
Abstract
General knowledge of dioxygen-activating mononuclear non-heme iron(II) enzymes containing a 2-His-1-carboxylate facial triad has significantly expanded in the last few years, due in large part to the extensive library of crystal structures that is now available. The common structural motif utilized by this enzyme superfamily acts as a platform upon which a wide assortment of substrate transformations are catalyzed. The facial triad binds a divalent metal ion at the active site, which leaves the opposite face of the octahedron available to coordinate a variety of exogenous ligands. The binding of substrate activates the metal center for attack by dioxygen, which is subsequently converted to a high-valent iron intermediate, a formidable oxidizing species. Herein, we summarize crystallographic and mechanistic features of this metalloenzyme superfamily, which has enabled the proposal of a common but flexible pathway for dioxygen activation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nature typically employs metal centers within enzymes to activate molecular oxygen, carrying out crucial transformations involved in metabolism, mammalian physiology, and biodegradation processes [1–9]. A specific subset of these enzymes consists of those having mononuclear non-heme iron(II) active sites that catalyze a wide variety of reactions, including the hydroxylation of aliphatic C–H bonds, the epoxidation of C–C double bonds, the cis-dihydroxylation of arene double bonds, heterocyclic ring formation, and oxidative aromatic ring cleavage. These reactions appear to represent a range of substrate transformations that is even broader than that associated with oxidative heme enzymes [10].
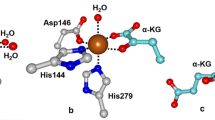
Crystallographic data on this superfamily of enzymes (Table 1) accumulated only within the past 10 years [11–20] have established the occurrence of a common structural motif that binds the divalent iron center, coined the 2-His-1-carboxylate facial triad [21, 22]. This motif consists of three endogenous protein ligands arranged at the vertices of one triangular face of an octahedron, which anchor the iron to the enzyme (Fig. 1). This three-pronged mode of attachment is in contrast to that for heme enzymes, which, by their very nature, must have the four equatorial sites occupied by the porphyrin. One axial site is then occupied by a variable amino acid residue that affixes the metalloporphyrin in the active site; this leaves only one coordination site available for an exogenous ligand, which must be dioxygen or one of its reduced derivatives. On the other hand, the metal centers of the non-heme iron enzymes have three coordination sites opposite the 2-His-1-carboxylate facial triad available for binding exogenous ligands such as O2, substrate, and/or cofactor that provide the protein with the flexibility with which to tune the reactivity of the iron(II) center.

The 2-His-1-carboxylate facial triad exemplified by the resting state of deacetoxycephalosporin C synthase (DAOCS), an α-ketoglutarate (α-KG) dependent mononuclear non-heme iron enzyme (1RXF.pdb)
The 2-His-1-carboxylate facial triad thus serves as a versatile platform for dioxygen activation. An impressive assortment of substrate transformations is catalyzed by a diverse collection of mononuclear non-heme iron(II) enzymes with distinct requirements for catalysis. Accordingly, they have been classified into five families in Fig. 2. The extradiol-cleaving catechol dioxygenases catalyze the aromatic ring cleavage of catechols at the C–C bond adjacent to the enediol group via a four-electron oxidation that incorporates both atoms of dioxygen into the product [6]. Rieske dioxygenases catalyze the cis-dihydroxylation of an arene double bond in which NADH serves as the two-electron source; furthermore both atoms of O2 are incorporated into the substrate, forming a cis-diol product [5]. The third class represents a remarkably versatile group of enzymes that require as the electron source a 2-oxoacid cosubstrate, typically α-ketoglutarate (α-KG), which undergoes oxidative decarboxylation during the reaction. The oxidizing equivalents produced therefrom can be used to effect C–H hydroxylation, oxygen-atom transfer, heterocyclic ring formation, or desaturation reactions [8, 9]. In yet another class are the aromatic amino acid hydroxylases, which utilize the tetrahydrobiopterin cofactor (BH4) as the electron source to hydroxylate the rings of aromatic amino acid residues; these enzymes are responsible for the rate-determining step in the formation of the neuronal signaling agents serotonin and the catecholamines [6, 7]. The last group represents a “catch-all” category for oxidases that at present includes isopenicillin N synthase (IPNS) [23], the ethylene-forming enzyme 1-aminocyclopropane-1-carboxylic acid oxidase (ACCO) [20, 24–26], and fosfomycin epoxidase [27, 28].

