
Abstract
Serine hydroxymethyltransferase 2 (SHMT2) converts serine into glycine in the mitochondrial matrix, transferring a methyl group to tetrahydrofolate. SHMT2 plays an important role in the maintenance of one-carbon metabolism. Previously, we found a negative correlation between the serine concentration and the progression of fatty liver disease (FLD). However, little is known about the role of SHMT2 in hepatic lipid metabolism. We established SHMT2 knockdown (KD) mouse primary hepatocytes using RNA interference to investigate the role of SHMT2 in lipid metabolism. SHMT2 KD hepatocytes showed decreased lipid accumulation with reduced glycine levels compared to the scramble cells, which was restored upon reintroducing SHMT2. SHMT2 KD hepatocytes showed downregulation of the mTOR/PPARɣ pathway with decreased gene expression related to lipogenesis and fatty acid uptake. Pharmacological activation of mTOR or PPARɣ overexpression blocked the inhibitory effect of SHMT2 KD on lipid accumulation. We also showed that glycine activated mTOR/PPARɣ signaling and identified glycine as a mediator of SHMT2-responsive lipid accumulation in hepatocytes. In conclusion, silencing SHMT2 in hepatocytes ameliorates lipid accumulation via the glycine-mediated mTOR/PPARɣ pathway. Our findings underscore the possibility of SHMT2 as a therapeutic target of FLD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fatty liver disease (FLD) is characterized by excess lipid accumulation in hepatocytes. There are two types of fatty liver disease: alcoholic fatty liver disease (AFLD) and non-alcoholic fatty liver disease (NAFLD). The global prevalence of NAFLD is constantly increasing (15% in 2005 to 25% in 2010) in parallel with the worldwide increase of obesity and type II diabetes mellitus (Younossi et al. 2016). Besides, FLD can progress to advanced liver diseases such as liver fibrosis, cirrhosis, and hepatocellular carcinoma. Despite clinical and public health significance of FLD, the underlying mechanism remains poorly understood, and there are no approved medications currently.
Recent studies have reported the role of amino acids in the management of liver diseases. Serine is a non-essential amino acid that participates in the cellular metabolic process linking the folate and methionine cycles (Fig. 1). The serine synthesis pathway supports cell proliferation by providing precursors for the biosynthesis of proteins, nucleotides, phospholipids, and glutathione (Ducker and Rabinowitz 2017). Serine hydroxymethyltransferase (SHMT) is a pyridoxal phosphate-dependent enzyme that catalyzes the folate-dependent serine/glycine interconversion. There are two SHMT genes in the human genome: SHMT1 encodes the cytoplasmic isozyme (SHMT1), and SHMT2 encodes the mitochondrial isozyme (SHMT2) (Garrow et al. 1993). SHMT2 is involved in the synthesis of mitochondrial thymidine monophosphate. However, the primary function of the enzyme is to provide one-carbon units from serine. In the folate cycle, SHMT2 converts tetrahydrofolate (THF) into methylene-THF, which is further reduced to 5-methyl-THF or converted into 10-formyl-THF. Demethylation of methylene-THF produces a one-carbon unit that is donated to the methionine cycle via the methylation of homocysteine. Therefore, SHMT2 is a critical enzyme linking glycolysis and the serine synthesis pathway with one-carbon metabolism (Locasale 2013).

Serine synthesis pathway and one-carbon metabolism. Serine is synthesized de novo using the glycolytic intermediate 3-phosphoglycerate (3-PG), which is catalyzed by 3-phosphoglycerate dehydrogenase (PHGDH). Serine provides one-carbon units to the folate cycle while producing glycine. Serine hydroxymethyltransferase (SHMT) is a pyridoxal phosphate-dependent enzyme that catalyzes the folate-dependent serine/glycine interconversion
Serine is synthesized de novo in mammalian cells by channeling the glycolytic intermediate 3-phosphoglycerate into the serine synthesis pathway, which is catalyzed by 3-phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), and phosphoserine phosphatase (PSPH), with PHGDH playing a pivotal role (Fig. 1; Amelio et al. 2014). Recently we demonstrated the critical roles of PHGDH and serine in lipid metabolism and fatty liver disease (FLD). We found decreased hepatic PHGDH expression and serine concentrations in steatosis animal models. We also found a significant negative correlation between the serine concentration and liver fat fraction in patients with FLD. Transgenic overexpression of PHGDH in mice attenuated high-fat diet (HFD)-induced fatty liver (Sim et al. 2020). Reduced NAD+ levels and SIRT1 activity were observed in the HFD-fed mice, Phgdh−/− mouse embryonic fibroblasts incubated in serine-free medium and mouse primary hepatocytes incubated in the presence of free fatty acids, which were reversed by PHGDH (Sim et al. 2020, 2019). Supplementation with serine attenuates the development of fatty liver in experimental animals by activating homocysteine metabolism and decreasing the production of reactive oxygen species via the glutathione antioxidant system (Sim et al. 2015; Zhou et al. 2018).
