
Abstract
Ubiquitin–proteasome system (UPS) plays a crucial role in many cellular processes such as cell cycle, proliferation and apoptosis. Aberrant activation of UPS may result in cellular transformation or other altered pathological conditions. Previous studies have shown that metal-based complexes could inhibit proteasome activity and induce apoptosis in certain human cancer cells. In the current study, we report that the cadmium and copper complexes with heterocycle-ornithine Schiff base are potent inhibitors of proteasomal chymotrypsin-like (CT-like) activity, leading to induction of apoptosis in cancer cells. Two novel copper-containing complexes and two novel cadmium-containing complexes with different heterocycle-ornithine Schiff base structures as ligands were synthesized and characterized. We found that complexes Cu1, Cd1 and Cd2 show proteasome-inhibitory activities in human breast cancer MDA-MB-231 and human prostate cancer LNCaP cells, resulting in the accumulation of p27, a natural proteasome substrate and other ubiquitinated proteins, followed by the induction of apoptosis. Our results suggest that metal complexes with heterocycle-ornithine Schiff base have proteasome-inhibitory capabilities and have the potential to be developed into novel anticancer drugs.
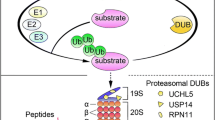
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ubiquitin–proteasome system (UPS) is a pivotal cellular pathway for protein homeostasis. It controls intracellular protein degradation and therefore regulates variety of key cellular processes such as cell cycle, proliferation, differentiation, apoptosis and immune response. Its target proteins include a broad array of regulatory proteins that are crucial for cell cycle progression, cell development and differentiation, DNA damage response and tumorigenesis. Therefore, a cell could modulate its protein expression patterns in response to a change in physiological conditions and UPS. Thus, UPS plays a critical role in normal health and disease states [1, 2]. Importantly, as UPS plays a key role in carcinogenesis and tumor progression, it has therefore been extensively studied as a novel molecular target for the development of new anticancer agents [3, 4].
The proteasome is a massive protease complex with multiple catalytic activities responsible for degrading a vast array of cellular proteins. To be degraded by the proteasome, these target proteins should be first tagged with at least 4 ubiquitin (Ub) molecules, which can then direct the substrate proteins to the 26S proteasome for their destruction. The 20S proteasome, the core subunit of the 26S proteasome complex, has at least three distinct catalytic activities, i.e., caspase/PGPH-like, trypsin-like and chymotrypsin-like activity. Several studies have shown that the inhibition of the proteasomal chymotrypsin-like activity results in the accumulation of various target proteins leading to the induction of apoptosis in various types of cancer cells [5, 6].
Metal-containing agents have been used in clinic for many years, e.g., cisplatin, a platinum-containing compound, is used in the chemotherapy for the treatment of various cancers [7–10]. However, patients show severe adverse effects like nephrotoxicity, ototoxicity, or electrolyte disturbance when treated with cisplatin-based chemotherapy. They also eventually develop drug resistance limiting the clinical use of this drug [11–13]. Therefore, many researchers have been trying to design, synthesize, and characterize new potential metal-based anticancer drugs to reduce toxicity, overcome resistance and improve clinical effectiveness [14–16].
A Schiff base possesses a carbon–nitrogen double bond (–C=N–R with R = aryl or alkyl group) as a functional group that is formed by condensation of an aldehyde or ketone with a primary amine. Schiff bases coordinate via the lone pair of the nitrogen atom from the –C=N–R moiety and additional functional groups leading to stabilization of many metals in various oxidation states [17]. Previously, it has been shown that the metal complexation with a Schiff base ligand improves the anticancer properties of the complex [18, 19]. Our previous work has been focused on the biological activity of Schiff base complexes and we have shown that many of these complexes have significant antitumor activity, associated with proteasome inhibition [20–23].
In the current study, we hypothesized that synthetic forms of Cu(II) and Cd(II) with heterocycle-ornithine Schiff base ligands might have proteasome-inhibitory and apoptosis-inducing activities in cancer cells. To test this hypothesis, we synthesized two novel cooper-containing complexes, Cu(C16H17N2O3)2·2H2O (Cu1) (C16H17N2O3=2-hydroxy-1-naphthaldehyde-l-ornithine) and Cu(C12H15N2O4)2·2H2O (Cu2) (C12H15N2O4=2,4-dihydroxybenzaldehyde-l-ornithine) and two novel cadmium-containing complexes, Cd(C16H17N2O3)2·2H2O (Cd1) (C16H17N2O3=2-hydroxy-1-naphthaldehyde-l-ornithine) and Cd(C12H15N2O4)2·2H2O (Cd2) (C12H15N2O4=2,4-dihydroxybenzaldehyde-l-ornithine) (Fig. 1) with different heterocycle-ornithine Schiff base structures as ligands and characterized them by various assays, including IR, UV, elemental analysis, 1H NMR analysis, thermogravimetric analysis and molar conductivity analysis. In the current study, we characterized and assessed the synthesized copper and cadmium complexes and compared their abilities to inhibit the proliferation and induce apoptosis of MDA-MB-231 breast cancer and LNCaP human prostate cancer cells. Of the complexes tested, Cu1, Cd1 and Cd2 could inhibit cellular proteasomal chymotrypsin-like activity and induce apoptosis in these breast and prostate cancer cells.