Reactions catalyzed by each of the five families of mononuclear non-heme iron enzymes containing a 2-His-1-carboxylate facial triad. Dioxygen is labeled to display the fate of each oxygen atom
Not surprisingly, extensive primary sequence homology exists within each family of mononuclear non-heme iron(II) enzymes containing a 2-His-1-carboxylate facial triad (Table 1) [13, 29–35]. However, these sequence motifs differ among the families, suggesting a convergent evolutionary pattern that has favored this structural motif. Herein, we summarize structural features of the active sites from these five families of dioxygen-activating mononuclear iron enzymes that reveal a common but flexible mechanism for dioxygen activation.
Active site structures
There are currently an astounding number of X-ray crystal structures available for dioxygen-activating mononuclear non-heme iron(II) enzymes (more than 125 structures of 30 different enzymes as of January 2005). These structures, some of which reflect a single enzyme at different stages of its catalytic cycle, definitively establish the 2-His-1-carboxylate facial triad as the common active site motif for all five enzyme families. The active site of a resting enzyme is exemplified by the α-KG dependent enzyme deacetoxycephalosporin C synthase (DAOCS, Fig. 1). On one face of this octahedral metal center is the 2-His-1-carboxylate triad, a monoanionic ligand set for binding iron(II), with three solvent molecules occupying the opposite face. At this stage, the metal center is unreactive toward dioxygen.
Examples from each enzyme family shown in Fig. 3 demonstrate that formation of an enzyme–substrate complex affects the coordination chemistry of the active site iron. For extradiol dioxygenases (i.e. 2,3-dihydroxybiphenyl 1,2-dioxygenase, BphC) and IPNS (Fig. 3a, c, respectively), their respective substrates bind directly to the iron(II) center, whereas for the α-KG dependent enzymes, such as clavaminate synthase (CAS), the cofactor α-KG coordinates to the metal center and the substrate is bound in close proximity (Fig. 3d).Footnote 1 For the Rieske dioxygenases, such as naphthalene dioxygenases (NDO), and the pterin-dependent hydroxylases, such as phenylalanine hydroxylase, neither substrate nor cofactor (in the latter case) binds directly to the metal center (Fig. 3b, e, respectively); however, in these scenarios the carboxylate of the facial triad acts as a bidentate ligand. In all five examples shown in Fig. 3, the iron center becomes five-coordinate at this stage of catalysis, a conclusion corroborated by magnetic circuilar dichroism (MCD) studies performed on a number of these enzymes [1, 3]. Substrate binding thus primes the iron active site for attack by dioxygen.

Mononuclear non-heme iron enzymes containing a 2-His-1-carboxylate facial triad that have been crystallized in a state that is poised for attack by O2. Representative members from each enzyme family include a the extradiol-cleaving catechol dioxygenase 2,3-dihydroxybiphenyl 1,2-dioxygenase (BphC–DHBP), b the Rieske dioxygenase naphthalene 1,2-dioxygenase (NDO–naphthalene), c isopenicillin N synthase (IPNS–ACV), d the α-KG dependent enzyme clavaminate synthase (CAS–α-KG–PCV), and e the pterin-dependent hydroxylase phenylalanine hydroxylase (PheOH–BH 4 –THA).