The serine/threonine kinase mammalian target of rapamycin (mTOR) senses insulin and nutrients and regulates cell proliferation, autophagy, and energy homeostasis (Liu and Sabatini 2020). mTOR is the catalytic subunit of two distinct multiprotein complexes, mTORC1 and mTORC2. The eukaryotic initiation factor 4E-binding proteins (4E-BPs) and ribosomal protein S6 kinases (S6Ks) are downstream of mTORC1. When active, mTORC1 facilitates lipogenesis via sterol regulatory element-binding protein-1 (SREBP1) and peroxisome proliferator-activated receptor ɣ (PPARɣ). SREBP1 is a transcription factor that regulates the expression of lipogenic genes in the liver (Bakan and Laplante 2012). mTORC1 increases the nuclear translocation of SREBP1 and promotes SREBP1 transcription in an S6K1-dependent manner (Düvel et al. 2010; Porstmann et al. 2008). PPARγ also appears to be a steatogenic transcription factor in the liver by regulating lipogenesis and fatty acid uptake (Skat-Rørdam et al. 2019). FLD patients show dysregulation of mTORC1 and increased SREBP1 and PPARɣ expression (Pettinelli and Videla 2011).
Based on the unique role of PHGDH in lipid metabolism and the critical role of SHMT2 in one-carbon metabolism, we investigated the role of SHMT2 in cellular lipid metabolism using an RNA interference system in mouse primary hepatocytes. SHMT2 knockdown (KD) hepatocytes showed decreased lipid accumulation with reduced glycine levels compared to the scramble cells, which were restored upon reintroduction of SHMT2. The lipid-lowering effect was mediated by inactivating the mTOR/PPARɣ signaling pathway, and the effects relied on the presence of glycine. The results suggest that both serine biosynthesis via PHGDH and serine catabolism by SHMT2 play roles in maintaining cellular homeostasis of lipid metabolism.
Materials and methods
Cell culture and chemicals
Male C57BL/6 mice (8- to 10-week-old; 20–24 g) were purchase from Orient Bio (Seongnam, Korea). Mice were anesthetized with Alfaxan (Jurox, Kansas City, MO), and mouse primary hepatocytes were isolated by two-step perfusion with calcium and magnesium-free Hanks' salt solution followed by a William's E medium containing collagenase type IV (Cat-no: C5138, Sigma-Aldrich, MI, USA). Hepatocytes were plated in William's E medium (Cat-no: W4125, Sigma-Aldrich, MI, USA) containing 10% fetal bovine serum, 100 nM dexamethasone, and 100 nM insulin, and allowed to adhere to the collagen-coated dish (Cat-no: CB40236, Thermo Fisher, MA, USA). After 4 h adhesion, cells were washed with PBS and replaced with William's E medium containing 10% FBS before the indicated treatments. Cells were incubated at 37 °C with air containing 5% CO2. The stock solution of glycine (Cat-no: G8790, Sigma-Aldrich, MI, USA) and MHY1485 (Cat-no: S7811, Selleckchem, TX, USA) were prepared in sterile water and DMSO, respectively, which were further diluted to the desired concentrations using the cell culture medium. All animal experiments were approved by the Institutional Animal Care and Use Committee of Seoul National University.
Generation of CRISPR/Cas9-mediated knockout cells
For CRISPR/Cas9-mediated knockout, single-guide RNA was designed using the MIT CRISPR design tool (http://crispr.mit.edu/). Single Guide Oligonucleotides used in this study were as follows: sgScramble (F): 5′-GCACTACCAGAGCTAACTCA-3′, sgScramble (R): 5′-TGAGTTAGCTCTGGTAGTGC-3′, sgSHMT2 (F): 5′-GTTGCTGTGCTGAGCCCGAA-3′, sgSHMT2 (R): 5′-TTCGGGCTCAGCACAGCAAC-3′. The CRISPR/Cas9 vector, pSpCas9(BB)-2A-GFP (PX458), was purchased from Addgene (#48138). sgSHMT2 and sgScramble oligos were inserted into the PX458 using Bbs I, Alkaline Phosphatase, and T4 ligase (Thermo Fisher, MA, USA) and propagated with JM109 competent cells (Ran et al. 2013). HepG2 cells were transfected with sgScramble, sgSHMT2-1/2 inserted PX458 using Lipofectamine 2000. After 2 days, cells were sorted by FACS ARIA III (BD Biosciences, CA, USA) using FITC filter. Only EGFP positive cells were collected and cultured in very low density for 2 weeks. Grown colonies were picked and screened by Western blot analysis.
Oxygen consumption rate measurement
According to the manufacturer's instruction, oxygen consumption rate (OCR) was measured using a Seahorse XFe96 analyzer (Agilent Technologies, CA, USA). Cells were treated with oligomycin (1.5 μM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP; 1 μM), and a mixture of antimycin A/rotenone (0.5 μM) for the measurement of key parameters of mitochondrial respiration.
