10):1278–1289. https://doi.org/10.1107/S0907444912027047
Wang ZM, Ho JX, Ruble JR, Rose J, Rüker F, Ellenburg M, Murphy R, Click J, Soistman E, Wilkerson L, Carter DC (2013) Structural studies of several clinically important oncology drugs in complex with human serum albumin. Biochim Biophys Acta 1830(12):5356–5374. https://doi.org/10.1016/j.bbagen.2013.06.032
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791. https://doi.org/10.1002/jcc.21256
Dassault Systèmes BIOVIA (2016) Discovery studio modeling environment, Release 2017, San Diego
Jurisevic M, Arsenijevic A, Pantic J, Gajovic N, Milovanovic J, Milovanovic M, Poljarevic J, Sabo T, Vojvodic D, Radosavljevic GD, Arsenijevic N (2018) The organic ester O,O′-diethyl-(S,S)-ethylenediamine-N,N′-di-2-(3-cyclohexyl)propanoate dihydrochloride attenuates murine breast cancer growth and metastasis. Oncotarget 9(46):28195–28212. https://doi.org/10.18632/oncotarget.25610
Lee JO, Kang MJ, Byun WS, Kim SA, Seo IH, Han JA, Moon JW, Kim JH, Kim SJ, Lee EJ, In Park S, Park SH, Kim HS (2019) Metformin overcomes resistance to cisplatin in triple-negative breast cancer (TNBC) cells by targeting RAD51. Breast Cancer Res 21(1):115. https://doi.org/10.1186/s13058-019-1204-2
Wilson WD, Ratmeyer L, Zhao M, Strekowski L, Boykin D (1993) The search for structure-specific nucleic acid-interactive drugs: effects of compound structure on RNA versus DNA interaction strength. Biochemistry 32(15):4098–4104. https://doi.org/10.1021/bi00066a035
Englert K, Hendi R, Robbs PH, Rees NV, Robinson APG, Tucker JHR (2020) Cisplatin adducts of DNA as precursors for nanostructured catalyst materials. Nanoscale Adv 2(10):4491–4497. https://doi.org/10.1039/D0NA00528B
Dhar S, Nethaji M, Chakravarty AR (2005) Effect of charge transfer bands on the photo-induced DNA cleavage activity of [1-(2-thiazolylazo)-2-naphtholato]copper(II) complexes. J Inorg Biochem 99(3):805–812. https://doi.org/10.1016/j.jinorgbio.2004.12.014
Salive ME, Cornoni-Huntley J, Phillips CL, Guralnik JM, Cohen HJ, Ostfeld AM, Wallace RB (1992) Serum albumin in older persons: relationship with age and health status. J Clin Epidemiol 45(3):213–221. https://www.jclinepi.com/article/0895-4356(92)90081-W/pdf
Chaves OA, da Silva VA, Sant’Anna CMR, Ferreiraa ABB, Ribeiro TAN, De-Carvalho MG, Cesarin-Sobrinho D, Netto-Ferreira JC (2017) Binding studies of lophirone B with bovine serum albumin (BSA): combination of spectroscopic and molecular docking techniques. J Mol Struct 1128:606–611. https://doi.org/10.1016/j.molstruc.2016.09.036
Shi JH, Pan DQ, Wang XX, Liu TT, Jiang M, Wang Q (2016) Characterizing the binding interaction between antimalarial artemether (AMT) and bovine serum albumin (BSA): spectroscopic and molecular docking methods. J Photochem Photobiol B 162:14–23. https://doi.org/10.1016/j.jphotobiol.2016.06.025
Shi JH, Zhou KL, Lou YY, Pan DQ (2018) Multi-spectroscopic and molecular modeling approaches to elucidate the binding interaction between bovine serum albumin and darunavir, a HIV protease inhibitor. Spectrochim Acta A Mol Biomol Spectrosc 188:362–371. https://doi.org/10.1016/j.saa.2017.07.040
Psomas G, Kessissoglou DP (2013) Quinolones and non-steroidal anti-inflammatory drugs interacting with copper(II), nickel(II), cobalt(II) and zinc(II): structural features, biological evaluation and perspectives. Dalton Trans 42(18):6252–6276. https://doi.org/10.1039/C3DT50268F
Rajendiran V, Karthik R, Palaniandavar M, Stoeckli-Evans H, Periasamy VS, Akbarsha MA, Srinag BS, Krishnamurthy H (2007) Mixed-ligand copper(II)-phenolate complexes: effect of coligand on enhanced DNA and protein binding, DNA cleavage, and anticancer activity. Inorg Chem 46(20):8208–8221. https://doi.org/10.1021/ic700755p