A key species in catalysis is the enzyme–substrate–(cofactor)–O2 complex, and the crystal structure of such a species should reveal how the reactants are arranged around the metal center just prior to the initiation of turnover. To date there are three structures with an end-on bound NO serving as a surrogate for O2 (Fig. 4), wherein NO can bind end-on trans to a carboxylate residue as in BphC (Fig. 4a) [37] and IPNS (Fig. 4b) [14] or trans to a histidine residue as in CAS (Fig. 4c) [38]. There is a structure of only one O2 adduct available (Fig. 5), showing a side-on O2 for NDO poised to attack the target double bond on indole [18]. These structures suggest that the 2-His-1-carboxylate facial triad provides the metal center with the flexibility to bind dioxygen in a number of modes, depending on the type of reaction to be catalyzed. This is a feature that distinguishes these non-heme iron enzymes from their heme counterparts and may account for the greater range of oxidative transformations that are catalyzed by this enzyme superfamily.

Mononuclear non-heme iron enzymes containing a 2-His-1-carboxylate motif that have been crystallized in the presence of a substrate and NO (an O2 analog) include a BphC (BphC–DHBP–NO), b IPNS (IPNS–ACV–NO), and c CAS (CAS–α-KG–A1–NO)

The only crystallographically characterized O2 complex of a mononuclear non-heme iron enzyme containing a 2-His-1-carboxylate facial triad is that of NDO–indole–O 2 . Note that an aspartate residue (D205) connects the Rieske [2Fe–2S] center (located in a neighboring subunit) to the mononuclear iron active site.
Mechanistic implications
Figure 6 shows a generalized mechanism for dioxygen-activating mononuclear non-heme iron(II) enzymes containing a 2-His-1-carboxylate motif based on the growing structural database for this superfamily and corresponding spectroscopic [1, 3] and computational [39–42] studies. The starting point is the iron(II) center of the resting enzyme that is coordinated to the facial triad on one face of its octahedral coordination sphere, with three readily displaceable ligands (shown as solvent molecules) on the opposite face (Fig. 6a). As well established by the MCD studies of Solomon and coworkers [1, 3] and supported by several crystal structures, substrate and/or cofactor binding results in the formation of a five-coordinate iron(II) center (Figs. 3, 6b) that is poised to bind O2. For the extradiol dioxygenases, IPNS, and α-KG dependent enzymes, coordination of an anionic substrate or cofactor to the iron center is observed. The replacement of a neutral solvent molecule with an anionic ligand should decrease the FeIII/FeII reduction/oxidation potential and render the iron center more susceptible to oxidation by O2. On the other hand, for the pterin-dependent hydroxylases and Rieske dioxygenases, neither the substrate nor the electron donating cofactor coordinates to the metal center; instead the carboxylate of the facial triad becomes a bidentate ligand. It would appear that this change is sufficient to lower the barrier for O2 binding in these active sites. Despite these differences among the five enzyme classes, the Fe–O2 adduct in all cases does not form until the substrate and/or cofactor is present, a mechanistic strategy that helps to protect an enzyme from self-inactivation. Regardless of whether it binds near or directly to the iron(II) center, substrate binding acts as a trigger to open the sixth coordination site for O2, thus priming the metal center for attack by dioxygen [1–3].

A general mechanistic pathway catalyzed by a mononuclear non-heme iron enzyme containing a 2-His-1-carboxylate facial triad. a The iron(II) center of the resting form of the enzyme is anchored to the active site by the 2-His-1-carboxylate facial triad (depicted by the triangular base); on the opposite face are three readily displaceable ligands, depicted here as water molecules. b The addition of substrate displaces the sixth ligand and primes the metal center for c attack by O2, which is d reduced to the peroxide oxidation level and e further activated to generate a high-valent oxidizing species. This intermediate is responsible for transforming the prime substrate into product, which is released from the active site, and a the resting form of the active site is subsequently regenerated. Refer to Figs. 3, 4, and 5 to view the various possible ligands that can occupy the remaining two coordination sites of the complexes depicted in b–e.
The next step in the proposed mechanism involves the binding of O2 to the metal center (Fig. 6c). The crystal structures displayed in Fig. 3 show that the 2-His-1-carboxylate facial triad can accommodate reaction schemes with molecular oxygen bound opposite to any of the three protein residues, suggesting that the identity of the trans ligand is not crucial for the activation of O2. Whether the trans ligand may modulate the reactivity of the bound O2 is an intriguing question that is worth investigating.
