
Abstract
The active sites of metalloenzymes that catalyze O2-dependent reactions generally contain iron or copper ions. However, several enzymes are capable of activating O2 at manganese or nickel centers instead, and a handful of dioxygenases exhibit activity when substituted with cobalt. This minireview summarizes the catalytic properties of oxygenases and oxidases with mononuclear Mn, Co, or Ni active sites, including oxalate-degrading oxidases, catechol dioxygenases, and quercetin dioxygenase. In addition, recent developments in the O2 reactivity of synthetic Mn, Co, or Ni complexes are described, with an emphasis on the nature of reactive intermediates featuring superoxo-, peroxo-, or oxo-ligands. Collectively, the biochemical and synthetic studies discussed herein reveal the possibilities and limitations of O2 activation at these three “overlooked” metals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As attested by the articles in this special issue, the on-going tale of O2 activation by metalloenzymes has featured two metals as the principal protagonists: iron (both heme and nonheme) and copper. Indeed, our fundamental understanding of biological O2 activation has been shaped, in large part, through the study of a handful of enzymatic prototypes, such as cytochrome P450, methane monoxygenases, tyrosinase, and the α-ketoglutarate-dependent dioxygenases. Because of this, it is easy to forget that other transitions metals have had distinguished “bit-parts” in this 60-year-old saga. Manganese, cobalt, and nickel are redox-active elements that occupy positions adjacent to iron and copper on the periodic table, and all three metals are capable of activating O2 in biological environments. For example, many organisms employ manganese-dependent oxidases to degrade the toxic metabolite oxalate [1], and a manganese-containing catechol dioxygenase catalyzes a critical step in the catabolism of aromatic compounds [2]. Certain soil bacteria are able to degrade the compound quercetin—a flavonol released into the environment by decomposing plant material—due to a nickel-dependent dioxygenase [3]. And while no known enzyme requires cobalt to activate O2 in vivo, cobalt-substituted dioxygenases generated in vitro have exhibited robust activities that rival the native enzymes [4].
Furthermore, an impressive number of synthetic Mn, Co, and Ni complexes have been reported that harness the power of O2 to perform oxidative transformations. Some of these complexes were prepared with biomimetic intent, whereas others were generated solely for the purpose of developing novel O2-dependent chemistry. Regardless, investigations into the reactivities and spectroscopic properties of these complexes have illuminated key features of the relevant enzymes. Synthetic studies have also revealed unique aspects of O2 activation at Mn, Co, and Ni sites that suggest alternative “plot lines” that were not pursued in the evolution of metal-dependent oxygenases.
The goal of the present minireview is to summarize recent advances in our understanding of O2 activation at nonheme mononuclear Mn, Co, and Ni centers in both biological and synthetic contexts (manganese-containing ribonucleotide reductases, which feature a dinuclear active site, are treated elsewhere in this special issue). The scope of the review is limited to (bio)chemical systems that involve direct reaction of the metal center with O2. Thus, we have excluded enzymes that employ reduced O2-derivatives (O ·−2 , H2O2) as substrates, such as catalase (Mn) or superoxide dismutase (Mn, Ni), along with those that operate by substrate activation mechanisms (e.g., manganese-containing lipoxygenases, and nickel acireductone dioxygenase). Before delving into a discussion of specific enzymes and related models, we will first consider general aspects of the O2 activation mechanisms commonly employed by these three transition metals, as elucidated through the study of synthetic complexes.
Dioxygen activation pathways for mononuclear Mn, Co, and Ni complexes
Manganese
As illustrated in Scheme 1, mononuclear Mn sites are capable of supporting an assortment of superoxo-, (hydro)peroxo-, and oxo-bound intermediates similar to those observed in heme and nonheme iron enzymes. Superoxomanganese(III) complexes are often proposed as the first intermediates formed upon reaction of Mn(II) with O2, although well-characterized examples are rare due to generally short lifetimes. By employing stopped-flow absorption spectroscopy at reduced temperatures, Kovacs and Rybak-Akimova observed the putative superoxomanganese(III) intermediate (complex 1 in Fig. 1) generated by treatment of a five-coordinate Mn(II)-thiolate complex with O2 [5, 6]. This species quickly reacts with a second equivalent of Mn(II) to afford a dimanganese(III) complex bridged by a trans-μ-1,2-peroxide ligand. Lang et al. succeeded in obtaining a crystal structure of a Mn(III) complex (2) featuring a superoxide ligand in an end-on (η1) binding mode (Fig. 1) [7]. The unique stability of 2 is attributed to the steric bulk of the calix [4] arene supporting ligand, which prevents dimerization. The Mn/O2 unit exhibits an unusual linear geometry (Mn–O–O bond angle of 180°) due to π···π and electrostatic interactions between the superoxide anion and NMe +3 -substituted phenyl rings of the calix [4] arene “bowl”. The lengthy Mn−O(O) bond of 2.444 Å indicates that the superoxide ligand is only weakly bound to the Mn(III) center.

Dioxygen reactivity of mononuclear Mn species

Synthetic nonheme Mn(III) complexes with side-on (η2) peroxide ligands—first reported by Kitajima and Moro-oka in 1994 [8]—are generally prepared by treatment of Mn(II) precursors with superoxide or H2O2 (Scheme 1) [9, 10]. The Borovik group, however, was able to derive a peroxomanganese(III) complex (3; Fig. 2) from direct reaction of a Mn(II) complex with O2 in the presence of an H-atom donor, which presumably serves to reduce the transient superoxomanganese(III) intermediate [11–13]. Regardless of the ancillary ligand, η2-peroxomanganese(III) complexes are rather unreactive and conversion to a dioxomanganese (V) species has not been achieved. The [Mn3+(η2-O2)]+ unit is sufficiently nucleophilic to perform the deformylation of aldehydes (e.g., cyclohexanecarboxaldehyde) to produce formate and oxidized products [11, 12, 14–17]. A recent study of a bispidine-supported [Mn3+(η2-O2)]+ complex revealed an alternative deformylation mechanism that involves the initial hydrogen-atom transfer from the aldehyde substrate to the peroxide ligand [18].

The peroxide bond is also activated by treatment with Lewis acids; for example, addition of Mn(II) has been shown to yield bis(μ-oxo-)dimanganese(III,IV) “diamond cores” via O–O bond cleavage [19]. Similarly, Jackson and coworkers demonstrated that the η2-peroxomanganese(III) complex [Mn(O2)(Me2EBC)]+ decays in the presence of Mn(II) starting material to yield a mixture of Mn(IV)-oxo and Mn(III)-OH species (Me2EBC = 4,11-dimethyl-1,4,8,11-tetraaza-bicyclo[6.6.2]hexadecane) [20]. Recently, Nam et al. trapped an end-on manganese(III)-hydroperoxide intermediate (5) via addition of acid to [Mn(η2-O2)(14-TMC)]+ (4) [14], where 14-TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (Fig. 2) [21]. Reactivity studies indicated that protonation enhances the ability of the manganese peroxide moiety to perform electrophilic chemistry, such as oxygen atom transfer (OAT) reactions.
The long-standing interest in high-valent oxomanganese species arises, in large part, from their relevance to putative intermediates in the oxygen-evolving mechanism of photosystem II. An impressive number of binuclear Mn(IV)2 and Mn(III)Mn(IV) complexes with μ-oxide ligands have been reported, and some were prepared via aerobic oxidation of Mn(II) precursors [22–26]. This reaction likely occurs by the initial formation of a dimanganese(III)-peroxide intermediate, followed by O–O bond cleavage (Scheme 1). Interestingly, synthetic chemists have yet to trap a mononuclear Mn(IV)-oxocomplex using O2 as the sole oxidant. Nearly, all of the currently available oxomanganese complexes, such as 6 in Fig. 3, were prepared by treating Mn(II) precursors with O-atom transfer reagents, such as iodosylbenezene (PhIO), peroxy acids, or H2O2 [27–32]. Borovik and coworkers, however, have developed an alternative route to mononuclear high-valent oxomanganese complexes. Using a tripodal ligand capable of stabilizing terminal oxometal units through multiple H-bonding interactions (Fig. 3), a monomeric oxomanganese(III) complex (7) was generated via reaction with O2 [33]. Stepwise oxidation of this complex by ferrocenium yielded Mn(IV)-oxo and Mn(V)-oxo derivatives (8 and 9, respectively), as confirmed by EPR spectroscopy and DFT calculations [34–36].