Nile red staining and confocal microscopy
We performed a Nile Red assay to examine the effect of SHMT2 knockdown on fatty acid-induced lipid accumulation. Mouse primary hepatocytes were plated in 96 black-sided clear bottom well plates at 1 × 104 cells per well. After stabilization, cells were treated with chemical or siRNA as indicated and then incubated with oleic acid 200 μM in a serum-free medium containing 1% fatty acid-free BSA for 24 h. Cells were fixed with 4% paraformaldehyde and then stained with 5 µg/mL Hoechst 33258 (Cat-no: 94403, Sigma-Aldrich, MI, USA) and 0.5 µg/mL Nile red solution (Cat-no: 19123, Sigma-Aldrich, MI, USA). Images were captured with a Leica TCS8 confocal microscope. We used Cytation 3 cell imaging microplate reader (BioTek, VT, USA) for quantitative analysis. Hoechst 33258 fluorescence was measured with an excitation of 341 nm and an emission of 452 nm, while Nile red fluorescence was determined using a 488 nm excitation and a 550 emission. Differences in cell number were corrected using Hoechst 33258 fluorescence to normalize the Nile red signal in each well.
Determination of triglycerides
According to the manufacturer's protocols, triglyceride concentrations in the cells were measured using Triglyceride Quantification Kit (Cat-no: MAK266, Sigma-Aldrich, MI, USA).
Transient transfection
siRNA for SHMT2 (siSHMT2), pCMV-PPARɣ, and pCMV-SHMT2 were purchased at Bioneer (Cat-no: #108037-2, Daejeon, South Korea), Korea Human Gene Bank (Cat-no: mMU002547, Daejeon, South Korea), and Origene (Cat-no: RC204239, MD, USA), respectively. Mouse primary hepatocytes were transfected with siSHMT2 using RNAiMax (Cat-no: 13778075, Invitrogen, CA, USA). According to the manufacturer's instructions, SHMT2 KD hepatocytes and SHMT2 KO cells were transfected with pCMV- PPARɣ and pCMV-SHMT2 using Lipofectamine 2000, respectively. Transfection efficiency was determined by Western blot analysis.
Western blot analysis
Cell pellets were lysed on ice for 30 min in lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate) and centrifuged at 12,000 rpm for 15 min. Supernatants were collected, electrophoresed on 6–8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto polyvinylidene fluoride (PVDF) membrane (Millipore, MA, USA). Western blots were probed with the following antibodies: ACC (#3662), p-ACC (#3661), AMPK (#2532), p-AMPK (#2535), S6K (#2708), p-S6K (#9205), mTOR (#2972), p-mTOR (#2971), SHMT2 (#12762), and GAPDH (#2118) from Cell Signaling Technology, and PPARɣ (sc-7273), SREBP1 (sc-366) and β-actin (sc-47778) from Santa Cruz Biotechnology (TX, USA). Western blots were developed using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Cat-no: 34580, Thermo Fisher, MA, USA).
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
For PCR amplification, total RNA was extracted by Easy-BlueTM Toal RNA extraction kit (Cat-no: 17061, Intron Biotechnology, Seongnam, South Korea). The cDNA was prepared with QuantiTect Reverse Transcription Kit (Cat-no: 205313, Qiagen, Hilden, Germany). qRT-PCR was performed on a SteponeTM Real-time PCR system (Applied Biosystems, CA, USA) using iTaqTM Universal SYBR Green Supermix kit (Cat-no: 1725121, Bio-rad, CA, USA) according to the manufacturer's protocol. The primer sequences are as follows: β-actin (F): 5′-CTGGGTATGGAATCCTGTGG-3′, β-actin (R): 5′-TGCATTTGCCAATGTCTAGC-3′, Shmt1 (F): 5′-TTCACAAAATCCACGCTCG-3′, Shmt1 (R): 5′-GCTGTAAACCTCGGCATC-3′, Shmt2 (F): 5′-CCCGGTACTACACCGATACA-3′, Shmt2 (R): 5′-GACCAGCTGACCACATCTCT-3′, Pparɣ (F): 5′-AGACCACTCGCATTCCTTT-3′, Pparɣ (R): 5′-CACAGACTCGGCACTCAAT-3′, Cebpα (F): 5′-CAAGAACAGCAACGAGTACCG-3′, Cebpα (R): 5′-GTCACTGGTCAACTCCAGCAC-3′, Cd36 (F): 5′-CCATTCCTCAGTTTGGTTCC-3′, Cd36 (R): 5′-TGCATTTGCCAATGTCTAGC-3′, Fabp4 (F): 5′-TCTCCAGTGAAAACTTCGAT-3′, Fabp4 (R): 5′-TTACGCTGATGATCATGTTG-3′, Srebpf1c (F): 5′-TGGAGACATCGCAAACAAG-3′, Srebpf1c (R): 5′-GGTAGACAACCAGCCGCATC-3′, Acaca (F): 5′-AGCAGATCCGCAGCTTG-3′, Acaca (R): 5′-ACCTCTGCTCGCTGAGTGC-3′, Fasn (F) 5′-TTCCGTCACTTCCAGTTAGAG-3′, Fasn (R): 5′-TTCAGTGAGGCGTAGTAGACA-3′, Scd1 (F): 5′-TGACCTGAAAGCCGAGAA-3′, Scd1 (R): 5′-ATGTGCCAGCGGTACTCA-3′.