Chemical structures of cadmium and copper complexes Cu1, Cd1, Cu2 and Cd2
Materials and methods
Materials
All the chemicals were used without further purification in this work. 2,4-Dihydroxybenzaldehyde and 2-hydroxy-1-naphthaldehyde were purchased from Acros and l-Ornithine monohydrochloride was purchased from Aladdin. Dimethyl sulfoxide (DMSO) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO). All compounds were made as 50 mM stocks in DMSO and stored at 4 °C. DMEM/F12 (1:1), RPMI-1640 and penicillin/streptomycin were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Aleken Biologicals (Nash, TX, USA). The fluorogenic peptide substrate Suc-LLVY-AMC (for the CT-activity assay) was purchased from Calbiochem (San Diego, CA). Mouse monoclonal antibody against human poly (ADP-ribose) polymerase (PARP), mouse monoclonal antibodies against ubiquitin (P4D1) and p27 (F-8), goat polyclonal antibody against β-actin (C-11) and all secondary antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA).
Elemental analyses were carried out on a Carlo Erba 1106 full-automatic trace organic elemental analyzer. Infrared spectra were recorded as KBr pellets on a Nicolet 170SX spectrophotometer in the 4,000–400 cm−1 region. The UV spectra were performed on a Unicam UV2 spectrometer. 1H NMR spectra were obtained on a Bruker AVANCE III (600-MHz) spectrometer. Thermogravimetric measurements were made using a PerkinElmer TGA7 instrument. The heating rate was programmed to be 10 °C min−1 with a protecting stream of N2 flowing at a rate of 40 mL min−1. Molar conductivity was measured with a WTWLF model 330 conductivity meters, using prepared solution of the complex in DMSO.
Synthesis of complexes
l-Ornithine monohydrochloride (2.0 mmol) and potassium hydroxide (1.5 mmol) were dissolved in 30 mL of methanol with magnetic stirring, and then 2-hydroxy-1-naphthaldehyde or 2,4-dihydroxybenzaldehyde (2.0 mmol) was added dropwise to the solution. The reaction mixture was heated to 60 °C with stirring and then refluxed for 5 h to get a bright yellow transparent solution. Next, the solution of M(OAc)2·nH2O [M = Cd(II) and Cu(II)] (1.0 mmol) in methanol (10 mL) was added, and the mixture was stirred and refluxed at 60 °C for 4 h to yield a precipitate, which was filtered off, to produce the final complexes. The synthesis routes of complexes Cu1, Cd1, Cu2 and Cd1 are shown in Scheme 1.

The synthesis routes of complexes
Cu1: yield, 85 %; Anal. Calc. for Cu1 {%, [Cu(C16H17N2O3)2·2H2O], FW = 670.2123 g mol−1}; C, 57.34; H, 5.71; N, 8.36. Found (%): C, 57.73; H, 5.30; N, 8.68. UV: λ max (nm): 243, 325. IR data (KBr, cm−1): 3,419.68, υ(–OH); 1,621.79, υ(–C=N–); 1,568.24, υas(COO−); 1,356.47, υs(COO−); 506.71, υ(Cu–N); 427.68, υ(Cu–O). 1H NMR (DMSO-d6, 600 MHz; s, singlet; d, doublet; t, triplet): δ (ppm) 9.871 (2H, s, CH=N); 9.037 (2H, s, –OH); 7.847 (2H, t, –Ph–H); 7.560 (2H, s, –Ph–H); 6.947 (2H, d, –Ph–H); 6.792 (2H, t, –Ph–H); 6.492 (4H, m, –Ph–H); 4.677 (4H, s, –NH2); 3.16 (2H, d, –CH); 3.021 (4H, t, –CH2); 2.651 (4H, t, –CH2); 2.230 (4H, t, –CH2). TG analysis: residue 12.11 % (calculated 11.87 %, CuO). Molar conductivity, Λm (S cm2 mol−1): 9.42.
Cd1: yield, 85 %; Anal. Calc. for Cd1 {%, [Cd(C16H17N2O3)2·2H2O], FW = 719.0773 g mol−1}; C, 53.25; H, 5.32; N, 7.79. Found (%): C, 53.43; H, 5.05; N, 8.01. UV: λ max (nm): 247, 323. IR data (KBr, cm−1): 3,443.89, υ(–OH); 1,629.56, υ(–C=N–); 1,552.12, υas(COO−); 1,351.47, υs(COO−); 505.14, υ(Cu–N); 447.31, υ(Cu–O). 1H NMR (DMSO-d6, 600 MHz; s, singlet; d, doublet; t, triplet): δ (ppm) 9.863 (2H, s, CH=N); 9.041 (2H, s, –OH); 7.967 (2H, t, –Ph–H); 7.561 (2H, s, –Ph–H); 6.834 (2H, d, –Ph–H); 6.798 (2H, t, –Ph–H); 6.572 (4H, m, –Ph–H); 4.699 (4H, s, –NH2); 3.213 (2H, d, –CH); 3.078 (4H, t, –CH2); 2.758 (4H, t, –CH2); 2.239 (4H, t, –CH2). TG analysis: residue 18.36 % (calculated 17.88 %, CdO). Molar conductivity, Λm (S cm2 mol−1): 8.31.