The three adjacent coordination sites available for exogenous ligand binding opposite the 2-His-1-carboxylate facial triad provide a convenient mechanism for juxtaposing dioxygen and its initial target, either the substrate itself or the cosubstrate, such that they are in the proper orientation to react. This juxtaposition is nicely illustrated in the crystal structure of the enzyme complex IPNS–δ-(L-α-aminoadipoyl)-L-cysteinyl-D-valine (ACV)–NO (Fig. 4b), where the oxygen atom of the O2 surrogate NO is equidistant from the valine nitrogen and the cysteinyl β-carbon of the substrate ACV, from which two hydrogen atoms are abstracted and between which a bond forms. This mechanistic feature is also utilized by extradiol dioxygenases (Fig. 4a) and α-KG dependent enzymes (Fig. 4c), thereby providing a convenient mechanism for O2 reduction by substrate or cofactor. Although neither substrate nor cofactor binds to the iron center of the pterin-dependent hydroxylases, BH4 is close enough to the iron center to form an iron–O2–pterin intermediate, which is analogous to the intermediates proposed for extradiol dioxygenases, IPNS, and α-KG dependent enzymes. In the case of the Rieske dioxygenases, it is proposed that O2 binding to the iron(II) center is followed immediately by electron transfer from the nearby reduced Rieske [2Fe–2S] cluster to generate an FeIII(η2–O2) species [18]. This species is the only dioxygen-bound intermediate in the superfamily to be crystallographically observed thus far (Fig. 5). Whatever the mechanistic details, O2 is reduced to the peroxide oxidation level (Fig. 6d) at this stage of the mechanism in all enzyme classes.
O–O bond cleavage occurs in the next step of the mechanistic pathway. For the extradiol and Rieske dioxygenases, substrate oxidation may occur either directly from the peroxo intermediate or via prior cleavage of the O–O bond to form a high-valent intermediate. Independent density functional theory (DFT) calculations support one scenario or the other for the extradiol-cleaving dioxygenases [39, 41], but direct mechanistic evidence is lacking. For the Rieske dioxygenases, DFT calculations favor direct attack by the iron(III)-side-on-peroxo species owing to the high energy calculated for the putative HO–FeV=O intermediate [40], a species that is related to the active species in related OsO4, RuO4, and MnO4− systems known to effect cis-dihydroxylation of olefins [43–45]. However, there is some experimental evidence in favor of prior O–O bond cleavage to form an oxoiron(V) oxidant. It has been reported that 18O from H 182 O is incorporated into the indanol product formed by the hydroxylation of indane by toluene dioxygenases [5]; this observation requires an oxidant capable of 18O exchange with water. So the involvement of high-valent iron-oxo species is uncertain in these two enzyme classes.
On the other hand, oxoiron(IV) intermediates are implicated from substrate analogue studies for IPNS [13], α-KG dependent enzymes [8], and pterin-dependent hydroxylases [7] (Fig. 6e) as the species responsible for attacking the prime substrate and transforming it into product. Direct evidence for a high-valent iron intermediate has recently been obtained for the α-KG dependent enzyme taurine/α-KG dioxygenases (TauD). A high-spin oxoiron(IV) center with a UV absorption near 320 nm has been trapped in the reaction of the TauD–α-KG–taurine complex with O2 and characterized by Mössbauer (ΔEQ=−0.88 mm/s and δ=0.31 mm/s) [46], resonance Raman (νFe=O=821 cm−1) [47], and extended X-ray absorption fine structure (rFe=O=1.62 Å) [48] techniques. Furthermore, the rate of decay of this intermediate is strongly retarded by deuterium isotope substitution of the target C–H bond (kinetic isotope effect approximately 28–50) [49]. Thus, by extension, the oxoiron(IV) species is postulated to be the oxidant in many of the diverse transformations carried out by this superfamily of enzymes.