Two mononuclear Mn(V)-oxo species have been generated by aerobic oxidation of Mn(III) complexes bearing highly anionic, square-planar macrocycles similar to porphyrins. The corrolazine-based oxomanganese(V) complex of Goldberg was prepared by a free-radical mechanism initiated by photoactivation of the Mn(III) precursor in the presence of O2 [37, 38]. The tetraamido-based oxomanganese(V) complex of Nam and Fukuzumi is proposed to arise from homolytic O–O cleavage of a manganese(IV) (hydro)peroxide intermediate formed after O2 binding to Mn(III) [39]. While notable, these mechanisms differ from manganese-based O2 activation at nonheme sites in nature, which begin by reaction of O2 with Mn(II) centers.
Cobalt
The O2-activation landscape of cobalt is summarized in Scheme 2. The ability of square-planar Co(II) complexes featuring porphyrin, phthalocyanine, tetrazamacrocyclic, and Schiff-base (e.g., salen) ligands to reversibly bind O2 has been studied extensively for decades, and the findings are described in several helpful reviews [40–43]. These Co/O2 adducts, which consist of a low-spin Co(III) center bound to a η1-superoxide ligand (10 in Fig. 4), have often served as models of Fe/O2 species in dioxygen carrier proteins; yet the former exhibits greater stability, on average, due to the kinetic inertness of Co(III). In addition, numerous Co(II)-salen catalysts have been developed for the aerobic oxidation of phenols and olefins [44–48]. The key step in the catalytic cycle generally involves H-atom transfer (HAT) from a weak O–H or C–H bond to the cobalt(III)-bound superoxide ligand, thereby initiating a radical-based oxidation mechanism [49, 50]. When supported by neutral amine-based ligands, mononuclear superoxocobalt(III) intermediates are usually not observed due to fast reaction with a second Co center, thereby yielding dicobalt(III) complexes with μ-1,2-(su)peroxide ligands [51–54].

Dioxygen reactivity of mononuclear Co species

In 1990, Theopold and coworkers reported the crystal structure of a mononuclear cobalt(II) complex (11) featuring a side-on (η2) superoxide ligand [55]. The complex was prepared via oxidative addition of O2 to a low-valent cobalt(I) center supported by the hydrotris(3-tert-butyl-5-methyl-pyrazolyl)borate (TptBu,Me) ligand (Fig. 4). When a less sterically bulky Tp ligand was employed, the [Co(η2-O2)] adduct dimerized at low temperatures to yield a dicobalt(II) complex with two trans-μ-η1:η1 superoxide ligands (Scheme 2) [57]. The O–O bond lengths measured by X-ray crystallography for these Tp-based complexes range between 1.26 and 1.36 Å, suggesting that the putative Co(II)-superoxo-units possess partial Co(III)-peroxide character. In certain cases, treatment of the mononuclear [Co(η2-O2)(Tp)] adduct with a second Co(I) equivalent yielded a dicobalt(III) bis(μ-oxo) species, which likely arises from O–O homolysis of a dicobalt(II) μ-η2:η2-peroxide intermediate [58, 59]. Other structurally characterized [Co2(μ-O)2]2+ complexes derived from reaction with O2 have featured bidentate β-diketiminato [60], guanidinato [61], or monodeprotonated ureayl ligands [62]. Hikichi et al. have shown that dicobalt(III) bis(μ-oxo) intermediates are potent oxidants capable of hydroxylating the isopropyl substituents of the Tp ligand [59, 63, 64].
Mononuclear peroxocobalt(III) complexes have been prepared by one of two routes: (1) direct reaction of Co(I) centers with O2 or (2) treatment of Co(II) precursors with H2O2 under basic conditions. Meyer et al. used the former approach to prepare [Co(O2)(TIMENxyl)]+ (12; Fig. 5), where TIMENxyl is a tripodal N-heterocyclic carbene ligand [65, 66]. The strong σ-donating ability of the carbene groups favors a peroxocobalt(III) configuration over the superoxocobalt(II) alternative adopted by Tp-based complexes. Nam and coworkers followed the H2O2/NEt3 route to prepare complexes, such as complex 13 in Fig. 5, featuring X-TMC macrocycles of varying sizes (X = 12, 13, 14, or 15, where X is the number of atoms in the cyclam ring) [67, 68]. In all structurally characterized examples, the peroxide ligand coordinates to Co(III) in a side-on (η2) fashion with an O–O bond distance of ~1.43 Å. The η2-peroxocobalt(III) complexes are sluggish oxidants with limited ability to perform hydroxylation, HAT, or OAT reactions; however, they are competent to perform oxidative nucleophilic reactions with aldehydes [67–69]. Nam et al. have shown that treatment of [Co(η2-O2)]+ complexes with acids provides a spectroscopically observable cobalt(III)-hydroperoxide species capable of both electrophilic and nucleophilic oxidation reactions [68, 70]. When the 15-TMC ligand is employed, decay of the cobalt(III)-hydroperoxide species is accompanied by hydroxylation of the methyl group of the TMC ligand [68]. Mechanistic evidence suggests that the hydroxylation is performed by a short-lived oxocobalt(IV) or oxylcobalt(III) intermediate (not observed) arising from O–O bond homolysis (Scheme 2).

Although cobalt complexes with terminal oxoligands are frequently invoked as transient intermediates in the decay of cobalt-(alkyl/hydro)peroxide species, attempts to isolate such an entity have not been successful to date [55, 57–59, 71–74]. This shortcoming is not surprising given the intrinsic instability of terminal oxometal complexes of groups 9–11 (often referred to as the “oxowall” for tetragonal structures) [75]. The nearest analogs of these elusive species are a handful of mononuclear Co(III) and Co(IV) complexes featuring [Co−O−M] units, where M is a redox-inactive metal ion like Sc(III). For example, Ray and coworkers found that treatment of [Co2+(TMG3tren)(OTf)]+ with a derivative of PhIO triggers hydroxylation of the tripodal TMG3tren supporting ligand (TMG3tren = tris[2-(N-tetramethyguanidyl)ethyl]amine) [76]. The oxocobalt species (14) in Fig. 6 was trapped by addition of Sc3+ at low temperatures. Data gathered using EPR and X-ray absorption spectroscopies suggested that 14 features a [Co4+(μ-O)Sc3+] core with an S = 3/2 ground state, although [Co3+(μ-O•)Sc3+] and [Co3+(μ-OH)Sc3+] assignments have also been proposed [73, 77]. Using a similar approach, Ray and Nam generated a series of S = 1/2 [Co4+(μ-O)Mn+] complexes (15 M; M = Sc3+, Y3+, Ce3+, Zn2+) via reaction of PhIO with the square-planar [Co3+(TAML)]− complex (TAML = tetraamido macrocyclic ligand) in the presence of redox-inactive metal ions (Fig. 6) [78]. The 15 M complexes are capable of performing both HAT and OAT reactions, with reactivity increasing with the Lewis acidity of the redox-inactive metal ion.

Nickel
Nickel(II) complexes are generally unreactive towards O2 unless electron-rich ligands are employed to depress the Ni2+/3+ redox potential. For example, Kimura and Martell reported a series of nickel(II) complexes with dioxopentaaza macrocyclic ligands capable of catalyzing oxygen insertion reactions under aerobic conditions [79–81]. Although never isolated, superoxonickel(III) species were proposed as the active intermediates in the oxygenation mechanisms. Nickel(II)-thiolate complexes are also known to react with O2 to afford the corresponding metallosulfoxides and metallosulfones. Mechanistic evidence suggests that these reactions involve direct reaction of O2 with the thiolate ligand without oxidation of the nickel(II) center [82].
Much of the nickel-dioxygen chemistry reported in the past 15 years has harnessed the enhanced O2 reactivity of electron-rich Ni(I) precursors. In 2001, Riordan described the synthesis of a dinickel(III) bis(μ-oxo) complex (16; Fig. 7) from the reaction of O2 with [Ni+(CO)(PhTttBu)], where PhTttBu = phenyltris((tert-butylthio)methyl)borate [83–85]. Resonance Raman (rR) and X-ray absorption spectroscopies affirmed the presence of the [Ni 3+2 (μ-O)2] core [86]. The formation of 16 was presumed to follow the conventional superoxo→peroxo→bis(μ-oxo) pathway involving the initial formation of a 1:1 Ni:O2 adduct, followed by reaction with a second Ni(I) equivalent and O–O bond homolysis. In support of this mechanism, a monomeric η 2-superoxonickel(II) species (17; Fig. 7) was obtained by reaction of O2 with [Ni+(CO)(PhTtAd)], which features bulky adamantyl (Ad) groups on the PhTt supporting ligand to prevent dinucleation [87]. Treatment of 17 with [Ni+(CO)(PhTttBu)] afforded the asymmetric dimer, [(PhTtAd)Ni3+(μ-O)2Ni3+(PhTttBu)].