Cu2: yield, 85 %; Anal. Calc. for Cu2 {%, [Cu(C12H15N2O4)2·2H2O], FW = 602.0937 g mol−1}; C, 47.87; H, 5.69; N, 9.31. Found (%): C, 48.54; H, 5.58; N, 9.57. UV: λ max (nm): 237, 316. IR data (KBr, cm−1): 3,454.59, υ(–OH); 1,621.03, υ(–C=N–); 1,563.46, υas(COO−); 1,362.47, υs(COO−); 1,229.58, υ(Ar–O); 522.42, υ(Cu–N); 426.69, υ(Cu–O). 1H NMR (DMSO-d6, 600 MHz; s, singlet; d, doublet; t, triplet): δ (ppm) 9.347 (2H, s, CH=N); 9.034 (4H, s, –OH); 8.847–6.392 (6H, t, –Ph–H); 4.537 (4H, s, –NH2); 3.675 (2H, s, –CH); 3.031 (4H, t, –CH2); 2.551 (4H, t, –CH2); 2.325 (4H, t, –CH2). TG analysis: residue 13.87 % (calculated 13.21 %, CuO). Molar conductivity, Λm (S cm2 mol−1): 12.13.
Cd2: yield, 85 %; Anal. Calc. for Cd2 {%, [Cd(C12H15N2O4)2·2H2O], FW = 650.9587 g mol−1}; C, 44.28; H, 5.26; N, 8.60. Found (%): C, 44.13; H, 5.01; N, 8.21. UV: λ max (nm): 231, 321. IR data (KBr, cm−1): 3,401.53, υ(–OH); 1,632.17, υ(–C=N–); 1,599.74, υas(COO−); 1,362.07, υs(COO−); 1,229.07, υ(Ar–O); 512.37, υ(Cu–N); 426.86, υ(Cu–O). 1H NMR (DMSO-d6, 600 MHz; s, singlet; d, doublet; t, triplet): δ (ppm) 9.473 (2H, s, CH=N); 9.048 (1H, s, –OH); 8.616–6.213 (6H, t, –Ph–H); 4.307 (4H, s, –NH2); 3.674 (2H, s, –CH); 3.138 (4H, t, –CH2); 2.560 (4H, t, –CH2); 2.420 (4H, t, –CH2). TG analysis: residue 19.65 % (calculated 19.72 %, CdO). Molar conductivity, Λm (S cm2 mol−1): 13.48.
Cell culture and whole cell extract preparation
MDA-MB-231 human breast cancer cells and LNCaP human prostate cancer cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). MDA-MB-231 231 human breast cancer cells were cultured in DMEM/F-12 (1:1) and LNCaP human prostate cancer cells were cultured in RPMI-1640 medium. All media were supplemented with 10 % FBS, 100 μg/mL streptomycin and 100 U/mL penicillin (Life Technologies, Carlsbad, CA USA). All cells were maintained in a humidified atmosphere containing 5 % CO2 at 37 °C. The cells were treated as indicated, harvested, washed with phosphate-buffered saline (PBS), lysed in lysis buffer [50 mM tris(hydroxymethyl)aminomethane Tris–HCl, pH 8.0, 150 mM NaCl, 0.5 % NP40], vortexed at 4 °C for 30 min, and centrifuged at 12,000×g for 14 min [24]. The supernatants were collected as whole cell extracts and used for the measurement of chymotrypsin-like activity and Western blot analysis, as previously described [25].
Cell proliferation assay
The effect of each cadmium complex on cell proliferation was determined by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay. In brief, MDA-MB-231 human breast cancer and LNCaP human prostate cancer cells were seeded in triplicates in a 96-well plate and incubated at 37 °C until 70–80 % confluent, then treated with the indicated concentration of each complex for 24 h. Media was then removed and MTT solution (1 mg/mL) was added followed by a 2-h incubation period. MTT was then removed and 100 μL DMSO was added to dissolve formazan, the metabolized MTT product, followed by measuring the absorbance values on a Victor 3 multi-label plate reader (PerkinElmer Wellesley, MA).
In vitro proteasomal activity assay
A MDA-MB-231 cell extract (10 μg) was incubated in 100 μL of assay buffer (20 mM Tris–HCl, pH 7.5) and 20 μM of chymotrypsin-like substrate Suc-LLVY-AMC, using different concentrations of copper and cadmium complexes Cu1, Cu2, Cu2 and Cd2 or DMSO as a vehicle control at 37 °C for 2 h. Following incubation, proteasome CT-like activity was measured using the Wallac Victor 3 Multi-label Counter with an excitation filter of 365 nm and emission filter of 460 nm.
Proteasomal CT-like activity assay using human breast cancer cells and human prostate cancer cells
MDA-MB-231 human breast cancer and LNCaP human prostate cancer cells were treated as indicated, lysed and protein concentrations measured by the Bio-Rad Protein Assay (Bio-Rad Hercules, CA). Whole cell extracts (10 μg) were incubated for 2 h at 37 °C in 100 μL of assay buffer (20 mM Tris–HCl, PH 7.5) with 20 μM fluorogenic peptide substrate Suc-LLVY-AMC. Proteasomal CT-like activity was measured using the Wallac Victor 3 Multi-label Counter with an excitation filter of 365 nm and emission filter of 460 nm.
Western blot assay
Proteins (30 μg) from whole cell extracts were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane, followed by incubation with antibodies against specified proteins and visualization with an enhanced chemiluminescence reagent (Denville Scientific Metuchen, NJ), as previously described [26].
Cellular morphology analysis
Morphological changes in the cancer cells after indicated treatments were observed using a Zeiss (Thornwood, NY) Axiovert 25 microscope with phase contrast, as previously described [27].