Swanepoel B, Nitulescu GM, Olaru OT, Venables L, van de Venter M (2019) Anti-cancer activity of a 5-aminopyrazole derivative lead compound (BC-7) and potential synergistic cytotoxicity with cisplatin against human cervical cancer cells. Int J Mol Sci 20(22):5559. https://doi.org/10.3390/ijms20225559
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717. https://doi.org/10.1038/srep42717
Pfeffer CM, Singh ATK (2018) Apoptosis: a target for anticancer therapy. Int J Mol Sci 19(2):448. https://doi.org/10.3390/ijms19020448
Velma V, Dasari SR, Tchounwou PB (2016) low doses of cisplatin induce gene alterations, cell cycle arrest, and apoptosis in human promyelocytic leukemia cells. Biomark Insights 11:113–121. https://doi.org/10.4137/BMI.S39445
Lopez J, Tait SW (2015) Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer 112(6):957–962. https://doi.org/10.1038/bjc.2015.85
Zaman S, Wang R, Gandhi V (2014) Targeting the apoptosis pathway in hematologic malignancies. Leuk Lymphoma 55(9):1980–1992. https://doi.org/10.3109/10428194.2013.855307
Park MS, De Leon M, Devarajan P (2002) Cisplatin induces apoptosis in LLC-PK1 cells via activation of mitochondrial pathways. J Am Soc Nephrol 13(4):858–865. https://doi.org/10.1681/asn.v134858
Weinlich R, Oberst A, Beere HM, Green DR (2017) Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18(2):127–136. https://doi.org/10.1038/nrm.2016.149
Xu Y, Ma HB, Fang YL, Zhang ZR, Shao J, Hong M, Huang CJ, Liu J, Chen RQ (2017) Cisplatin-induced necroptosis in TNFα dependent and independent pathways. Cell Signal 31:112–123. https://doi.org/10.1016/j.cellsig.2017.01.004
Gao CY, Zelenka PS (1997) Cyclins, cyclin-dependent kinases and differentiation. BioEssays 19(4):307–315. https://doi.org/10.1002/bies.950190408
Casimiro MC, Crosariol M, Loro E, Li Z, Pestell RG (2012) Cyclins and cell cycle control in cancer and disease. Genes Cancer 3(11–12):649–657. https://doi.org/10.1177/1947601913479022
Wang B, Wang Z, Han L, Gong S, Wang Y, He Z, Feng Y, Yang Z (2019) Prognostic significance of cyclin D3 expression in malignancy patients: a meta-analysis. Cancer Cell Int 19:158. https://doi.org/10.1186/s12935-019-0865-3
Chi Y, Huang S, Liu M, Guo L, Shen X, Wu J (2015) Cyclin D3 predicts disease-free survival in breast cancer. Cancer Cell Int 15:89. https://doi.org/10.1186/s12935-015-0245-6
Pang W, Li Y, Guo W, Shen H (2020) Cyclin E: a potential treatment target to reverse cancer chemoresistance by regulating the cell cycle. Am J Transl Res 12(9):5170–5187
Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, Bedrosian I, Knickerbocker C, Toyofuku W, Lowe M, Herliczek TW, Bacus SS (2002) Cyclin E and survival in patients with breast cancer. N Engl J Med 347(20):1566–1575. https://doi.org/10.1056/nejmoa021153
Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, Sun P, Tan CK, Hengst L, Slingerland J (2007) p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell 128(2):281–294. https://doi.org/10.1016/j.cell.2006.11.049
Katayose Y, Kim M, Rakkar AN, Li Z, Cowan KH, Seth P (1997) Promoting apoptosis: a novel activity associated with the cyclin-dependent kinase inhibitor p27. Cancer Res 57(24):5441–5445
Hoffman B, Liebermann DA (2008) Apoptotic signaling by c-MYC. Oncogene 27(50):6462–6472. https://doi.org/10.1038/onc.2008.312
Madden SK, de Araujo AD, Gerhardt M, Fairlie DP, Mason JM (2021) Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c-Myc. Mol Cancer 20(1):3. https://doi.org/10.1186/s12943-020-01291-6