Serine, glycine, and AICAR measurement
Cells were washed with ice-cold PBS and harvested with ice-cold 80% methanol. The lysates were centrifuged at 10,000g for 10 min at 4 °C. Supernatants were collected and diluted further with 80% methanol. Quantitative analysis of serine, glycine, and AICAR was performed using liquid chromatography–tandem mass spectrometry (LC–MS/MS). The LC–MS/MS system consisted of a Prominence™ UFLC system with a parallel LC-20ADXR pump, autosampler, and column oven (Shimadzu, Kyoto, Japan) and an API 4000 LC–MS/MS system equipped with a Turbo V IonSpray source (Applied Biosystems, CA, USA). Serine/glycine or AICAR was separated on a C18 Thermo AQUASIL (2.1 mm × 150 mm, 3 µm) or a C18 ACQUITY UPLC® HSS T3 (2.1 mm × 100 mm, 1.8 µm), respectively. The mobile phase was followed the gradient of solvent A (deionized water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid) at a flow rate of 0.2 mL/min. The mass spectrometer was operated in multiple reaction monitoring (MRM) in the positive ion electrospray mode. Peak detection was showed in the MRM mode at m/z 106 → 60 for serine, m/z 76 → 76 for glycine, m/z 109 → 62 for 13C3 serine (internal standard for serine and glycine), m/z 259 → 110 for AICAR, and m/z 302 → 170 for 2-chloroadenosine (internal standard for AICAR).
Statistical analysis
All data were expressed as mean ± SD. Statistical analysis was performed using Student's t-test or one-way analysis of variance (ANOVA) where appropriate. Differences between groups were considered to be statistically significant at p < 0.05. Multiple comparisons were evaluated by one-way ANOVA followed by Tukey's multiple comparison test (GraphPad Prism 7.0; GraphPad Software, Inc., CA, USA).
Results
Silencing SHMT2 ameliorates oleic acid-induced lipid accumulation in mouse primary hepatocytes
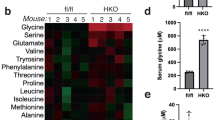
Previously, we investigated the role of PHGDH and serine metabolism in the development of FLD (Sim et al. 2020). As we found a negative correlation between the serine concentration and FLD progression, we wondered whether SHMT2 inhibition would increase the intracellular serine concentration and protect the hepatocytes from lipid accumulation. To test this, we established SHMT2 KD hepatocytes by RNA interference. Mouse primary hepatocytes were isolated and stabilized for 4 h and then transfected with SHMT2 siRNA for 72 h. Upon transfection, SHMT2 protein expression was downregulated in SHMT2 KD hepatocytes (Fig. 2a). To assess the siRNA specificity, we measured SHMT1 and SHMT2 mRNA levels. As expected, SHMT2 mRNA was decreased by 85%, with unaltered SHMT1 mRNA levels (Fig. 2a). Given the observations that SHMT2 affects mitochondrial electron transport chain components (Minton et al. 2018), we assessed the respiratory profile in SHMT2 KD hepatocytes using a Seahorse Flux Analyzer. SHMT2 KD hepatocytes showed similar oxygen consumption rate (OCR) signatures at every phase measured (Fig. 2b). Mitochondrial SHMT2 converts serine into glycine, a major glycine source in cell proliferation (Fig. 2c) (Jain et al. 2012). Consistent with SHMT2 function, the intracellular glycine concentration was decreased significantly by 40.4%, from 39.11 ± 4.73 to 23.32 ± 2.39 nmol/mg protein, while the serine concentration was unchanged in SHMT2 KD hepatocytes. Next, we examined the effect of SHMT2 inhibition on lipid metabolism. SHMT2 inhibition abrogated oleic acid-induced lipid accumulation, as determined by Nile red staining, and reduced oleic acid-induced triglycerides accumulation (Fig. 2d). Similar results were obtained in CRISPR/Cas9 system-mediated SHMT2 knockout HepG2 cells (SHMT2 KO; Fig. 2e). The lipid-lowering effect disappeared when we reintroduced SHMT2 into SHMT2 KO cells by transient transfection (Fig. 2e). These data led to our hypothesis that SHMT2 downregulation plays a role in lipid metabolism independent of the serine concentration.

SHMT2 knockdown reduces lipid accumulation in mouse primary hepatocytes. a Mouse primary hepatocytes were transiently transfected with siSHMT2 for 72 h. Western blot analysis examined the protein level of SHMT2 to confirm SHMT2 knockdown. SHMT1 and SHMT2 mRNA levels were measured by qRT-PCR. Bars represent means ± SD (n = 4). b Mouse primary hepatocytes were transiently transfected with siSHMT2 for 72 h. Mitochondrial function was analyzed by measuring OCR using a Seahorse XF Cell Mito Stress Test Kit. All data represent the mean ± SD (n = 3). c A brief schematic of serine/glycine metabolism by SHMT2. d Mouse primary hepatocytes were transfected with siSHMT2 for 48 h and then incubated with 200 μM oleic acid for 24 h. Nile red staining measured the lipid content of cells. Representative images are shown, and each bar represents the mean ± SD (n = 5 for Nile red; n = 3 for triglycerides). e SHMT2 KO cells were transfected with pCMV-SHMT2 for 48 h and then incubated with 200 μM oleic acid for 24 h. Western blot analysis examined the expression of SHMT2, and Nile red staining measured lipid content of cells. Each bar represents the mean ± SD (n = 4). **p < 0.01, ***p < 0.001 with the comparisons labeled
Silencing SHMT2 does not induce AICAR accumulation and AMPK activation
SHMT2 generates 10-formyl-THF, which is required for the conversion of 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) into 5-formyl-AICAR (FAICAR) during purine synthesis (Fig. 3a). AICAR activates AMP-activated protein kinase (AMPK), a master regulator in glucose and lipid metabolism (Viollet et al. 2006). Therefore, we asked whether AICAR accumulates in SHMT2 KD hepatocytes and plays a pivotal role in lipid metabolism. However, SHMT2 inhibition did not increase the intracellular AICAR level in hepatocytes (Fig. 3b). In line with this, SHMT2 inhibition did not change the level of AMPK phosphorylation (Fig. 3c).