Results
Characterization of copper and cadmium complexes
The structural elucidation of the complexes is supported by infrared spectroscopy (IR spectra). There are wide and strong bands at 3,400–3,500 cm−1 in the spectra of compounds Cu1, Cd1, Cu2 and Cd2 which are assigned to H2O absorption. The IR spectra also shows sharp bands at 1,621–1,633 cm−1 corresponding to υ(–C=N–) in compounds Cu1, Cd1, Cu2 and Cd2 [28]. The evidence of the complexation of oxygen and nitrogen is obtained from the appearance of new bands at 426–448 cm−1 and 505–523 cm−1 which is assignable to υ(M–O) and υ(M–N) for the complexes. The difference between the value of υas(COO−) and υs(COO−) is greater than 200 cm−1, thus confirming that carboxylic radical is in the form of monodentate in the coordination complex.
The specifics of the 1H NMR studies are described in the ‘Materials and methods’ section. From the 1H NMR spectra analysis, we conclude that the complexes consist of coordination between the ligands and the M (II) ion. The four complexes give a singlet at 9.037 (2H), 9.041 (2H), 9.034 (4H) and 9.048 (4H) ppm, assigned to the protons (–OH), respectively. So the hydrogen atom of phenolic hydroxyl was found to still be present in the complexes Cu1, Cu2, Cd1 and Cd2. In addition, the complexes Cu1, Cd1, Cu2 and Cd2 also show a singlet at 4.677 (4H), 4.699 (4H), 4.537 (4H) and 4.307 (4H) ppm, assigned to the protons (–NH2), respectively, which indicates that the hydrogen atoms of –NH2 were still be present in the complexes. However, the hydrogen atom of –COOH was in fact displaced by a metal ion. Consequently, the newly synthesized complexes were indeed formed by coordination with the metal ion, a conclusion further supported by IR output data.
The UV–Vis absorption spectra for Cd1, Cd2, Cu1 and Cu2 complexes, dissolved in DMSO, were obtained and recorded in the 200–500 nm range. For the complexes Cu1 and Cd2, the λ max values, 231–247 nm and 316–325 nm can be attributed to the π–π* and n–π* transition of ligands, respectively. Furthermore, the absorption bands tend to shift towards longer wavelengths, which can also be ascribed to the metal-to-ligand charge transfer transitions taking place.
TG analysis of these complexes was recorded in the range of 25–800 °C. The residue rates of the metal complexes were 12.11–19.65 %, respectively, consistent with the calculated values (11.87–19.72 %).
All of the compounds are soluble in DMSO and stable in air. The molar conductivities (Λm) of Cu2, Cd1 and Cd2 in DMSO were 9.42, 12.13, 8.32 and 13.48 S cm2 mol−1, respectively. As the molar conductivities were less than 35 S cm2 mol−1 [29], these complexes are considered to be nonelectrolytes and are quite stable in culture media [3, 35, 36]. This data suggest that the active species are metal complexes.
Metal complexes have been the research interest of our lab and we have previously synthesized and tested various metal complexes [3, 28, 35, 36]. The synthesis routes of complexes Cu1, Cd1, Cu2 and Cd2 that are discussed here are shown in the Scheme 1. Methanol was used as solvent during these syntheses. In these types of syntheses, redox reaction rarely takes place. Also, we used M(OAc)2·nH2O [M = Cd(II) and Cu(II)] to synthesize the metal complexes. Therefore, we can say with certainty that the copper derivatives are Cu(II) complexes.
Complexes Cu1, Cd1, Cu2 and Cd2 inhibit proliferation of MDA-MB-231 human breast cancer cells and LNCaP human prostate cancer cells
Previously we reported that some Cd(II) and Cu(II) complexes could inhibit proliferation of cancer cells [30]. In the current study to examine whether the four novel Schiff base complexes, Cu1, Cd1, Cu2 and Cd2 (Fig. 1), are capable of inhibiting the proliferative abilities, MDA-MB-231 cells and LNCaP cells were treated with each compound at 20, 40 and 60 μM for 24 h, followed by MTT assay. Cells treated with DMSO were used as a control. For MDA-MB-231 cells, we found that Cu1, Cd1 and Cd2 had similar growth-inhibitory activity, resulting in 95, 99 and 54 % inhibition at 60 μM, respectively (Fig. 2a). However, Cu2 showed only 13 % inhibition at 60 μM after 24 h of treatment (Fig. 2a). When LNCaP cells were used, we found that Cu1, Cd1 and Cd2 showed similar pattern of cell growth inhibition, resulting in 83, 93 and 64 % inhibition at 60 μM after 24 h of treatment, respectively (Fig. 2b). However, Cu2 induced less than 14 % inhibition at 60 μM after 24 h of treatment.

3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay for MDA-MB-231 cells (a) and LNCap cells (b). The cells were treated with Cu1, Cd1, Cu2 and Cd2 complexes for 24 h at various concentrations as indicated. After 24 h, the medium was removed, and the cells were treated with MTT solution, as described in “Experimental” section. Dimethyl sulfoxide (DMSO) was used as a control
Complexes Cu1, Cd1, Cu2 and Cd2 inhibit chymotrypsin-like activity of the proteasome in vitro under cell-free conditions
To investigate whether these copper and cadmium complexes can inhibit proteasomal activities, we incubated Cu1, Cd1, Cu2 and Cd2 at various concentrations with a breast cancer MDA-MB-231 cell extract for 2 h, with DMSO treatment as a control. The results clearly indicated that the compounds Cu1, Cd1, Cu2 and Cd2 were the most potent against proteasomal chymotrypsin-like activity under cell-free conditions (Fig. 3). Therefore, these complexes could inhibit chymotrypsin-like activity of the cell-free proteasome in a concentration-dependent manner and that Cu2 might not be able to get into cells. For the later experiments, we therefore used only Cu1, Cd1 and Cd2.