Left Schematic representations of η 2-superoxonickel(II) and dinickel(III) bis(μ-oxo) complexes (17 and 16, respectively) prepared by Riordan and coworkers [83, 87]. Right X-ray crystal structures of complexes 18 and 19 generated by Driess et al. The 18-crown-6 ether in the structure of 19 was omitted for clarity [88, 90]
More recently, Driess et al. reported the crystal structure of a square-planar η2-superoxonickel(II) complex (18; Fig. 7) bearing a β-diketiminato ligand prepared by reaction of a Ni(I) precursor with O2 [88, 89]. The observed O–O bond distance of 1.35 Å is characteristic of a superoxide ligand. One-electron reduction of 18 using elemental potassium provided an unusual structure (19) in which a μ-η2:η2-peroxide ligand with an O–O bond distance of 1.47 Å serves as a bridge between the nickel(II) center and a potassium ion solvated by 18-crown-6 ether [90]. Reaction of 18 with a Cu(I) complex was shown to trigger O–O bond cleavage leading to formation of a heterobimetallic [Cu3+(μ-O)2Ni3+] core [91].
The N-tetramethylcyclam (TMC) framework is capable of supporting both superoxonickel(II) and peroxonickel(III) units, depending on the size of the TMC ring. End-on (η1) superoxonickel(II) complexes have been synthesized using 13-TMC and 14-TMC [92, 93]. Riordan and coworkers observed that oxygenation of [Ni+(14-TMC)]+ at low temperature generates a metastable dinickel(II) species (20; Fig. 8) with a bridging μ-1,2-peroxide ligand, as determined by rR and DFT analyses [94, 95]. Under conditions of excess O2, the mononuclear complex [Ni2+(η1-O2)(14-TMC)]+ (21) is the dominant product [92]. The same complex was also prepared by addition of excess H2O2 and NEt3 to [Ni2+(14-TMC)]2+. Remarkably, reaction of [Ni2+(12-TMC)]2+ with the same H2O2/NEt3 combination yielded a η2-peroxonickel(III) complex instead of the expected η1-superoxonickel(II) species, highlighting the ability of TMC ancillary ligands to modulate the geometric and electronic structures of mononuclear [NiO2]+ intermediates [10, 96]. Nam and others have shown that [Ni3+(η2-peroxo)]+ complexes, like their Mn and Co congeners, perform the nucleophilic deformylation of aldehydes [96, 97]. By contrast, end-on and side-on superoxonickel(II) complexes are electrophilic and participate in OAT reactions with triphenylphosphine [93].

The only crystallographically characterized example of a nickel-hydroperoxide complex was recently reported by Gade and coworkers. These researchers found that reaction of O2 with a pincer-based Ni(I) complex (Fig. 9) yielded a η1-superoxonickel(II) complex (22) that exists in equilibrium with its dinickel(II) μ-1,2-peroxo-bridged counterpart (23) [98]. Treatment of the latter with H2O2 provided complex 24 and the resulting crystal structure revealed a square-planar Ni(II) center bound to an end-on hydroperoxide donor. Aerobic decomposition of 24 is accompanied by autoxidation of the pincer ligand to yield a novel alkylperoxo-metallacyclic complex [99].

Series of superoxo-, peroxo-, and hydroperoxo-nickel(II) complexes generated by Gade et al. using a chiral pincer ligand [98]
The same factors that make it challenging to isolate mononuclear oxocobalt species (vide supra) have also prevented, at least so far, definitive characterization of a nickel complex featuring a terminal oxoligand. Transient oxo/oxyl nickel species are likely involved in alkane hydroxylation reactions catalyzed by Ni(II) complexes in the presence of the oxidant m-chloroperbenzoic acid (mCPBA) [100, 101]. The proposed catalytic mechanism involves the initial formation of an acylperoxo-nickel(II) species, followed by O–O bond homolysis to yield the active oxo-nickel(III) or oxylnickel(II) intermediate. Ray and coworkers have succeeded in trapping a metastable complex (25) that arises from oxidation of [Ni2+(TMG3tren)(OTf)2] with mCPBA at −30 °C [102]. Data gathered using EPR and UV–vis spectroscopies indicate that this species consists of a Ni(III) center bound to a terminal oxo- or hydroxo-ligand, although a conclusive structural determination was not possible due to its instability and low yield of formation (~15 %). Company et al. followed a similar procedure to generate a putative oxylnickel(III) species (26) supported by a tetradentate bisamidate ligand [103, 104]. Analysis with X-ray absorption spectroscopy (XAS) revealed a nickel–oxygen distance of 1.88 Å, and the preponderance of spectroscopic and computational data favored an oxylnickel(III) configuration rather than the oxonickel(IV) alternative. While these results are certainly promising, the assignment of 26 as an oxylnickel(III) species remains tentative and additional characterization with structural and spectroscopic methods is required to confirm this hypothesis (Fig. 10).

Dioxygen activation at mononuclear Mn, Co, and Ni enzymes and model complexes
Manganese oxalate-degrading enzymes
The accumulation of oxalic acid—a byproduct of metabolic pathways in plants, fungi, and microbes—has harmful consequences for nearly all organisms. To mitigate the toxic effects of this organic acid, nature has evolved two classes of manganese-dependent enzymes that degrade oxalate via different reactions [1, 105, 106]. Oxalate oxidase (OxOx) catalyzes the oxidation of oxalate to two moles of CO2 (Eq. 1). Dioxygen serves as the electron and proton acceptor in this process, resulting in concomitant formation of H2O2. In contrast, oxalate decarboxylase (OxDC) catalyzes the conversion of oxalate to formate and CO2 (Eq. 2). Even though the OxDC reaction is merely a disproportionation, the enzyme requires both manganese and O2 for activity [107]. The OxOx family plays the dominant role in oxalate catabolism in plants [108], whereas the OxDC family is more common in fungi and bacteria [1]. Humans lack these oxalate-degrading enzymes and thus excessive amounts of dietary oxalate (hyperoxaluria) can lead to formation of calcium oxalate stones in the kidney [109].
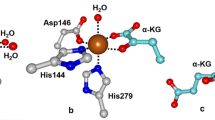
Both OxOx and OxDC belong to the functionally diverse superfamily of cupin proteins, and the close structural and sequence similarities between the two enzymes point to a common evolutionary ancestor [110, 111]. A high-resolution X-ray crystal structure published in 2000 revealed that barley OxOx is a homohexamer in which each monomer contains one manganese ion within its jellyroll β-barrel domain [112]. Crystal structures of bacterial OxDC’s from Bacillus subtilis display a similar homohexameric arrangement [113, 114]. However, each OxDC subunit consists of two cupin domains and, therefore, possesses two nearly identical manganese-binding sites, suggesting that OxDC might have arisen from gene duplication of OxOx [113]. Based on structural and mutagenesis studies, it appears that catalytic activity largely occurs at the N-terminal site, while the role of the C-terminal site remains ambiguous [114–117]. The active sites of OxOx and OxDC both feature a six-coordinate manganese(II) center bound to one glutamate and three histidine side chains, in addition to two water molecules in a cis arrangement (Fig. 11) [112, 113].

Manganese active site of OxOx from Hordeum vulgare (barley) [112]. PDB 1FI2
The catalytic cycles of OxOx and OxDC are initiated by coordination of oxalate to the Mn(II) center, displacing one or both of the H2O molecules found in the resting state. It is generally assumed that oxalate binds in a monodentate fashion, and this hypothesis is supported by crystal structures of OxOx featuring the substrate-analog glycolate [118] and a cobalt-substituted OxDC/oxalate complex [119]. Oxalate coordination serves to lower the Mn3+/2+ redox potential, thus facilitating formation of a Mn/O2 adduct with η1-superoxomanganese(III) character [120]. In the OxOx/OxDC mechanisms favored by both Richards and Bornemann, O2 binding is followed by a key proton-coupled electron transfer (PCET) step involving deprotonation of the oxalate ligand and transfer of an electron from oxalate to the nascent Mn(III) center (Scheme 3) [114, 121, 122]. The end-result is a putative superoxomanganese(II)-(oxalate radical anion) intermediate common to both OxOx and OxDC. Because one-electron oxidation of oxalate dramatically weakens its C–C bond [123], this intermediate undergoes spontaneous decarboxylation to generate CO2 and a manganese-bound formyl radical anion. At this point, the mechanisms of OxOx and OxDC diverge. The formyl radical either couples with the superoxo-ligand to yield a manganese(II)-peroxycarbonate species (OxOx) or undergoes protonation by a conserved second-sphere Glu162 residue to afford a manganese(III)-formate intermediate (OxDC) [114, 121, 122]. In support of this proposal, Bornemann and coworkers demonstrated that B. subtilis OxDC can be converted into an oxidase by mutating four residues (including Glu162) in the flexible lid of the N-terminal site [124]. Furthermore, radical-trapping EPR experiments have confirmed the presence of superoxo- and oxalate-derived radicals during catalysis [124–126], and the OxDC mechanism shown in Scheme 3 is consistent with heavy-atom isotope effect measurements [122, 127].

Proposed catalytic mechanisms for OxOx and OxDC
The publication of the enzymatic structures has stimulated the development of synthetic models of the OxOx and OxDC active sites. In 2005, Berreau and coworkers prepared a mononuclear Mn(II) complex (27) with a N3O donor set provided by the chelate bpppa (Fig. 12), in which the oxygen donor is provided by the 2-amido substituent of one of the pyridine rings [128]. Attempts to generate the oxalate-bound form of 27 afforded a dimanganese(II) complex, [Mn2+(μ2-oxalate)(bpppa)2]2+, that features a bridging oxalate dianion. Subsequently, Berreau et al. generated a series of [Mn2+(N3O)(X)(MeOH)]+ complexes (X = Cl−, Br−, I−) using bpppa and a related derivative to mimic the 3His/1Glu coordination of the enzymes [129]. No reactivity studies with O2 were reported. The Pecoraro and Chavez groups have both prepared structural models of the resting active sites using 1,4,7-triazacyclononane (TACN) ligands with pendant acetate groups [130, 131]. Chavez found that complex 28 (Fig. 12) binds oxalate in solution and exposure to O2 results in catalytic decomposition of oxalate to CO2 (2 equiv. of CO2 per oxalate ion). However, further analysis found that the catalytically active species is likely [Mn3+(oxalate)2]−, not complex 28 [130].