Silencing SHMT2 does not induce AICAR accumulation and AMPK activation. a A brief overview of folate-mediated one-carbon metabolism. SHMT2 inhibition blocks purine synthesis and also promotes an increase in AICAR levels by glycine depletion. b Mouse primary hepatocytes were transiently transfected with siSHMT2 for 72 h. LC–MS/MS analyzed intracellular AICAR from mouse primary hepatocytes. Each bar represents the mean ± SD (n = 5). c Western blot analysis examined the phosphorylation and protein expression level of AMPK in SHMT2 KD hepatocytes. Shown on the right is the densitometric quantification of the Western blot analysis. Each bar represents the mean ± SD (n = 3)
Silencing SHMT2 downregulates PPARɣ via the mTOR/S6K pathway
To understand the molecular mechanism by which silencing SHMT2 inhibits lipid accumulation, we investigated the effect of SHMT2 inhibition on gene expression, emphasizing genes associated with lipid metabolism. SHMT2 KD hepatocytes showed down-regulation of several genes responsible for hepatic fatty acid uptake and lipogenesis. SHMT2 inhibition decreased mRNA expression of Pparɣ, Srebf1c, and its target gene Cebpα, Cd36, Fabp4, Acaca, Fasn, and Scd1 (Fig. 4a). Since PPARɣ and SREBP1 are regulated by the mTOR pathway (Bakan and Laplante 2012; Li et al. 2014), we determined the expression and phosphorylation of mTOR and S6K in SHMT2 KD hepatocytes. In line with the mRNA level, the phosphorylation of mTOR and S6K were decreased, resulting in a significant decrease in PPARɣ protein expression (Fig. 4b). However, SHMT2 inhibition did not change the SREBP1 protein levels (data not shown).

Silencing SHMT2 decreases lipid accumulation mediated by the mTOR/PPARɣ pathway. a Mouse primary hepatocytes were transiently transfected with siSHMT2 for 72 h. qRT-PCR analyzed the indicated transcripts. Each bar represents the mean ± SD (n = 4). b Western blot analysis examined the expression of the indicated protein. Each bar represents the mean ± SD (n = 4). c Isolated hepatocytes were transfected as described above and then treated with MHY1485 1 µM for 24 h. Western blot analysis detected the expression of proteins. Shown on the right is the densitometric quantification of the Western blot analysis (n = 3). d Mouse primary hepatocytes were transfected with siSHMT2 for 48 h and then incubated with 200 μM oleic acid in the presence of DMSO or MHY1485 1 µM for 24 h. Nile red staining measured the lipid content of cells. Each bar represents the mean ± SD (n = 3). e Mouse primary hepatocytes were transfect-ed with siSHMT2 for 48 h and then transfected with pCMV-PPARɣ for 24 h. Western blot analysis confirmed the transfection efficiency, and Nile red staining measured the lipid content of cells. Each bar represents the mean ± SD (n = 3). f SHMT2 KO cells were transfected with pCMV-SHMT2 for 48 h to reintroduce SHMT2. Western blot analysis examined the expression of PPARɣ in SHMT2 KO cells after the reintroduction of SHMT2 by transient transfection. One representative of three independent experiments with similar results is shown. *p < 0.05; **p < 0.01; ***p < 0.001 with the comparisons labeled
SHMT2 KD hepatocytes were treated with the mTOR activator MHY1485 for 24 h to determine whether mTOR/S6K plays a role in PPARɣ downregulation. While SHMT2 inhibition reduced the phosphorylation of mTOR/S6K and decreased the PPARɣ protein level, MHY1485 treatment substantially restored the phosphorylation of mTOR/S6K with increased PPARɣ protein expression (Fig. 4c). Interestingly, MHY1485 treatment increased lipid accumulation in SHMT2 KD hepatocytes, as measured by Nile red staining (Fig. 4d). PPARɣ overexpression also restored lipid accumulation in SHMT2 KD cells (Fig. 4e). Consistent with the results in SHMT2 KD hepatocytes, SHMT2 overexpression restored the PPARɣ protein expression in SHMT2 KO cells (Fig. 4f). These results show that SHMT2 KD suppresses the lipid accumulation in hepatocytes via the mTOR/PPARɣ pathway.