Inhibition of CT-like activity under cell-free condition. A MDA-MB-231 cell extract (10 μg) was incubated with DMSO or various concentrations of Cu1, Cd1, Cu2 and Cd2 for 2 h, followed by the proteasomal chymotrypsin-like activity assay
Concentration-dependent proteasome inhibition and apoptosis induction in MDA-MB-231 human breast cancer cells by complexes Cu1, Cd1 and Cd2
We found that our complexes Cu1, Cd1 and Cd2 were able to inhibit the chymotrypsin-like activity of cell-free proteasome in vitro (Fig. 3). To test whether they were able to inhibit tumor cellular proteasome activity, MDA-MB-231 cells were treated with various concentrations (5–60 μM) of Cu1, Cd1 and Cd2 and DMSO as control for 24 h. Cell lysates were prepared after denoted treatments as described in “Materials and methods” section and used for measurement of proteasome activity by evaluating the levels of cellular proteasomal chymotrypsin-like activity, accumulation of proteasome target protein, p27, and other ubiquitinated proteins.
All compounds at 10 μM showed about 25 % inhibition of proteasome CT-like activity, and when the doses of these compounds were increased to 40 μM, Cd1 showed 90 % inhibition,where as Cu1 and Cd2 showed about 50 % inhibition (Fig. 4a). In addition, the accumulation of proteasome substrate, p27 and ubiquitinated proteins was observed in MDA-MB-231 cells treated with Cu1, Cd1 and Cd2 in a concentration-dependent manner (Fig. 4b), consistent with the proteasome activity inhibition (Fig. 4a). Interestingly, we observed morphological changes (shrunken and rounded up cell appearance) in the cancer cells after treatment with the test compounds (Fig. 4c) along with PARP cleavage (Fig. 4b), indicating that the cancer cells are undergoing apoptosis. The MDA-MB-231 cells began to show morphological changes when they were treated with 20 μM of Cu1 and Cd2 and 10 μM of Cd1 (Fig. 4c). Furthermore, the cleaved PARP fragment p85 appeared at 20 μM and 40 μM of Cd1 and Cd2 (Fig. 4b). Interestingly, we also observed a reduction in the p116 full-length PARP leading to its disappearance at the 40 μM of Cu1. Therefore, these results show that, complexes Cu1, Cd1 and Cd2 can inhibit proteasome activity and subsequently induce apoptosis in breast cancer cells in a concentration-dependent manner.

Dose–response experiment in MDA-MB-231 cells. MDA-MB-231 cells were treated with either DMSO or indicated concentrations of complexes Cu1, Cd1 and Cd2 for 24 h. This was followed by measuring inhibition of the cellular proteasomal CT-like activity using the fluorescent substrate Suc-LLVY-AMC (a), Western blot analysis using specific antibodies to PARP, p27, ubiquitin and β-actin (as loading control) (b), and detection of morphological changes (c)
Concentration-dependent proteasome inhibition and apoptosis induction in LNCaP human prostate cancer cells by complexes Cu1, Cd1 and Cd2
Similarly, to investigate whether these complexes have concentration-dependent effects on LNCaP cells, we treated the cells with various concentrations (5–60 μM) of Cu1, Cd1 and Cd2 with DMSO as a solvent control for 24 h. The results indicate that at 10 μM, the compounds Cu1, Cd1 and Cd2 were able to cause about 30 % inhibition of the proteasomal CT-like activity (Fig. 5a). However, at 60 μM, Cu1, Cd1 and Cd2 showed potent inhibition of proteasomal CT-like activity. (Fig. 5a). Consistent with that, the accumulation of ubiquitinated proteasomal substrate proteins and p27 was also observed in a concentration-dependent manner when LNCaP Cells were treated with Cu1, Cd1 and Cd2 (Fig. 5b).

Dose–response experiment in LNCap cells. LNCap cells were treated with either DMSO or indicated concentrations of complexes Cu1, Cd1 and Cd2 for 24 h. This was followed by measuring inhibition of the cellular proteasomal CT-like activity using the fluorescent substrate Suc-LLVY-AMC (a), Western blot analysis using specific antibodies to PARP, p27, ubiquitin and β-actin (as loading control) (b), and detection of morphological changes (c)
When assessing PARP cleavage in characterizing the apoptosis-inducing ability of these compounds in LNCaP cells, we observed that the PARP cleavage fragment p85 appeared at 10 and 20 μM of Cd1 with a reduction in the p116, the full-length PARP at 40 μM dose of Cu1 and Cd2 (Fig. 5b). Morphological changes, indicative of cellular apoptosis, were observed at the 20 and 40 μM concentrations in accordance with the cleavage and disappearance of PARP (Fig. 5c). These results demonstrate that the complexes Cu1, Cd1 and Cd2 have the ability to inhibit the proteasome and induce apoptosis in a concentration-dependent manner in LNCaP cells.