Extradiol catechol dioxygenases
The aerobic degradation of biomolecules by soil bacteria requires a variety of metal-containing dioxygenases that incorporate both atoms of O2 into the product [132–134]. Extradiol catechol dioxygenases (ECDOs) catalyze the oxidative scission of the C–C bond adjacent to the vicinal dihydroxy unit of catechol-containing substrates, as shown in Eq. 3 [135, 136]. While the majority of ECDOs feature a mononuclear iron(II) active site, homoprotocatechuate-2,3-dioxygenases (HPCDs) from Bacillus brevis [
137
] and Arthrobacter globiformis (Mn-MndD) [138] contain a manganese(II) center instead. Iron-containing HPCD (Fe-HPCD) and Mn-MndD exhibit 83 % sequence identity and crystallographic studies revealed that the active-site structures are nearly superimposable to beyond 15 Å from the metal centers [139, 140]. In the resting states of both enzymes, the divalent metal ion is bound to one glutamate and two histidine residues in a facial geometry (the so-called 2-His-1-carboxylate facial triad [141]); three labile H2O ligands complete the metal coordination sphere.
Regardless of the active-site metal, all ECDOs are thought to follow a common mechanism (Scheme 4) that has been elucidated through a combination of crystallographic, spectroscopic, kinetic, and computational studies [132, 142–144]. The catalytic cycle begins with displacement of the H2O molecules by the monodeprotonated substrate, which coordinates in a bidentate fashion. The resulting M(II) center is then capable of binding O2 in the vacant site adjacent to the bound substrate. On the basis of experimental and computational evidence, it has been proposed that formation of the M/O2 adduct is coupled to oxidation of the bound substrate, yielding a superoxo-M(II)-semiquinone (SQ) species (Scheme 4) [145–151]. Studies of relevant model complexes have confirmed that such an intermediate is feasible due to the “noninnocent” nature of dioxolene ligands [152–157]. However, spectroscopic studies of Fe-HPCD employing mutant enzyme and/or unactivated substrate have failed to detect a catalytically viable Fe(II)/SQ species [158–160], and a recent density functional theory (DFT) analysis of the iron-based mechanism favored a superoxo-Fe(III)-catecholate description instead [161]. Thus, the precise electronic structure of the M/O2 adduct remains a matter of dispute, and we will return to this topic below. Nevertheless, it is clear that the next step in the catalytic cycle involves formation of a metal(II)-alkylperoxide intermediate, followed by Criegee rearrangement with ring insertion of an oxygen atom. Hydrolysis of the resulting lactone finally yields the aliphatic product (Scheme 4).

Proposed catalytic mechanism for extradiol catechol dioxygenase
During the past decade, important insights into the O2 activation mechanism of ECDOs have been gained through the preparation of enzymes reconstituted with a nonphysiological metal in the active site. Since these studies were the subject of a recent review article published in this journal [4], we will provide only a cursory overview of the major findings here. In a seminal 2008 paper, the Que and Lipscomb groups described a methodology for “swapping” the metal ions Fe-HPCD and Mn-MndD, thereby generating manganese-substituted HPCD (Mn-HPCD) and iron-substituted MndD (Fe-MndD) [140]. Subsequent efforts by the same groups yielded cobalt-substituted HPCD (Co-HPCD) [162]. Comparison of HPCD crystal structures collected for the wild-type (WT) and metal-substituted enzymes revealed that the active-site structures are identical regardless of the metal ion present (Mn, Fe, or Co) [140, 162]. Kinetic measurements determined that the steady-state catalytic parameters for the four Fe- and Mn-containing enzymes (Fe-HPCD, Mn-MndD, Fe-MndD, and Mn-HPCD) were quite similar, indicating that metal-substitution has little effect on enzyme activity [140]. This result is surprising, because the Mn3+/2+ redox potential is intrinsically higher than the Fe3+/2+ potential by approximately 0.7 V [163], and the identical first and second coordination spheres of HPCD and MndD preclude the possibility of redox tuning by the active site. If the mechanism required one-electron transfer from the M(II) ion to O2, one would expect Mn-HPCD and Mn-MndD to be less active than their iron-containing counterparts, not equally active. To rationalize this apparent inconsistency, the authors argued that the ECDO mechanism does not, in fact, require a well-defined change in metal oxidation state upon O2 binding; instead, the metal ion merely serves to shuttle an electron from the catecholate substrate to O2, giving rise to the elusive O ·−2 /M(II)/SQ intermediate in a concerted step [140, 164].
The metal-substituted HPCDs have been utilized in attempts to trap and characterize reactive intermediates. The reaction of Mn-HPCD with O2 generates two short-lived intermediates that were observed by EPR spectroscopy in samples prepared via rapid freeze-quench experiments [165]. EPR analysis indicated that the first-formed species (Int1) consists of a Mn(III) center bound to an unidentified radical, perhaps superoxide or semiquinone. The later-forming intermediate (Int2) is an Mn(II) species and was assigned to the alkylperoxide intermediate in Scheme 4. These results would seem to contradict the theory, described above, that the oxidation state of the metal center does not change during ECDO catalysis. However, the low yield of Int1 (5 %) and a lack of structural information make it difficult to draw firm mechanistic conclusions.
The kinetic parameters measured for Co-HPCD differ from those reported for Fe- and Mn-HPCD in ways that are informative. The Co3+/2+ redox potential is predicted to be ~ 1.15 V higher than the Fe3+/2+ potential for ions in the same coordination environment, which accounts for the very low affinity of Co-HPCD for O2 under turnover conditions (K O2 M = 1200 μM, compared with ~50 μM for Fe- and Mn-HPCD) [162]. However, this diminished O2 affinity is partially offset by a larger k cat-value for Co-HPCD under conditions of O2 saturation; indeed, k cat is highly dependent on the solution concentration of O2. Taken together, these results suggest that the rate-determining step is O2-binding/activation for Co-HPCD, whereas product release is known to be rate-limiting for Fe-HPCD. A possible explanation for these mechanistic differences became apparent in EPR studies of Co-HPCD using an electron-poor substrate, 4-nitrocatechol (4NC) [166]. While the Co-HPCD/4NC complex features a high-spin (S = 3/2) Co(II) center, exposure to O2 affords a low-spin (S = 1/2) signal characteristic of a superoxocobalt(III) species. This spin-state change may impose a kinetic barrier to O2 binding in Co-HPCD that is not present in Fe- and Mn-HPCD, where the metal ions remain high-spin throughout the catalytic cycle. A superoxocobalt(III) species is not observed when the native substrate is employed. It was proposed that the more activated substrate rapidly transfers an electron to the Co(III) center to yield a putative high-spin O ·−2 /Co(II)/SQ species [166]. This hypothesis found support in a recent QM/MM study of the Co-HPCD mechanism by Lai and coworkers [167].
By studying both wild-type and metal-substituted enzymes with a variety of experimental and computational methods, a largely consistent picture of O2 activation in ECDOs has emerged. The broad similarities in catalysis between Mn-, Fe-, and Co-HPCD, despite the vastly different redox potentials of the respective metal ions, clearly indicate that O2 binding requires a significant amount of electron transfer from the catecholate substrate, although partial or transient oxidation of the metal centers may also be involved [4]. On-going efforts to trap and characterize catalytic intermediates will likely provide additional insights into the mechanisms of these finely-tuned enzymes.
Attempts to generate structural and functional models of the manganese-dependent ECDOs have been rather limited, lagging far behind biomimetic studies of iron-containing ring-cleaving dioxygenases [132, 168, 169]. Que and coworkers used neutral, tetradentate N4 chelates to prepare six-coordinate Mn-MndD models featuring a monoanionic 4-nitrocatecholate ligand, yet O2 reactivity experiments were not pursued [170]. Other ECDO models have been generated by binding dianionic catecholate ligands to Mn(III) centers [171–176]. The resulting complexes often exhibit valence tautomerization between Mn(III)-catecholate and Mn(II)-semiquinone configurations (Fig. 13), where the latter resembles the O ·−2 /Mn(II)/SQ species proposed for the enzymatic mechanism. Several groups have demonstrated that six-coordinate Mn(II)/SQ complexes serve as intermediates in the catalytic oxidation of catechols to benzoquinones under aerobic conditions (i.e., catechol oxidase activity) [171–173]. Hikichi and coworkers also observed small amounts of intradiol and extradiol ring-cleavage products upon reaction of the five-coordinate complex [Mn2+(DTBSQ)(TpiPr2)] (29; Fig. 13) with O2 (DTBSQ = 3,5-di-tert-butylsemiquinonate) [176].