The properties of the oxoiron(IV) intermediate of TauD can be compared with those of synthetic oxoiron(IV) complexes that have been characterized within the same time frame [50–55], including one that has been characterized crystallographically [50]. Interestingly, the TauD intermediate shares similar νFe=O and rFe=O values with the model complexes, but the TauD intermediate is high-spin iron(IV), while all the synthetic complexes are low-spin. DFT studies suggest that the ground-state bonding properties of the Fe=O bond should not be greatly affected by spin state [56], in agreement with the experimental results, but more work needs to be done to uncover subtleties that may affect the reactivity of the oxoiron(IV) center.
Perspective
Over the past few years the number of structurally characterized enzymes containing an iron center bound by the 2-His-1-carboxylate facial triad has grown dramatically. Members of this superfamily of enzymes continue to be found throughout aerobic life and are responsible for carrying out an impressive array of transformations that require dioxygen activation. The observation of Fe–O2 [18] and FeIV=O [46] intermediates for two of these enzymes, coupled with corresponding advances in trapping synthetic intermediates [2], represent a significant step forward toward understanding the mechanisms of dioxygen activation by this superfamily. However, for almost all of the 2-His-1-carboxylate iron enzymes, the nature of the oxidizing species remains unproven and, in some cases, enigmatic at best. As studies of these enzymes progress, we expect to see common threads in the mechanisms for dioxygen activation. Undoubtedly, these mechanistic insights will help define how one of nature’s recurring metal binding motifs is able to promote dioxygen activation for a variety of reactions.
Notes
A notable exception to this generalization is the case of DAOCS in which α-KG and penicillin were found in recent structures of respective binary complexes to bind in overlapping regions of the active site; it would thus appear that a ternary DAOCS–α-KG–penicillin complex could not form [36].
Abbreviations
- α-KG:
-
α-Ketoglutarate
- 2,3-CTD:
-
Catechol 2,3-dioxygenase
- 4,5-PCD:
-
Protocatechuate 4,5-dioxygenase
- A1:
-
Deoxyguanidinoproclavaminate
- ACCO:
-
1-Aminocyclopropane-1-carboxylic acid oxidase
- ACV:
-
δ-(L-α-Aminoadipoyl)-L-cysteinyl-D-valine
- ANS:
-
Anthocyanidin synthase
- AtsK:
-
Alkylsulfatase
- BH4:
-
6(R)-L-erythro-5,6,7,8-Tetrahydrobiopterin
- BphC:
-
2,3-Dihydroxybiphenyl 1,2-dioxygenase
- CarC:
-
Carbapenem synthase
- CAS:
-
Clavaminate synthase
- DAOCS:
-
Deacetoxycephalosporin C synthase
- DFT:
-
Density functional theory
- DHBD:
-
Synonymous with BphC
- DHBP:
-
2,3-Dihydroxybiphenyl
- EXAFS:
-
Extended X-ray absorption fine structure
- FIH-1:
-
Factor-inhibiting hypoxia-inducible factor-1
- HGO:
-
Homogentisate 1,2-dioxygenase
- HPCD:
-
Homoprotocatechuate 2,3-dioxygenase
- HPPD:
-
4-Hydroxyphenylpyruvate dioxygenase
- IPNS:
-