Time-dependent proteasome inhibition and apoptosis induction in MDA-MB-231 human breast cancer cells by complexes Cu1, Cd1 and Cd2
If proteasome inhibition due to complexes Cu1, Cd1 and Cd2 was responsible for apoptosis induction, we would expect the proteasomal inhibition to be the earlier event to apoptosis. To test this idea, MDA-MB-231 breast cancer cells were treated with 40 μM of Cu1, Cd1 and Cd2 for indicated time points, followed by the measurement of proteasome inhibition and induction of apoptosis. In this kinetic experiment, we found that the proteasome inhibition by Cu1, Cd1 and Cd2 started as early as 2 h, as evident by a 10, 60 and 13 % respective decrease in the proteasome activity level (Fig. 6a) and an increase in the levels of ubiquitinated proteins (Fig. 6b). Consistent with the results for proteasome inhibition, the levels of proteasome target protein, p27, were increased in a time-dependent manner (starting at 4 h; Fig. 6b). In the same kinetic experiment, the cleaved PARP fragment p85 appeared 24 h after treatment with Cd2 (Fig. 6b). We also observed a reduction in the full-length PARP which disappeared at the 24 h of treatment with Cu1 and Cd1 (Fig. 6b). Furthermore, gradually increasing apoptotic morphological changes in cancer cells were detected starting from 8 h of treatment with each complex (Fig. 6c). These results suggest that apoptosis induced by Cu1, Cd1 and Cd2 is a consequential event of their proteasome inhibition.

Kinetic effects of cadmium complexes Cu1, Cd1 and Cd2 in MDA-MB-231 cells. MDA-MB-231 cells were treated with 40 μM of these complexes for the indicated period. This was followed by measuring inhibition of the proteasomal CT-like activity using the fluorescent substrate Suc-LLVY-AMC (a), Western blot analysis using specific antibodies to PARP, p27, ubiquitin and β-actin (as loading control) (b), and detection of morphological changes (c)
Time-dependent proteasome inhibition and apoptosis induction in LNCaP human prostate cancer cells by complexes Cu1, Cd1 and Cd2
To investigate whether these complexes show a similar pattern of time-dependent proteasome inhibition in LNCaP cells, we treated the cells with Cu1, Cd1 and Cd2 using the same experimental conditions as described in “Time-dependent proteasome inhibition and apoptosis induction in MDA-MB-231 human breast cancer cells by complexes Cu1, Cd1 and Cd2” section. In this kinetic experiment, we found that the proteasome inhibition in LNCaP cells started as early as 2 h, as evident by a 7, 52 and 8 % respective decrease in the proteasome activity level (Fig. 7a) with enhanced levels of ubiquitinated proteins after treatment with Cu1, Cd1 and Cd2 (Fig. 7b). Consistent with proteasome inhibition, the level of proteasome target protein, p27, was increased in a time-dependent manner (starting at 4 h; Fig. 7b). In the same kinetic experiment, we also observed a reduction in the p116 full-length PARP which disappeared at the 24 h of treatment with Cu1, Cd1 and Cd2 (Fig. 7b). Furthermore, apoptotic morphological changes were detected after 12 h of treatment with each complex, also increasing gradually as time progressed (Fig. 7c). These results suggest that apoptosis induced by Cu1, Cd1 and Cd2 is a consequential event of proteasome inhibition.

Kinetic effects of cadmium complexes Cu1, Cd1 and Cd2 in LNCaP cells. LNCap cells were treated with 40 μM of these complexes for the indicated period. This was followed by measuring inhibition of the proteasomal CT-like activity using the fluorescent substrate Suc-LLVY-AMC (a), Western blot analysis using specific antibodies to PARP, p27, ubiquitin and β-actin (as loading control) (b), and detection of morphological changes (c)
Discussion
Some of the important apoptosis-inducing antineoplastic drugs like cisplatin have a metallic center, such as heavy metal compound, platinum. These types of drugs have been used in clinic to treat various human cancers for years. However, the clinical use of cisplatin is restricted due to dose-dependent toxicity and the development of resistance [11–13]. These shortcomings have triggered a search for other metal-based compounds that show lower toxicity, higher selectivity, and a broader spectrum of activity [14–16]. As a result, compounds containing gold, copper, zinc, etc. showed potent tumor proteasome-inhibitory activity [20–27].
The proteasome has been established as an anticancer drug target due to the approval of bortezomib for the treatment of multiple myeloma and mantle cell lymphoma. Unfortunately, some patients are intrinsically resistant to it or eventually develop resistance to it. In addition, bortezomib has shown little or no effect on solid tumors. Therefore, we designed metal-based proteasome inhibitors as potential anticancer agents that could be developed for the treatment of solid tumors like breast and prostate cancer [38]. Proteasome inhibition might be effective strategies in the anticancer therapy due to the fact that the cancer cells are much more dependent on these processes as compared to normal cells and proteasome inhibition leads to apoptosis selectively in cancer cells [31–33]. Furthermore, it has also been reported that some copper and cadmium complexes are capable of inhibiting proteasome and induce apoptosis in cancer cells [20, 21, 25, 34, 35]. We synthesized novel cadmium- or copper-containing complexes based on amino acid Schiff base to further explore the strategy to incorporate heavy metals into amino acid Schiff base. To study the potential anticancer effects of novel cadmium- and copper-containing complexes and to investigate the mechanism by which these novel complexes can inhibit tumor cell proliferation, we tested their biological activity in MDA-MB-231 human breast cancer cells and LNCaP human prostate cancer cells.