Cobalt dioxolene complexes have attracted considerable scrutiny due to their valence tautomeric behavior [177], yet the O2 reactivity of such complexes has not been examined until recently. In an extension of their manganese studies, Hikichi et al. found that [Co2+(DTBSQ)(Tp Me2)] serves as a catalyst for the aerobic oxidation of ortho- and para-hydroquinones with simultaneous formation of H2O2 [178]. More recently, Riordan and coworkers reported that the five-coordinate complex [Co2+(DTBSQ)(PhTttBu)] (30; Fig. 13) reacts with O2 to afford the intradiol cleavage product in 16 % yield [179].
Quercetin dioxygenases
The aerobic breakdown of plant-derived flavonols by fungi and some bacteria is an important process in soil environments [180, 181]. The first step in the catabolism of quercetin (3,5,7,3′,4′-pentahydroxyflavone) involves dioxygenolytic cleavage of the 3-hydroxyflavone ring to yield a depside product and carbon monoxide (Eq. 4). This reaction is catalyzed by quercetin dioxygenases (QDOs), which belong to the cupin superfamily of enzymes [182]. The best-studied QDOs have been isolated from fungi and require copper for activity. Crystallographic studies revealed that the active site of fungal QDO from Aspergillus japonicus (QDOAj) features a mononuclear Cu(II) center bound to the three His residues and one water molecule [183]. In contrast, an X-ray structure of bacterial QDO from Bacillus subtilis (QDOBs) featured two monoiron(II) active sites per subunit [184]. Both iron centers are pentacoordinate with four protein ligands (one Glu and three facial His) and one H2O ligand (Fig. 14). However, in the site closest to the C-terminus, the Glu ligand is only weakly bound with an Fe–OGlu distance of 2.44 Å. While QDOBs is thought to be an iron-containing dioxygenase in vivo, it exhibits equal or greater activity with other transition-metal ions, including Mn, Co, Ni, and Cu [184]. The QDO from Streptomyces sp. strain FLA (QDOFLA) is similarly “promiscuous”, displaying catalytic activity following the order Ni > Co > Mn (strangely, QDOFLA is inactive with ferrous iron) [3, 185]. Crystallographic studies indicate that the active sites of QDOBs (Fe) and QDOFLA (Ni) provide similar metal coordination environments regardless of metal ion identity [184, 186]. Structures of the enzyme-substrate complexes revealed that the deprotonated substrate coordinates in a monodentate manner via the O3 atom [183, 186].

X-ray crystal structures of the substrate-bound active site of QDOFLA (left) and complex 31 (right)
The precise role of the metal center in the O2 activation mechanism of QDOs has been the subject of much debate [187]. The fact that QDO activity is largely independent of the redox potential of the active-site metal makes it unlikely that the catalytic cycle involves electron transfer from the M(II) center to O2. Moreover, flavonolate anions are known to react with O2 to yield QDO-type products in the absence of redox-active ions, leading some to suggest that the metal ion does not bind O2 at all and simply serves to deprotonate the substrate [188]. As shown in Scheme 5a, such a mechanism could proceed via direct addition of O2 to the flavonolate ring, or outer-sphere electron transfer to yield superoxide and substrate radicals. In either case, the resulting cyclic peroxide species would decompose via concerted O–C and O–O bond cleavages, affording the depside and CO products. Recently, however, Dobbek and coworkers reported strong evidence in favor of a metal-based O2 activation mechanism. After exposing crystals of the substrate-bound enzyme to O2 for short periods of time, the resulting X-ray structure revealed the presence of a Ni/O2 adduct in the QDOFLA active site [186]. The O2 ligand is coordinated in a side-on fashion with Ni–O and O–O bond distances of 2.4 and 1.3 Å, respectively. Based on their results, Dobbek et al. proposed the ECDO-like mechanism shown in Scheme 5b, in which O2 binding yields a superoxo-Ni(II)-(flavonol radical) intermediate, followed by O–C bond formation. A similar mechanism was derived from DFT studies performed by Siegbahn [189]. While it is possible that the crystallographically observed Ni/O2 is not catalytically relevant, this study provides unambiguous proof that O2 is capable of coordinating to the metal center of a QDO.

Two possible catalytic mechanisms for quercetin dioxygenase
The majority of QDO model complexes prepared to date contain copper-flavonolate units, and these efforts have been reviewed by Kaizer et al. [190]. In light of the ability of QDOs to operate with non-native metals, several groups have prepared metal(II)-flavonolate complexes using other first-row transition metals [191–196]. In the past few years, Sun and coworkers have reported a series of QDO models (M = Mn, Fe, Co, Ni, Cu, and Zn) supported by a tetradentate N3O chelate featuring a benzoate donor which closely resembles the enzymatic coordination environments (see complex 31 in Fig. 14) [197–200]. Unlike the enzyme, however, the flavonolate ligands in Sun’s models coordinate in a bidentate manner to afford six-coordinate structures. While reaction rates vary with metal ion, each of these complexes reacts with O2 at elevated temperatures to yield CO and oxidized ring-cleavage products. Berreau and coworkers also demonstrated that irradiation of metal(II)-flavonolate complexes (M = Mn, Co, Ni, Cu) in the presence of O2 triggers QDO-like reactivity [195].
There are numerous similarities between QDOs and nickel-containing acireductone dioxygenase (Ni-ARD) which catalyzes an “exit reaction” from the methionine salvage pathway (Scheme 6) [187, 201–203]. Both enzymes generate CO as a product and contain mononuclear active sites featuring 3His/1Glu coordination of a divalent metal ion [204]. Like QDO, Ni-ARD exhibits nearly full activity with non-native metal ions, such as Co(II) and Mn(II) [204, 205]. Even more remarkably, an iron-containing ARD (Fe-ARD) is also involved in the methionine salvage pathway, yet the iron and nickel enzymes catalyze different dioxygenation reactions with the same substrate (Scheme 6) [205]. However, the metal ions of both ARD enzymes are not redox active under catalytic conditions; instead, the mechanism proceeds via direct addition of O2 to the metal-bound substrate dianion [206]. Thus, the Fe- and Ni-ARDs are examples of substrate activation by a metalloenzyme, not O2 activation. It has been proposed that the unique regioselectivity of the ARDs is due to distinct coordination modes of acireductone to the Fe(II) and Ni(II) centers (the so-called “chelate hypothesis”) [206, 207]. The chelate hypothesis, however, is not supported by biomimetic studies. The Berreau group has prepared synthetic models of the Fe- and Ni-ARD active sites that exhibit metal-dependent regioselectivity similar to that of the enzymes, even though the acireductone ligand adopts the same coordination mode in the Fe and Ni complexes [208–215]. Mechanistic studies indicated that the Fe- and Ni-ARD models react with O2 to generate a common triketone intermediate, and it is the subsequent reactivity of this triketone with the metal complexes and H2O that accounts for the observed regioselectivity. The chelate hypothesis has also been called into question by computational studies of the Fe- and Ni-ARD mechanisms [216].