Isopenicillin N synthase
- LigAB:
-
Synonymous with 4,5-PCD
- MCD:
-
Magnetic circular dichroism
- MndD:
-
MnII-dependent HPCD
- MPC:
-
Metapyrocatechase (synonymous with 2,3-CTD)
- NDO:
-
Naphthalene 1,2-dioxygenase
- PCV:
-
Proclavaminate
- PheOH:
-
Phenylalanine hydroxylase
- TauD:
-
Taurine/α-KG dioxygenase
- THA:
-
3-(2-Thienyl)-L-alanine
- TrpOH:
-
Tryptophan hydroxylase
- TyrOH:
-
Tyrosine hydroxylase
References
Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J (2000) Chem Rev 100:235–349
Costas M, Mehn MP, Jensen MP, Que L Jr (2004) Chem Rev 104:939–986
Solomon EI, Decker A, Lehnert N (2003) Proc Natl Acad Sci USA 100:3589–3594
Bugg TDH (2003) Tetrahedron 59:7075–7101
Wackett LP (2002) Enzyme Microb Tech 31:577–587
Vaillancourt FH, Bolin JT, Eltis LD (2004) In: Ramos J-L (ed) Pseudomonas. Kluwer/Plenum, New York, pp 359–395
Fitzpatrick PF (2003) Biochemistry 42:14083–14091
Hausinger RP (2004) Crit Rev Biochem Mol Biol 39:21–68
Schofield CJ, Zhang Z (1999) Curr Opin Struct Biol 9:722–731
Sono M, Roach MP, Coulter ED, Dawson JH (1996) Chem Rev 96:2841–2887
Han S, Eltis LD, Timmis KN, Muchmore SW, Bolin JT (1995) Science 270:976–980
Senda T, Sugiyama K, Narita H, Yamamoto T, Kimbara K, Fukuda M, Sato M, Yano K, Mitsui Y (1996) J Mol Biol 255:735–752
Roach PL, Clifton IJ, Fülöp V, Harlos K, Barton GJ, Hajdu J, Andersson I, Schofield CJ, Baldwin JE (1995) Nature 375:700–704
Roach PL, Clifton IJ, Hensgens CMH, Shibata N, Schofield CJ, Hajdu J, Baldwin JE (1997) Nature 387:827–830
Valegård K, van Scheltinga ACT, Lloyd MD, Hara T, Ramaswamy S, Perrakis A, Thompson A, Lee H-J, Baldwin JE, Schofield CJ, Hajdu J, Andersson I (1998) Nature 394:805–809
Zhang Z, Ren J, Stammers DK, Baldwin JE, Harlos K, Schofield CJ (2000) Nat Struct Biol 7:127–133
Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, Ramaswamy S (1998) Structure 6:571–586
Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S (2003) Science 299:1039–1042
Flatmark T, Stevens RC (1999) Chem Rev 99:2137–2160
Zhang Z, Ren J-s, Clifton IJ, Schofield CJ (2004) Chem Biol 11:1383–1394
Hegg EL, Que L Jr (1997) Eur J Biochem 250:625–629
Que L Jr (2000) Nat Struct Biol 7:182–184
Schofield CJ, Baldwin JE, Byford MF, Clifton I, Hajdu J, Hensgens C, Roach P (1997) Curr Opin Struct Biol 7:857–864
Rocklin AM, Tierney DL, Kofman V, Brunhuber NMW, Hoffman BM, Christoffersen RE, Reich NO, Lipscomb JD, Que L Jr (1999) Proc Natl Acad Sci USA 96:7905–7909
Zhou J, Rocklin AM, Lipscomb JD, Que L Jr, Solomon EI (2002) J Am Chem Soc 124:4602–4609
Rocklin AM, Kato K, Liu H-w, Que L Jr, Lipscomb JD (2004) J Biol Inorg Chem 9:171–182
Liu P, Liu A, Yan F, Wolfe MD, Lipscomb JD, Liu H-w (2003) Biochemistry 42:11577–11586
Liu P, Mehn MP, Yan F, Zhao Z, Que L Jr, Liu H-w (2004) J Am Chem Soc 126:10306–10312
Eltis LD, Bolin JT (1996) J Bacteriol 178:5930–5937
Spence EL, Kawamukai M, Sanvoisin J, Braven H, Bugg TDH (1996) J Bacteriol 178:5249–5256
Jiang H, Parales RE, Lynch NA, Gibson DT (1996) J Bacteriol 178:3133–3139
Prescott AG (1993) J Exp Bot 44:849–861