First, we measured the antiproliferative activities of these complexes by the MTT assay, and found that Cu1 Cd1 and Cd2 suppressed the proliferation of MDA-MB-231 human breast cancer cells in a concentration-dependent manner. In addition, we found that Cu1, Cd1 and Cd2 had similar growth-inhibitory activity, resulting in 95, 99 and 54 % inhibition of cell proliferation at 60 μM, respectively (Fig. 2a). However, Cu2 induced less than 13 % inhibition at 60 μM after 24 h of treatment. Secondly, we performed a cell-free proteasomal activity assay and found that all the four complexes potently inhibited the chymotrypsin-like activity of the cell-free proteasome with IC50 values of 6.1, 1.7, 3.7 and 2.6 μM, respectively (Fig. 3). As for the complex Cu2, although it could inhibit chymotrypsin-like activity of the cell-free purified proteasome in vitro, it could not inhibit cellular proteasomal activities. It might be possible that this compound is unable to enter the cancer cells. As Cu2 did not showed significant inhibition of cell proliferation in MDA-MB-231 and LNCaP cells, we decided not to study it further. Proteasome inhibition due to complexes Cu1 Cd1 and Cd2 was confirmed by the increased levels of the proteasome target protein p27 in dose and time dependent as shown by the Western blot analysis.
In this study, we also made observations about the structures and proteasome-inhibitory potential of various metal-containing complexes (Fig. 1). While Cd2 has a same amino acid Schiff base like Cu2, their activities differ greatly with only difference being the metal ion that is complexed (Fig. 2). We found that copper complexes with the same ligands have little activity, compared to Cadmium. This pattern of proteasome inhibition was correlated with the results in the MTT assay (Fig. 2). Similarly, Cd1 is more potent than Cu1. The inhibition of cell proliferation was strongly associated with the inhibition of CT-like activity of the proteasome (Fig. 3), and accumulation of proteasome substrate-ubiquitinated proteins (Figs. 4b, 5b, 6b, 7b). Furthermore, Cu1 Cd1 and Cd2 complexes also induced apoptosis in cancer cells as evident from PARP cleavage and phenotypic morphologic changes in cancer cells (Figs. 4c, 5c, 6c, 7c). We found that Cd-coordinating compounds were relatively more potent in their ability to inhibit MDA-MB-231, breast cancer cell proliferation [3].
To test whether the proteasome-inhibitory and apoptosis-inducing properties of these test compounds are limited to a particular cancer type, we investigated the effect of each complex in human prostate cancer cells (LNCaP) at the same treatments conditions. The results showed that the complexes Cd1, Cu1 and Cd2 were potent inhibitors of prostate cancer cell proliferation. These results were similar to those observed in the MDA-MB-231 breast cancer cells.
We synthesized many amino acid Schiff base metal complexes such as l-methionine, l-ornithine and l-tryptophan [28, 36]. We used a direct synthesis method. Unfortunately, we cannot get the amino acid Schiff base in its solid state. Therefore, we could not compare metal complexes activity with their amino acid Schiff base ligands. It has been shown that Schiff base possesses antivirus and antibacterial activities. Few researchers have shown that the Schiff base also has anticancer activities. It has also been shown that [21], the biological activities of metal complexes could be better than their Schiff base ligands. For example, the metal complex GVC is more sensitive than the Schiff base GV (for their structures, see Fig. 8).

Chemical structures of the complexes GV, GVC, Z-Cu3, Z-Cd3, P-C1, P-C2, J-C3 and J-C4 in some published paper
Previously, we have shown that several Schiff base–copper and –cadmium complexes (Fig. 8) could be proteasome inhibitors and induce apoptosis in human cancer cells [3, 35, 36]. Compared with the metal complexes Z-Cu3, Z-Cd3, P-C1, P-C2, J-C3 and J-C4 in some published paper, the metal complexes Cu1, Cd1 and Cd2 are more sensitive due to the possibility that these compounds are able to easily enter the cancer cells [3, 35, 36].
Use of copper to target cancer cells has been suggested for long time although with limited success. The major problem with this is the limited intracellular transportation of copper [37]. Although cadmium (Cd) is a major environmental contaminant and might lead to carcinogenesis, our results indicate that organic Cd complexes could be potent inhibitor of proteasomal chymotrypsin-like (CT-like) activity, leading to apoptosis induction in cancer cells.
We investigated the growth-inhibitory activity of four metal-based complexes along with their mechanism of action. Cd1, Cu1 and Cd2 are potent proteasome inhibitors and apoptosis inducers in human breast cancer cells and human prostate cancer cells. In conclusion, our study suggests that metal complexes could inhibit proteasomal activities and have the potential to be developed into novel anticancer drugs.