Reactions catalyzed by Ni- and Fe-containing ARD
Conclusions
Collectively, the studies discussed in this review highlight the relative capabilities of mononuclear Mn, Co, and Ni centers to activate O2 in both enzymatic and synthetic environments. There are close parallels between the O2 reaction pathways employed by manganese- and iron-containing systems. Both metals are able to access oxidation states between +2 and +5 under turnover conditions, permitting the formation of high-valent oxometal intermediates stable enough for spectroscopic and/or crystallographic characterization. However, as noted above, none of the mononuclear oxomanganese(IV) complexes in the literature have been prepared via direct reaction of a Mn(II) center with O2. The primary obstacle in the Mn(II) + O2 → Mn(IV)=O reaction appears to be the O–O bond homolysis step. In contrast to iron-based chemistry, there are no reported examples of mononuclear Mn(III)-OOR complexes (R = vacant, H, or alkyl) converting to an observable oxomanganese(IV) species. Instead, O–O homolysis generally requires a second metal center, resulting in dinuclear bis(μ-oxo) structures (Scheme 1). Because of this, high-valent oxomanganese species in O2-activating enzymes are only found in dinuclear active sites like ribonucleotide reductase, whereas mononuclear oxygenases involving an oxometal(IV) intermediate employ iron exclusively.
The O2 activating potential of mononuclear Co(II) and Ni(II) centers is limited by the inability of these ions to access the +4 oxidation state and stabilize terminal oxide ligands. Thus, it is not surprising that all known O2-activating enzymes containing Co or Ni employ mechanisms, such as those shown in Schemes 4 and 5, that do not require formation of a high-valent, oxometal intermediate. Instead, the divalent metal ions in ECDO and QDO simply serve as conduits of electron density between the activated substrate and O2. Nevertheless, synthetic Co and Ni complexes have displayed rich O2 chemistry that surpasses the narrow reactivity of these metals in biological systems. For example, numerous mononuclear superoxo- and peroxo-cobalt(III) species have been generated via reaction of O2 with Co(II) and Co(I) complexes. These Co/O2 adducts react with a second cobalt equivalent to afford dinuclear [Co 3+2 (μ-O)2] units (Scheme 2). Because Ni(II) centers exhibit a low affinity for O2 unless supported by highly anionic chelates, recent Ni/O2 chemistry has successfully employed Ni(I) precursors to generate superoxo-nickel(II) and dinickel(III) species (Fig. 7). Future synthetic efforts will undoubtedly expand the boundaries of O2 chemistry for Mn, Co, and Ni complexes, and biochemists will hopefully discover new and surprising metalloenzymes that employ one of these “bit-players” in the drama of O2 activation.
References
Svedruzic D, Jonsson S, Toyota CG, Reinhardt LA, Ricagno S, Lindqvist Y, Richards NGJ (2005) Arch Biochem Biophys 433:176–192
Que L Jr, Reynolds MF (2000) Met Ions Biol Syst 37:505–525
Nianios D, Thierbach S, Steimer L, Lulchev P, Klostermeier D, Fetzner S (2015) BMC Biochem 16:1–11
Fielding AJ, Lipscomb JD, Que L Jr (2014) J Biol Inorg Chem 19:491–504
Coggins MK, Sun X, Kwak Y, Solomon EI, Rybak-Akimova E, Kovacs JA (2013) J Am Chem Soc 135:5631–5640
Kovacs JA (2015) Acc Chem Res 48:2744–2753
Liu L-L, Li H-X, Wan L-M, Ren Z-G, Wang H-F, Lang J-P (2011) Chem Commun 47:11146–11148
Kitajima N, Komatsuzaki H, Hikichi S, Osawa M, Moro-oka Y (1994) J Am Chem Soc 116:11596–11597
Leto DF, Jackson TA (2014) J Biol Inorg Chem 19:1–15
Cho J, Sarangi R, Nam W (2012) Acc Chem Res 45:1321–1330
Shook RL, Gunderson WA, Greaves J, Ziller JW, Hendrich MP, Borovik AS (2008) J Am Chem Soc 130:8888–8889
Shook RL, Borovik AS (2010) Inorg Chem 49:3646–3660
Shook RL, Peterson SM, Greaves J, Moore C, Rheingold AL, Borovik AS (2011) J Am Chem Soc 133:5810–5817
Seo MS, Kim JY, Annaraj J, Kim Y, Lee Y-M, Kim S-J, Kim J, Nam W (2007) Angew Chem. Int Ed 46:377–380
Annaraj J, Cho J, Lee Y-M, Kim SY, Latifi R, de Visser SP, Nam W (2009) Angew Chem. Int Ed 48:4150–4153
Kang H, Cho J, Cho K-B, Nomura T, Ogura T, Nam W (2013) Chem Eur J 19:14119–14125
Geiger RA, Chattopadhyay S, Day VW, Jackson TA (2011) Dalton Trans 40:1707–1715
Barman P, Upadhyay P, Faponle AS, Kumar J, Nag SS, Kumar D, Sastri CV, de Visser SP (2016) Angew Chem. Int Ed 55:11091–11095
Leto DF, Chattopadhyay S, Day VW, Jackson TA (2013) Dalton Trans 42:13014–13025
Colmer HE, Howcroft AW, Jackson TA (2016) Inorg Chem 55:2055–2069
So H, Park YJ, Cho K-B, Lee Y-M, Seo MS, Cho J, Sarangi R, Nam W (2014) J Am Chem Soc 136:12229–12232
Mukhopadhyay S, Mandal SK, Bhaduri S, Armstrong WH (2004) Chem Rev 104:3981–4026
Gohdes JW, Armstrong WH (1992) Inorg Chem 31:368–373
Larson E, Lah MS, Li X, Bonadies JA, Pecoraro VL (1992) Inorg Chem 31:373–378
Pecoraro VL, Baldwin MJ, Gelasco A (1994) Chem Rev 94:807–826
Horwitz CP, Winslow PJ, Warden JT, Lisek CA (1993) Inorg Chem 32:82–88
Chen J, Lee Y-M, Davis KM, Wu X, Seo MS, Cho K-B, Yoon H, Park YJ, Fukuzumi S, Pushkar YN et al (2013) J Am Chem Soc 135:6388–6391
Wu X, Seo MS, Davis KM, Lee Y-M, Chen J, Cho K-B, Pushkar YN, Nam W (2011) J Am Chem Soc 133:20088–20091
Chen J, Yoon H, Lee Y-M, Seo MS, Sarangi R, Fukuzumi S, Nam W (2015) Chem Sci 6:3624–3632
Yoon H, Lee Y-M, Wu X, Cho K-B, Sarangi R, Nam W, Fukuzumi S (2013) J Am Chem Soc 135:9186–9194
Garcia-Bosch I, Company A, Cady CW, Styring S, Browne WR, Ribas X, Costas M (2011) Angew Chem. Int Ed 50:5648–5653
Engelmann X, Monte-Perez I, Ray K (2016) Angew Chem. Int Ed 55:7632–7649
Cook SA, Borovik AS (2015) Acc Chem Res 48:2407–2414
Parsell TH, Behan RK, Green MT, Hendrich MP, Borovik AS (2006) J Am Chem Soc 128:8728–8729
Gupta R, Taguchi T, Lassalle-Kaiser B, Bominaar EL, Yano J, Hendrich MP, Borovik AS (2015) Proc Natl Acad Sci USA 112:5319–5324
Taguchi T, Gupta R, Lassalle-Kaiser B, Boyce DW, Yachandra VK, Tolman WB, Yano J, Hendrich MP, Borovik AS (2012) J Am Chem Soc 134:1996–1999
Prokop KA, Goldberg DP (2012) J Am Chem Soc 134:8014–8017
Sahu S, Goldberg DP (2016) J Am Chem Soc 138:11410–11428
Hong S, Lee Y-M, Sankaralingam M, Vardhaman AK, Park YJ, Cho K-B, Ogura T, Sarangi R, Fukuzumi S, Nam W (2016) J Am Chem Soc 138:8523–8532
Niederhoffer EC, Timmons JH, Martell AE (1984) Chem Rev 84:137–203
Jones RD, Summerville DA, Basolo F (1979) Chem Rev 79:139–179
Busch DH, Alcock NW (1994) Chem Rev 94:585–623
Smith TD, Pilbrow JR (1981) Coord Chem Rev 39:295–383
Bailey CL, Drago RS (1987) Coord Chem Rev 79:321–332
Cozzi PG (2004) Chem Soc Rev 33:410–421
Nam W, Kim HJ, Kim SH, Ho RYN, Valentine JS (1996) Inorg Chem 35:1045–1049
Deng Y, Busch DH (1995) Inorg Chem 34:6380–6386
Simandi LI (2003) Catal Met Complexes 26:265–328
Bakac A (2000) J Am Chem Soc 122:1092–1097
Bakac A (1997) J Am Chem Soc 119:10726–10731
Lever ABP, Gray HB (1978) Acc Chem Res 11:348–355
Gavrilova AL, Qin CJ, Sommer RD, Rheingold AL, Bosnich B (2002) J Am Chem Soc 124:1714–1722
Spingler B, Scanavy-Grigorieff M, Werner A, Berke H, Lippard SJ (2001) Inorg Chem 40:1065–1066