Glycine is a potential mediator of the SHMT2 inhibition-induced lipid-lowering effect
Glycine has been proposed as an mTOR activator (Sun et al. 2016; Wang et al. 2014). A previous study indicated that SHMT2 overexpression promotes cell proliferation by activating the mTOR pathway via its metabolic product, glycine (Wang et al. 2019). As expected, exogenous glycine upregulated mTOR/S6K signaling in a dose-dependent manner (Fig. 5a), which increased PPARɣ mRNA and protein levels (Fig. 5b). We hypothesized that glycine depletion mediated the downregulation of the mTOR pathway in SHMT2 KD hepatocytes. SHMT2 inhibition reduced the intracellular glycine concentration by 40%, and this glycine depletion was abolished upon exogenous glycine treatment (Fig. 5c). Consistent with the intracellular glycine level, glycine-treated SHMT2 KD hepatocytes did not show decreased lipid accumulation (Fig. 5d). Glycine treatment also induced the phosphorylation of mTOR and S6K, leading to the increased PPARɣ protein expression (Fig. 5e). These data indicate that glycine, a metabolic product of SHMT2, mediates the lipid-lowering effect in SHMT2 KD hepatocytes via downregulation of the mTOR/PPARɣ pathway. Thus, SHMT2 regulates hepatic lipid metabolism by modulating the glycine-mediated mTOR/PPARɣ pathway.

Glycine blocks the inhibitory effects of SHMT2 knockdown on lipid accumulation. a Isolated hepatocytes were incubated with glycine for 24 h. Western blot analysis examined the expression and phosphorylation level of mTOR and S6K. Shown on the right is the densitometric quantification of the Western blot analysis (n = 3). b PPARɣ mRNA and protein expression were measured by qRT-PCR and Western blot analysis (n = 3). c Isolated hepatocytes were transfected as described above and then treated with glycine 0.5 mM for 24 h. LC–MS/MS analyzed intracellular glycine from mouse primary hepatocytes. Each bar represents the mean ± SD (n = 5). d Mouse primary hepatocytes were transfected with siSHMT2 for 48 h and then incubated with 200 μM oleic acid in the presence of glycine 0.5 mM for 24 h. Nile red staining measured the lipid content of cells. Each bar represents the mean ± SD (n = 3). e Isolated hepatocytes were treated as described above, and Western blot analysis was used to examine the expression of key proteins of the mTOR pathway. Shown on the right is the densitometric quantification of the Western blot analysis (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001 with the comparisons labeled
Discussion
The goal of this study is to investigate the role of SHMT2 in hepatic lipid metabolism. We showed that SHMT2 KD decreased glycine production and downregulated the mTOR/S6K/PPARɣ pathway with reduced gene expression related to lipogenesis and fatty acid uptake (Fig. 6). Pharmacological activation of mTOR by MHY1485 blocked the inhibitory effect of SHMT2 KD on lipid accumulation. Lipid accumulation was also not relieved by SHMT inhibition in PPARɣ-overexpressed SHMT2 KD hepatocytes. Treatment of SHMT2 KD hepatocytes with exogenous glycine upregulated the mTOR pathway, which abolished the inhibitory effect of SHMT2 on lipid accumulation.

The underlying mechanisms of SHMT2 inhibition-induced lipid lowering effect via glycine-mediated mTOR activation
To our knowledge, this is the first demonstration that SHMT2 regulates lipid metabolism in the liver. SHMT2 is a critical regulator in one-carbon metabolism and a negative prognostic biomarker in hepatocellular carcinoma, but few studies have examined the role of SHMT2 in lipid metabolism (Ji et al. 2019). It has been reported that SHMT2-null cells exhibit a significant reduction in OCR and loss of mitochondrially encoded proteins (Minton et al. 2018). Primary fibroblasts from patients with SHMT2 variants show impaired SHMT2 enzymatic function and mitochondrial redox metabolism malfunction (García-Cazorla et al. 2020). SHMT2 knockout mice also exhibit embryonic lethality due to mitochondria dysfunction in the fetal liver (Tani et al. 2019). Because mitochondria dysfunction provokes metabolic disturbances and can contribute to the development of FLD, these studies suggest a link between SHMT2 impairment and FLD. However, we did not observe the decrease of mitochondrial respiration in SHMT2 KD hepatocytes. The reason for the discrepancy is currently unknown, but it may be attributable to cell-type specificity. By contrast, SHMT2 impairment alters folate metabolism and purine biosynthesis in glioma cells and leads to the accumulation of AICAR via the reduced conversion of AICAR to FAICAR (Kim et al. 2015). We reported the protective role of serine in FLD progression via the modulation of homocysteine metabolism and SIRT1 activation (Sim et al. 2019, 2015). These results led us to speculate that serine or AICAR mediates the protective effect of SHMT2 inhibition on lipid accumulation. However, this study showed that SHMT2 inhibition decreased lipid accumulation with unaltered intracellular concentrations of AICAR and serine. We found that glycine, the primary product of SHMT2, plays a role in SHMT2-mediated lipid metabolism.
In addition, our study suggests the possible roles of SHMT2 in liver fibrosis and hepatocarcinogenesis. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) promotes liver fibrosis by activation of hepatic stellate cells (HCSs) with increased aerobic glycolysis (Mejias et al. 2020). A recent study demonstrated that mTOR activation promotes collagen synthesis in lung fibroblasts by increasing PFKFB3-mediated aerobic glycolysis (Hu et al. 2020). Because SHMT2 can regulate the mTOR pathway via glycine synthesis, SHMT2 might be considered as the therapeutic target for liver fibrosis. Moreover, SHMT2 is involved in cell proliferation and is a negative prognostic biomarker in hepatocellular carcinoma (Ji et al. 2019; Woo et al. 2016). The increased de novo lipogenesis in hepatocellular carcinoma is reflected in the higher expression of lipogenic genes such as Srebp1c and Scd1 (Budhu et al. 2013; Yamashita et al. 2009). Given the importance of metabolic reprogramming in hepatocarcinogenesis, our study suggests that SHMT2 plays a role in hepatocarcinogenesis by enhancing lipogenesis.