Abbreviations
- DMSO:
-
Dimethyl sulfoxide
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- PARP:
-
Poly(ADP-ribose) polymerase
- PBS:
-
Phosphate-buffered saline
- Suc-LLVY-AMC:
-
N-Succinyl-Leu–Leu-Val-Tyr-7-amino-4-methylcoumarin
- UPS:
-
Ubiquitin–proteasome system
References
Du W, Mei QB (2013) Acta Pharmacol Sin 34:187–188
Campello L, Esteve-Rudd J, Cuenca N, Martín-Nieto J (2013) J Mol Neurobiol 47:790–810
Zhang Z, Bi CF, Buac D, Fan YH, Zhang X, Zuo J, Zhang PF, Zhang N, Dong LL, Dou QP (2013) J Inorg Biochem 123:1–10
Adams J (2004) Nat Rev Cancer 4:349–360
Amici M, Forti K, Nobili C, Lupidi G, Angeletti M, Fioretti E, Eleuteri AM (2002) J Biol Inorg Chem 7:750–756
Seemüller E, Lupas A, Stock D, Löwe J, Huber R, Baumeister W (1995) Science 268:579–582
Kabolizadeh P, Engelmann BJ, Pullen N, Stewart JK, Ryan JJ, Farrell NP (2012) J Biol Inorg Chem 17:123–132
Freitas N, Gomes A, Porto G, Fernandes E (2010) J Biol Inorg Chem 15:1275–1283
Guo ZJ, Sadler PJ (1999) Angew Chem Int Ed 38:1513–1531
Selvakumaran M, Pisarcik DA, Bao R, Yeung AT, Hamilton TC (2003) Acad J Cancer Res 63:1311–1316
Baird RD, Kaye SB (2003) Eur J Cancer 39:2450–2461
Wang D, Lippard SJ (2005) Nat Rev Drug Discov 4:307–320
Hartmann JT, Lipp HP (2003) Expert Opin Pharmacother 4:889–901
Hartinger CG, Zorbas-Seifried S, Jakupec MA, Kynast B, Zorbas H, Keppler BK (2006) J Inorg Biochem 100:891–904
Yan YK, Melchart M, Habtemariam A, Sadler PJ (2005) Chem Commun 38:4764–4776
Ang WH, Dyson PJ (2006) Eur J Inorg Chem 20:4003–4018
Cozzi PG (2004) Chem Soc Rev 33:410–421
Holla BS, Veerendra B, Shivananda MK, Poojary B (2003) Eur J Med Chem 38:759–767
Creaven BS, Duff B, Egan DA, Kavanagh K, Rosair G, Thangella VR, Walsh M (2010) Inorg Chim Acta 363:4048–4058
Zhang X, Bi CF, Fan YH, Cui Q, Chen D, Xiao Y, Dou QP (2008) Int J Mol Med 22:677–682
Xiao Y, Bi CF, Fan YH, Cui C, Zhang X, Dou QP (2008) Int J Oncol 33:1073–1079
Adsule S, Barve V, Chen D, Ahmed F, Dou QP, Padhye S, Sarkar FH (2006) Med Chem 49:7242–7246
Padhye S, Yang HJ, Jamadar A, Cui QC, Chavan D, Dominiak K, McKinney J, Banerjee S, Dou QP, Sarkar FH (2009) Pharm Res 26:1874–1880
Motaghed M, Al-Hassan FM, Hamid SS (2014) Int J Mol Med 33:8–16
Daniel KG, Gupta P, Harbach RH, Guida WC, Dou QP (2004) Biochem Pharmacol 67:1139–1151
Chen D, Daniel KG, Chen MS, Kuhn DJ, Landis-Piwowar KR, Dou QP (2005) Biochenm Pharmacol 69:1421–1432
Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP (2005) Breast Cancer Res 7:897–908
Zhang N, Fan YH, Bi CF, Zuo J, Wang Q (2012) Russ J Coord Chem 38:349–352
Gong QJ, Jin WJ, Dong C (2000) Appl Chem 17:227–229
Li LH, Yang HJ, Chen D, Cui QC, Dou QP (2008) Toxicol Appl Pharmacol 229:206–214
Adams J (2003) Drug Discov Today 8:307–315
Dou QP, Li B (1999) Drug Resist Updat 2:215–223
Almond JB, Cohen GM (2002) Leukemia 16:433–443
Chen D, Cui QC, Yang HJ, Dou QP (2006) Cancer Res 66:10425–10433
Zhang PF, Bi CF, Schmitt SM, Li X, Fan YH, Zhang N, Dou QP (2014) Int J Mol Med. doi:10.3892/ijmm.2014.1838
Zuo J, Bi CF, Fan YH, Buac D, Nardon C, Daniel KG, Dou QP (2013) J Inorg Biochem 118:83–93
Xiao Y, Chen D, Zhang X, Cui QZ, Fan YH, Bi CF, Dou QP (2010) Int J Oncol 37:81–87
Buac D, Shen M, Schmitt S, Kona FR, Deshmukh R, Zhang Z, Neslund-Dudas C, Mitra B, Dou QP (2013) Curr Pharm Des 19:4025–4038
Acknowledgments
This research was supported by the National Natural Science Foundation of China to C.F. Bi (No. 21371161 and No. 21071134), the Specialized Research Fund for the Doctoral Program of Higher Education of China to Y. H. Fan (No. 20120132110015), the National Cancer Institute to Q. P. Dou (5R01CA127258-05, R21CA184788-01), the Natural Science Foundation of Shandong Province to X. Zhang (No. ZR2012BQ026), and a scholarship from Chinese Scholarship Council to Z. Y. Zhang.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhang, Z., Bi, C., Fan, Y. et al. l-Ornithine Schiff base–copper and –cadmium complexes as new proteasome inhibitors and apoptosis inducers in human cancer cells. J Biol Inorg Chem 20, 109–121 (2015). https://doi.org/10.1007/s00775-014-1219-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-014-1219-1