Borovok I, Landman O, Kreisberg-Zakarin R, Aharonowitz Y, Cohen G (1996) Biochemistry 35:1981–1987
Tan DSH, Sim T-S (1996) J Biol Chem 271:889–894
Kappock TJ, Caradonna JP (1996) Chem Rev 96:2659–2756
Valegård K, van Scheltinga ACT, Dubus A, Ranghino G, Öster LM, Hajdu J, Andersson I (2004) Nat Struct Mol Biol 11:95–101
Sato N, Uragami Y, Nishizaki T, Takahashi Y, Sazaki G, Sugimoto K, Nonaka T, Masai E, Fukuda M, Senda T (2002) J Mol Biol 321:621–636
Zhang Z, Ren J-s, Harlos K, McKinnon CH, Clifton IJ, Schofield CJ (2002) Febs Lett 517:7–12
Deeth RJ, Bugg TDH (2003) J Biol Inorg Chem 8:409–418
Bassan A, Blomberg MRA, Siegbahn PEM (2004) J Biol Inorg Chem 9:439–452
Siegbahn PEM, Haeffner F (2004) J Am Chem Soc 126:8919–8932
Bassan A, Borowski T, Siegbahn PEM (2004) Dalton Trans 20:3153–3162
Schröder M (1980) Chem Rev 80:187–213
Shing TKM, Tam EKW, Tai VW-F, Chung IHF, Jiang Q (1996) Chem Eur J 2:50–57
Lee DG, Chen T (1989) J Am Chem Soc 111:7534–7538
Price JC, Barr EW, Tirupati B, Bollinger JM Jr, Krebs C (2003) Biochemistry 42:7497–7508
Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP (2004) J Am Chem Soc 126:1022–1023
Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM Jr, Krebs C (2004) J Am Chem Soc 126:8108–8109
Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM Jr (2003) J Am Chem Soc 125:13008–13009
Rohde J-U, In J-H, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L Jr (2003) Science 299:1037–1039
Lim MH, Rohde J-U, Stubna A, Bukowski MR, Costas M, Ho RYN, Münck E, Nam W, Que L Jr (2003) Proc Natl Acad Sci USA 100:3665–3670
Kaizer J, Costas M, Que L Jr (2003) Angew Chem Int Ed 42:3671–3673
Kaizer J, Klinker EJ, Oh NY, Rohde J-U, Song WJ, Stubna A, Kim J, Münck E, Nam W, Que L Jr (2004) J Am Chem Soc 126:472–473
Balland V, Charlot M-F, Banse F, Girerd J-J, Mattioli TA, Bill E, Bartoli J-F, Battioni P, Mansuy D (2004) Eur J Inorg Chem 301–308
Rohde J-U, Torelli S, Shan X, Lim MH, Klinker EJ, Kaizer J, Chen K, Nam W, Que L Jr (2004) J Am Chem Soc 126:16750–16761
Decker A, Rohde J-U, Que L Jr, Solomon EI (2004) J Am Chem Soc 126:5378–5379
Sugimoto K, Senda T, Aoshima H, Masai E, Fukuda M, Mitsui Y (1999) Structure 7:953–965
Titus GP, Mueller HA, Burgner J, de Córdoba SR, Peñalva MA, Timm DE (2000) Nat Struct Biol 7:542–546
Serre L, Sailland A, Sy D, Boudec P, Rolland A, Pebay-Peyroula E, Cohen-Addad C (1999) Structure 7:977–988
Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC (1997) Nat Struct Biol 4:578–585
Kobe B, Jennings IG, House CM, Michell BJ, Goodwill KE, Santarsiero BD, Stevens RC, Cotton RGH, Kemp BE (1999) Nat Struct Biol 6:442–448
Erlandsen H, Kim JY, Patch MG, Han A, Volner A, Abu-Omar MM, Stevens RC (2002) J Mol Biol 320:645–661
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Koehntop, K.D., Emerson, J.P. & Que, L. The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J Biol Inorg Chem 10, 87–93 (2005). https://doi.org/10.1007/s00775-005-0624-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-005-0624-x