Mechanistically, our results indicated that glycine-mediated SHMT2-responsive lipid metabolism occurs via the mTOR pathway. Glycine is a non-essential amino acid needed to synthesize glutathione, creatine, and nucleic acids (Wang et al. 2013). Amino acids, including arginine, glutamine, leucine, and proline, regulate the mTOR pathway (Liu and Sabatini 2020). Recently, activation of the mTOR pathway was observed in glycine-treated C2C12 myotubes and pig intestinal epithelial cells, and glycine-mediated mTOR activation modulates protein synthesis and cell proliferation (Sun et al. 2016; Wang et al. 2014). These reports suggest that glycine enhances lipogenesis via the mTOR pathway. However, the serum glycine level is reduced in FLD patients (Bianchi et al. 2003; Gaggini et al. 2018). In line with epidemiological evidence, the Western diet impairs glycine biosynthesis in the liver, and glycine-based treatment attenuates FLD progression in animal models (Rom et al. 2020). Here, we report that the decline in glycine concentration suppressed the mTOR pathway in SHMT2 KD hepatocytes; conversely, exogenous glycine treatment upregulated the mTOR pathway, which blocked the inhibitory effect of SHMT2 on lipid accumulation. These results are consistent with studies of SHMT2-overexpressed primary hepatocytes, which suggest that glycine activates the mTOR pathway and facilitates liver regeneration (Wang et al. 2019). The alteration of SHMT2 activity can also affect one-carbon metabolism leading to the change of folate levels and methylation state of DNA (Locasale 2013). It has been reported that folic acid activates AKT/mTOR pathway during muscle cell differentiation (Hwang et al. 2015). In addition, DNMT3B, DNA methyltransferase, regulate mTORC2 component Rictor levels via miR-196b promoter methylation (Micevic et al. 2016). These studies suggest that the dysregulation of one-carbon metabolism can modulate mTOR activity in a folate- or methylation-dependent manner. Because SHMT2 KD did not change the folate levels in glioma cells, we can exclude the possibility that mTOR inactivation in SHMT2 KD cells is the folate-dependent manner (Kim et al. 2015). Although a recent study showed that silencing SHMT2 induces changes in DNA and histone methylation patterns, further study is needed to investigate whether SHMT2 modulates lipid metabolism through methylation-dependent mTOR regulation (Parsa et al. 2020).
The role of mTOR in promoting lipid synthesis has been well described. SREBP1 and PPARγ are downstream molecules mediating the effects of mTOR on hepatic lipid metabolism. The knockdown of Raptor, which is required for mTORC1 activity, inhibits Akt-induced SREBP1 expression and nuclear translocation (Porstmann et al. 2008). mTORC1 modulates the post-transcriptional regulation of SREBP1 by S6K (Düvel et al. 2010; Owen et al. 2012). PPARγ also mediates mTORC1-induced fatty acid uptake and lipogenesis in various cells and tissues (Li et al. 2014; Angela et al. 2016; Blanchard et al. 2012). Recently, mTORC2 has emerged as a critical regulator of lipid metabolism in the liver (Guri et al. 2017; Hagiwara et al. 2012; Yuan et al. 2012). Liver-specific Rictor KO mice show impaired insulin-stimulated hepatic lipogenesis and decreased hepatic expression of SREBP1 and PPARɣ (Hagiwara et al. 2012; Yuan et al. 2012). In this study, SHMT2 KD lowered glycine production and suppressed mTOR-mediated lipogenesis and fatty acid uptake in hepatocytes. We also found that glycine treatment induced a parallel increase in mTOR and PPARɣ in SHMT2 KD hepatocytes with enhanced lipid accumulation. These results suggest an association between mTOR and PPARɣ and the role of the mTOR/PPARɣ pathway in hepatic lipid metabolism. The increased lipid accumulation in PPARɣ-overexpressed SHMT2 KD hepatocytes supports this.