Cho YI, Joseph DM, Rose MJ (2013) Inorg Chem 52:13298–13300
Egan JW Jr, Haggerty BS, Rheingold AL, Sendlinger SC, Theopold KH (1990) J Am Chem Soc 112:2445–2446
Avdeef A, Schaefer WP (1976) J Am Chem Soc 98:5153–5159
Reinaud OM, Yap GPA, Rheingold AL, Theopold KH (1995) Angew Chem Int Ed Engl 34:2051–2052
Reinaud OM, Theopold KH (1994) J Am Chem Soc 116:6979–6980
Hikichi S, Akita M, Moro-oka Y (2000) Coord Chem Rev 198:61–87
Dai X, Kapoor P, Warren TH (2004) J Am Chem Soc 126:4798–4799
Jones C, Schulten C, Rose RP, Stasch A, Aldridge S, Woodul WD, Murray KS, Moubaraki B, Brynda M, La Macchia G et al (2009) Angew Chem. Int Ed 48:7406–7410
Larsen PL, Parolin TJ, Powell DR, Hendrich MP, Borovik AS (2003) Angew Chem Int Ed 42:85–89
Hikichi S, Yoshizawa M, Sasakura Y, Komatsuzaki H, Moro-Oka Y, Akita M (2001) Chem Eur J 7:5011–5028
Hikichi S, Komatsuzaki H, Akita M, Moro-oka Y (1998) J Am Chem Soc 120:4699–4710
Hu X, Meyer K (2005) J Organomet Chem 690:5474–5484
Hu X, Castro-Rodriguez I, Meyer K (2004) J Am Chem Soc 126:13464–13473
Cho J-H, Sarangi R, Kang H-Y, Lee J-Y, Kubo M, Ogura T, Solomon EI, Nam W-W (2010) J Am Chem Soc 132:16977–16986
Kim D, Cho J, Lee Y-M, Sarangi R, Nam W (2013) Chem Eur J 19:14112–14118
Jo Y, Annaraj J, Seo MS, Lee Y-M, Kim SY, Cho J, Nam W (2008) J Inorg Biochem 102:2155–2159
Tcho W-Y, Wang B, Lee Y-M, Cho K-B, Shearer J, Nam W (2016) Dalton Trans 24:14511–14515
Chavez FA, Mascharak PK (2000) Acc Chem Res 33:539–545
Chavez FA, Rowland JM, Olmstead MM, Mascharak PK (1998) J Am Chem Soc 120:9015–9027
Ray K, Heims F, Pfaff FF (2013) Eur J Inorg Chem 2013:3784–3807
Ray K, Pfaff FF, Wang B, Nam W (2014) J Am Chem Soc 136:13942–13958
Winkler JR, Gray HB (2012) Struct Bonding 142:17–28
Pfaff FF, Kundu S, Risch M, Pandian S, Heims F, Pryjomska-Ray I, Haack P, Metzinger R, Bill E, Dau H et al (2011) Angew Chem. Int Ed 50:1711–1715
Lacy DC, Park YJ, Ziller JW, Yano J, Borovik AS (2012) J Am Chem Soc 134:17526–17535
Hong S, Pfaff FF, Kwon E, Wang Y, Seo M-S, Bill E, Ray K, Nam W (2014) Angew Chem Int Ed 53:10403–10407
Kimura E, Machida R (1984) J Chem Soc Chem Commun 499–500
Kimura E, Sakonaka A, Machida R, Kodama M (1982) J Am Chem Soc 104:4255–4257
Chen D, Martell AE (1990) J Am Chem Soc 112:9411–9412
Grapperhaus CA, Darensbourg MY (1998) Acc Chem Res 31:451–459
Mandimutsira BS, Yamarik JL, Brunold TC, Gu W, Cramer SP, Riordan CG (2001) J Am Chem Soc 123:9194–9195
Kieber-Emmons MT, Riordan CG (2007) Acc Chem Res 40:618–625
Hikichi S, Yoshizawa M, Sasakura Y, Akita M, Moro-oka Y (1998) In 1998, Akita and Moro-oka reported the crystal structure of a similar dinickel(III) bis(μ-oxo) complex generated by treating a [Ni2 2+(μ-OH)2] precursor with H2O2. J Am Chem Soc 120:10567–10568
Schenker R, Mandimutsira BS, Riordan CG, Brunold TC (2002) J Am Chem Soc 124:13842–13855
Fujita K, Schenker R, Gu W, Brunold TC, Cramer SP, Riordan CG (2004) Inorg Chem 43:3324–3326
Yao S, Bill E, Milsmann C, Wieghardt K, Driess M (2008) Angew Chem. Int Ed 47:7110–7113
Yao S, Driess M (2012) Acc Chem Res 45:276–287
Yao S, Xiong Y, Vogt M, Gruetzmacher H, Herwig C, Limberg C, Driess M (2009) Angew Chem Int Ed 48:8107–8110
Kundu S, Pfaff FF, Miceli E, Zaharieva I, Herwig C, Yao S, Farquhar ER, Kuhlmann U, Bill E, Hildebrandt P et al (2013) Angew Chem Int Ed 52:5622–5626
Kieber-Emmons MT, Annaraj J, Seo MS, VanHeuvelen KM, Tosha T, Kitagawa T, Brunold TC, Nam W, Riordan CG (2006) J Am Chem Soc 128:14230–14231
Cho J, Kang HY, Liu LV, Sarangi R, Solomon EI, Nam W (2013) Chem Sci 4:1502–1508
Schenker R, Kieber-Emmons MT, Riordan CG, Brunold TC (2005) Inorg Chem 44:1752–1762
Kieber-Emmons MT, Schenker R, Yap GPA, Brunold TC, Riordan CG (2004) Angew Chem Int Ed 43:6716–6718
Cho J, Sarangi R, Annaraj J, Kim SY, Kubo M, Ogura T, Solomon EI, Nam W (2009) Nat Chem 1:568–572
Kim J, Shin B, Kim H, Lee J, Kang J, Yanagisawa S, Ogura T, Masuda H, Ozawa T, Cho J (2015) Inorg Chem 54:6176–6183
Rettenmeier CA, Wadepohl H, Gade LH (2015) Angew Chem Int Ed 54:4880–4884
Rettenmeier CA, Wadepohl H, Gade LH (2016) Chem Sci 7:3533–3542
Nagataki T, Ishii K, Tachi Y, Itoh S (2007) Dalton Trans 1120–1128
Nagataki T, Tachi Y, Itoh S (2006) Chem Commun 4016–4018
Pfaff FF, Heims F, Kundu S, Mebs S, Ray K (2012) Chem Commun 48:3730–3732
Corona T, Pfaff FF, Acuna-Pares F, Draksharapu A, Whiteoak CJ, Martin-Diaconescu V, Lloret-Fillol J, Browne WR, Ray K, Company A (2015) Chem Eur J 21:15029–15038
Corona T, Company A (2016) Chem Eur J 22:13422–13429
Baran EJ (2011) Adv Plant Physiol 12:369–389
Requena L, Bornemann S (1999) Biochem J 343:185–190
Tanner A, Bowater L, Fairhurst SA, Bornemann S (2001) J Biol Chem 276:43627–43634
Lane BG, Dunwell JM, Ray JA, Schmitt MR, Cuming AC (1993) J Biol Chem 268:12239–12242
Williams HE, Wandzilak TR (1989) J Urol 141:742–749
Dunwell JM, Khuri S, Gane PJ (2000) Microbiol Mol Biol Rev 64:153–179
Dunwell JM (1998) Biotechnol Genet Eng Rev 15:1–32
Woo E-J, Dunwell JM, Goodenough PW, Marvier AC, Pickersgill RW (2000) Nat Struct Biol 7:1036–1040
Anand R, Dorrestein PC, Kinsland C, Begley TP, Ealick SE (2002) Biochemistry 41:7659–7669
Just VJ, Stevenson CEM, Bowater L, Tanner A, Lawson DM, Bornemann S (2004) J Biol Chem 279:19867–19874
Just VJ, Burrell MR, Bowater L, McRobbie I, Stevenson CEM, Lawson DM, Bornemann S (2007) Biochem J 407:397–406
Moomaw EW, Angerhofer A, Moussatche P, Ozarowski A, Garcia-Rubio I, Richards NGJ (2009) Biochemistry 48:6116–6125
Angerhofer A, Moomaw EW, Garcia-Rubio I, Ozarowski A, Krzystek J, Weber RT, Richards NGJ (2007) J Phys Chem B 111:5043–5046
Opaleye O, Rose R-S, Whittaker MM, Woo E-J, Whittaker JW, Pickersgill RW (2006) J Biol Chem 281:6428–6433
Zhu W, Easthon LM, Reinhardt LA, Tu C, Cohen SE, Silverman DN, Allen KN, Richards NGJ (2016) Biochemistry 55:2163–2173
Zhu W, Wilcoxen J, Britt RD, Richards NGJ (2016) Biochemistry 55:429–434
Saylor BT, Reinhardt LA, Lu Z, Shukla MS, Nguyen L, Cleland WW, Angerhofer A, Allen KN, Richards NGJ (2012) Biochemistry 51:2911–2920
Reinhardt LA, Svedruzic D, Chang CH, Cleland WW, Richards NGJ (2003) J Am Chem Soc 125:1244–1252
Molt RW Jr, Lecher AM, Clark T, Bartlett RJ, Richards NGJ (2015) J Am Chem Soc 137:3248–3252
Burrell MR, Just VJ, Bowater L, Fairhurst SA, Requena L, Lawson DM, Bornemann S (2007) Biochemistry 46:12327–12336
Imaram W, Saylor BT, Centonze CP, Richards NGJ, Angerhofer A (2011) Free Radical Biol Med 50:1009–1015
Twahir UT, Stedwell CN, Lee CT, Richards NGJ, Polfer NC, Angerhofer A (2015) Free Radical Biol Med 80:59–66
Svedruzic D, Liu Y, Reinhardt LA, Wroclawska E, Cleland WW, Richards NGJ (2007) Arch Biochem Biophys 464:36–47
Fuller AL, Watkins RW, Dunbar KR, Prosvirin AV, Arif AM, Berreau LM (2005) Dalton Trans 1891–1896
Fuller AL, Watkins RW, Arif AM, Berreau LM (2006) Inorg Chim Acta 359:1282–1290
Pawlak PL, Panda M, Li J, Banerjee A, Averill DJ, Nikolovski B, Shay BJ, Brennessel WW, Chavez FA (2015) Eur J Inorg Chem 2015:646–655
Scarpellini M, Gaetjens J, Martin OJ, Kampf JW, Sherman SE, Pecoraro VL (2008) Inorg Chem 47:3584–3593