García-Gutiérrez L, Bretones G, Molina E, Arechaga I, Symonds C, Acosta JC, Blanco R, Fernández A, Alonso L, Sicinski P, Barbacid M, Santamaría D, León J (2019) Myc stimulates cell cycle progression through the activation of Cdk1 and phosphorylation of p27. Sci Rep 9(1):18693. https://doi.org/10.1038/s41598-019-54917-1
Bretones G, Delgado MD, León J (2015) Myc and cell cycle control. Biochim Biophys Acta 1849(5):506–516. https://doi.org/10.1016/j.bbagrm.2014.03.013
Song M, Bode AM, Dong Z, Lee MH (2019) AKT as a therapeutic target for cancer. Cancer Res 79(6):1019–1031. https://doi.org/10.1158/0008-5472.can-18-2738
Al-Bazz YO, Underwood JC, Brown BL, Dobson PR (2009) Prognostic significance of Akt, phospho-Akt and BAD expression in primary breast cancer. Eur J Cancer 45(4):694–704. https://doi.org/10.1016/j.ejca.2008.11.044
Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, Kuo MT (2012) Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res 72(10):2622–2633. https://doi.org/10.1158/0008-5472.can-11-3605
Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA (2003) Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia 17(3):590–603. https://doi.org/10.1038/sj.leu.2402824
Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C (2003) PI3K/Akt and apoptosis: size matters. Oncogene 22(56):8983–8998. https://doi.org/10.1038/sj.onc.1207115
Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z, Li W, Hu J, Lu C, Liu Y (2020) PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis 11(9):797. https://doi.org/10.1038/s41419-020-02998-6
Sanghvi CD, Olsen PM, Elix C, Peng SB, Wang D, Chen ZG, Shin DM, Hardcastle KI, MacBeth CE, Eichler JF (2013) Antitumor properties of five-coordinate gold(III) complexes bearing substituted polypyridyl ligands. J Inorg Biochem 128:68–76. https://doi.org/10.1016/j.jinorgbio.2013.07.014
Moretto J, Chauffert B, Ghiringhelli F, Aldrich-Wright JR, Bouyer F (2011) Discrepancy between in vitro and in vivo antitumor effect of a new platinum(II) metallointercalator. Invest New Drugs 29(6):1164–1176. https://doi.org/10.1007/s10637-010-9461-z
Kemp S, Wheate NJ, Pisani MJ, Aldrich-Wright JR (2008) Degradation of bidentate-coordinated platinum(II)-based DNA intercalators by reduced l-glutathione. J Med Chem 51(9):2787–2794. https://doi.org/10.1021/jm7016072
Acknowledgements
This work was funded by grants from the Ministry of Science, Technological Development and Innovation, Republic of Serbia (Agreements No. 451-03-47/2023-01/200111, 451-03-47/2023-01/200122, 451-03-9/2021-14/200111, 451-03-47/2023-01/200026), the bilateral project with PR China (06/2018), and the junior projects of the Faculty of Medical Sciences of the University of Kragujevac, Serbia (JP 02/20, JP 08/20, JP 16/19 and JP 10/18). This research has also received funding from the Serbian Academy of Sciences and Arts under project No. F128.
Author information
Authors and Affiliations
Contributions
Design of the study: NNA, GDR, JP, SR and MDŽ. Synthesis, characterization and investigation of the complexes-DNA/BSA interaction: AAF, SR, MBJ, GVJ and MDŽ. In vitro and in vivo investigation of anti-tumor activity: SZ, BSM, JA, GDR and JP. Drafting the manuscript: SZ, GDR, JP, MDŽ, SR and GVJ. All authors interpreted the data, contributed to discussion, reviewed and edited the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zornić, S., Simović Marković, B., Franich, A.A. et al. Characterization, modes of interactions with DNA/BSA biomolecules and anti-tumor activity of newly synthesized dinuclear platinum(II) complexes with pyridazine bridging ligand. J Biol Inorg Chem 29, 51–73 (2024). https://doi.org/10.1007/s00775-023-02030-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-023-02030-0