Costas M, Mehn MP, Jensen MP, Que L (2004) Chem Rev 104:939–986
Gibson DT, Parales RE (2000) Curr Opin Biotechnol 11:236–243
Parales RE, Haddock JD (2004) Curr Opin Biotechnol 15:374–379
Vaillancourt FH, Bolin JT, Eltis LD (2006) Crit Rev Biochem Mol Biol 41:241–267
Kovaleva EG, Lipscomb JD (2008) Nat Chem Biol 4:186–193
Que L Jr, Widom J, Crawford RL (1981) J Biol Chem 256:10941–10944
Whiting AK, Boldt YR, Hendrich MP, Wackett LP, Que L Jr (1996) Biochemistry 35:160–170
Vetting MW, Wackett LP, Que L Jr, Lipscomb JD, Ohlendorf DH (2004) J Bacteriol 186:1945–1958
Emerson JP, Kovaleva EG, Farquhar ER, Lipscomb JD, Que L (2008) Proc Natl Acad Sci USA 105:7347–7352
Koehntop KD, Emerson JP, Que L (2005) J Biol Inorg Chem 10:87–93
Lipscomb JD (2008) Curr Opin Struct Biol 18:644–649
Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD (2007) Acc Chem Res 40:475–483
Bugg TDH, Ramaswamy S (2008) Curr Opin Chem Biol 12:134–140
Arciero DM, Lipscomb JD (1986) J Biol Chem 261:2170–2178
Emerson JP, Wagner ML, Reynolds MF, Que L Jr, Sadowsky MJ, Wackett LP (2005) J Biol Inorg Chem 10:751–760
Sanvoisin J, Langley GJ, Bugg TDH (1995) J Am Chem Soc 117:7836–7837
Kovaleva EG, Lipscomb JD (2007) Science 316:453–457
Spence EL, Langley GJ, Bugg TDH (1996) J Am Chem Soc 118:8336–8343
Georgiev V, Borowski T, Blomberg MRA, Siegbahn PEM (2008) J Biol Inorg Chem 13:929–940
Siegbahn PEM, Haeffner F (2004) J Am Chem Soc 126:8919–8932
Pierpont CG (2011) Inorg Chem 50:9766–9772
Pierpont CG (2001) Coord Chem Rev 219:415–433
Bittner MM, Lindeman SV, Fiedler AT (2012) J Am Chem Soc 134:5460–5463
Bittner MM, Lindeman SV, Popescu CV, Fiedler AT (2014) Inorg Chem 53:4047–4061
Bittner MM, Kraus D, Lindeman SV, Popescu CV, Fiedler AT (2013) Chem Eur J 19:9686–9698
Baum AE, Lindeman SV, Fiedler AT (2016) Eur J Inorg Chem 2016:2455–2464
Mbughuni MM, Chakrabarti M, Hayden JA, Meier KK, Dalluge JJ, Hendrich MP, Munck E, Lipscomb JD (2011) Biochemistry 50:10262–10274
Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Munck E, Lipscomb JD (2010) Proc Natl Acad Sci USA 107:16788–16793
Mbughuni MM, Meier KK, Munck E, Lipscomb JD (2012) Biochemistry 51:8743–8754
Christian GJ, Ye SF, Neese F (2012) Chem Sci 3:1600–1611
Fielding AJ, Kovaleva EG, Farquhar ER, Lipscomb JD, Que L Jr (2011) J Biol Inorg Chem 16:341–355
Bratsch SG (1989) J Phys Chem Ref Data 18:1–21
Miller A-F (2008) Proc Natl Acad Sci USA 105:7341–7342
Gunderson WA, Zatsman AI, Emerson JP, Farquhar ER, Que L, Lipscomb JD, Hendrich MP (2008) J Am Chem Soc 130:14465–14467
Fielding AJ, Lipscomb JD, Que L (2012) J Am Chem Soc 134:796–799
Cao L, Dong G, Lai W (2015) J Phys Chem B 119:4608–4616
Bruijnincx PCA, Lutz M, Spek AL, Hagen WR, Weckhuysen BM, vanKoten G, Gebbink RJMK (2007) J Am Chem Soc 129:2275–2286
Bruijnincx PCA, van Koten G, Gebbink RJMK (2008) Chem Soc Rev 37:2716–2744
Reynolds MF, Costas M, Ito M, Jo D-H, Tipton AA, Whiting AK, Que L Jr (2003) J Biol Inorg Chem 8:263–272
Triller MU, Pursche D, Hsieh W-Y, Pecoraro VL, Rompel A, Krebs B (2003) Inorg Chem 42:6274–6283
Reddig N, Pursche D, Krebs B, Rompel A (2004) Inorg Chim Acta 357:2703–2712
Hitomi Y, Ando A, Matsui H, Ito T, Tanaka T, Ogo S, Funabiki T (2005) Inorg Chem 44:3473–3478
Banu KS, Chattopadhyay T, Banerjee A, Mukherjee M, Bhattacharya S, Patra GK, Zangrando E, Das D (2009) Dalton Trans 8755–8764
Caneschi A, Dei A (1998) Angew Chem. Int Ed 37:3005–3007
Komatsuzaki H, Shiota A, Hazawa S, Itoh M, Miyamura N, Miki N, Takano Y, Nakazawa J, Inagaki A, Akita M et al (2013) Chem Asian J 8:1115–1119
Tezgerevska T, Alley KG, Boskovic C (2014) Coord Chem Rev 268:23–40
Ikeda A, Hoshino K, Komatsuzaki H, Satoh M, Nakazawa J, Hikichi S (2013) New J Chem 37:2377–2383
Wang P, Yap GPA, Riordan CG (2014) Chem Commun 50:5871–5873
Iwashina T (2000) J Plant Res 113:287–299
Westlake DWS, Roxburgh JM, Talbot G (1961) Nature 189:510–511
Fetzner S (2012) Appl Environ Microbiol 78:2505–2514
Steiner RA, Kalk KH, Dijkstra BW (2002) Proc Natl Acad Sci USA 99:16625–16630
Gopal B, Madan LL, Betz SF, Kossiakoff AA (2005) Biochemistry 44:193–201
Merkens H, Kappl R, Jakob RP, Schmid FX, Fetzner S (2008) Biochemistry 47:12185–12196
Jeoung J-H, Nianios D, Fetzner S, Dobbek H (2016) Angew Chem. Int Ed 55:3281–3284
Allpress CJ, Berreau LM (2013) Coord Chem Rev 257:3005–3029
Kaizer J, Balogh-Hergovich E, Czaun M, Csay T, Speier G (2006) Coord Chem Rev 250:2222–2233
Siegbahn PEM (2004) Inorg Chem 43:5944–5953
Pap JS, Kaizer J, Speier G (2010) Coord Chem Rev 254:781–793
Grubel K, Rudzka K, Arif AM, Klotz KL, Halfen JA, Berreau LM (2010) Inorg Chem 49:82–96
Kaizer J, Barath G, Pap J, Speier G, Giorgi M, Reglier M (2007) Chem Commun 5235–5237
Pap JS, Matuz A, Barath G, Kripli B, Giorgi M, Speier G, Kaizer J (2012) J Inorg Biochem 108:15–21
Barath G, Kaizer J, Speier G, Parkanyi L, Kuzmann E, Vertes A (2009) Chem Commun 3630–3632
Grubel K, Marts AR, Greer SM, Tierney DL, Allpress CJ, Anderson SN, Laughlin BJ, Smith RC, Arif AM, Berreau LM (2012) Eur J Inorg Chem 2012:4750–4757
Matuz A, Giorgi M, Speier G, Kaizer J (2013) Polyhedron 63:41–49
Sun Y-J, Huang Q-Q, Li P, Zhang J-J (2015) Dalton Trans 44:13926–13938
Sun Y-J, Huang Q-Q, Zhang J-J (2014) Inorg Chem 53:2932–2942
Sun Y-J, Huang Q-Q, Zhang J-J (2014) Dalton Trans 43:6480–6489
Sun Y-J, Huang Q-Q, Tano T, Itoh S (2013) Inorg Chem 52:10936–10948
Maroney MJ, Ciurli S (2014) Chem Rev 114:4206–4228
Boer JL, Mulrooney SB, Hausinger RP (2014) Arch Biochem Biophys 544:142–152
Pochapsky TC, Ju T, Dang M, Beaulieu R, Pagani GM, OuYang B (2007) Met Ions Life Sci 2:473–500
Deshpande AR, Wagenpfeil K, Pochapsky TC, Petsko GA, Ringe D (2016) Biochemistry 55:1398–1407
Dai Y, Wensink PC, Abeles RH (1999) J Biol Chem 274:1193–1195
Dai Y, Pochapsky TC, Abeles RH (2001) Biochemistry 40:6379–6387
Pochapsky TC, Sondej-Pochapsky S, Ju T, Mo H, Al-Mjeni F, Maroney MJ (2002) Nat Struct Biol 9:966–972
Allpress CJ, Berreau LM (2014) Eur J Inorg Chem 2014:4642–4649
Allpress CJ, Grubel K, Szajna-Fuller E, Arif AM, Berreau LM (2013) J Am Chem Soc 135:659–668
Grubel K, Ingle GK, Fuller AL, Arif AM, Berreau LM (2011) Dalton Trans 40:10609–10620
Berreau LM, Borowski T, Grubel K, Allpress CJ, Wikstrom JP, Germain ME, Rybak-Akimova EV, Tierney DL (2011) Inorg Chem 50:1047–1057
Rudzka K, Grubel K, Arif AM, Berreau LM (2010) Inorg Chem 49:7623–7625
Grubel K, Fuller AL, Chambers BM, Arif AM, Berreau LM (2010) Inorg Chem 49:1071–1081
Szajna-Fuller E, Rudzka K, Arif AM, Berreau LM (2007) Inorg Chem 46:5499–5507
Szajna E, Arif AM, Berreau LM (2005) J Am Chem Soc 127:17186–17187
Sparta M, Valdez CE, Alexandrova AN (2013) J Mol Biol 425:3007–3018
Acknowledgements
The authors gratefully acknowledge financial support from the U.S. National Science Foundation (CHE-1056845) and Marquette University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fiedler, A.T., Fischer, A.A. Oxygen activation by mononuclear Mn, Co, and Ni centers in biology and synthetic complexes. J Biol Inorg Chem 22, 407–424 (2017). https://doi.org/10.1007/s00775-016-1402-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-016-1402-7