PPARɣ is abundantly expressed in adipose tissue (Skat-Rørdam et al. 2019). Although the basal hepatic PPARɣ level is low, PPARɣ mRNA expression is increased in animals fed an HFD and obese patients with steatosis and steatohepatitis (Pettinelli and Videla 2011; Morán-Salvador et al. 2011). PPARγ regulates lipid metabolism in the liver by enhancing lipogenesis and fatty acid uptake and facilitates lipid deposition in the liver (Morán-Salvador et al. 2011; Matsusue et al. 2003; Yu et al. 2003; Zhang et al. 2006). A crucial role of PPARγ in FLD progression is supported by the genetic ablation of PPARγ in the mouse liver. Hepatocyte‐specific PPARγ KO protects mice from the fatty liver with the downregulation of lipogenesis (Srebf1c, Scd1, and Acaca) and fatty acid uptake (Cd36 and Fabp4) (Morán-Salvador et al. 2011). Similarly, silencing hepatic PPARγ in ob/ob mice reduces lipid accumulation in the liver (Matsusue et al. 2003). Therefore, we investigated whether PPARγ mediates the lipid-lowering effect in SHMT2 KD hepatocytes. SHMT2 KD inhibited the expression of PPARγ and its target genes associated with lipogenesis and fatty acid uptake. Given the importance of PPARγ in the pathogenesis of fatty liver, PPARγ overexpression in SHMT2 KD hepatocytes blocked the inhibitory effect of SHMT2 KD on lipid accumulation. Moreover, a positive correlation between PPARγ and SREBP1 in obese patients with FLD suggests that PPARγ reinforces lipogenesis via SREBP1 upregulation (Pettinelli and Videla 2011). The downregulation of SREBP1 and its target genes in SHMT2 KD hepatocytes was consistent with the notion that PPARγ can induce lipogenesis by reinforcing SREBP1 in the development of FLD.
In summary, we demonstrated the roles of SHMT2 and glycine in hepatic lipid metabolism using SHMT2 KD primary hepatocytes. SHMT2 KD hepatocytes downregulated lipogenesis and fatty acid uptake via the mTOR/PPARɣ pathway. Exogenous glycine treatment of SHMT2 KD hepatocytes showed the correlations between glycine biosynthesis and the mTOR/PPARɣ pathway. Thus, SHMT2 may link glycine biosynthesis and the mTOR/PPARɣ pathway in the progression of FLD, suggesting that SHMT2 can be explored as a therapeutic target for FLD.
References
Amelio I, Cutruzzolá F, Antonov A, Agostini M, Melino G (2014) Serine and glycine metabolism in cancer. Trends Biochem Sci 39(4):191–198. https://doi.org/10.1016/j.tibs.2014.02.004
Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, Yokote K, Nakayama T (2016) Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARγ directs early activation of T cells. Nat Commun 7(1):13683. https://doi.org/10.1038/ncomms13683
Bakan I, Laplante M (2012) Connecting mTORC1 signaling to SREBP-1 activation. Curr Opin Lipidol 23(3):226–234. https://doi.org/10.1097/MOL.0b013e328352dd03
Bianchi G, Marchesini G, Brunetti N, Manicardi E, Montuschi F, Chianese R, Zoli M (2003) Impaired insulin-mediated amino acid plasma disappearance in non-alcoholic fatty liver disease: a feature of insulin resistance. Dig Liver Dis 35(10):722–727. https://doi.org/10.1016/s1590-8658(03)00416-x
Blanchard P-G, Festuccia WT, Houde VP, St-Pierre P, Brûlé S, Turcotte V, Côté M, Bellmann K, Marette A, Deshaies Y (2012) Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. J Lipid Res 53(6):1117–1125. https://doi.org/10.1194/jlr.M021485
Budhu A, Roessler S, Zhao X, Yu Z, Forgues M, Ji J, Karoly E, Qin LX, Ye QH, Jia HL, Fan J, Sun HC, Tang ZY, Wang XW (2013) Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 144(5):1066-1075.e1061. https://doi.org/10.1053/j.gastro.2013.01.054
Ducker GS, Rabinowitz JD (2017) One-carbon metabolism in health and disease. Cell Metab 25(1):27–42. https://doi.org/10.1016/j.cmet.2016.08.009
Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39(2):171–183. https://doi.org/10.1016/j.molcel.2010.06.022
Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, Ciociaro D, Abate ML, Gambino R, Cassader M, Bugianesi E, Gastaldelli A (2018) Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology 67(1):145–158. https://doi.org/10.1002/hep.29465
García-Cazorla À, Verdura E, Juliá-Palacios N, Anderson EN, Goicoechea L, Planas-Serra L, Tsogtbaatar E, Dsouza NR, Schlüter A, Urreizti R, Tarnowski JM, Gavrilova RH, Ruiz M, Rodríguez-Palmero A, Fourcade S, Cogné B, Besnard T, Vincent M, Bézieau S, Folmes CD, Zimmermann MT, Klee EW, Pandey UB, Artuch R, Cousin MA, Pujol A (2020) Impairment of the mitochondrial one-carbon metabolism enzyme SHMT2 causes a novel brain and heart developmental syndrome. Acta Neuropathol 140(6):971–975. https://doi.org/10.1007/s00401-020-02223-w
Garrow TA, Brenner AA, Whitehead VM, Chen XN, Duncan RG, Korenberg JR, Shane B (1993) Cloning of human cDNAs encoding mitochondrial and cytosolic serine hydroxymethyltransferases and chromosomal localization. J Biol Chem 268(16):11910–11916
Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, Jenoe P, Heim MH, Riezman I, Riezman H, Hall MN (2017) mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 32(6):807-823.e812. https://doi.org/10.1016/j.ccell.2017.11.011
Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Rüegg MA, Hall MN (2012) Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 15(5):725–738. https://doi.org/10.1016/j.cmet.2012.03.015
Hu X, Xu Q, Wan H, Hu Y, Xing S, Yang H, Gao Y, He Z (2020) PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab Investig 100(6):801–811. https://doi.org/10.1038/s41374-020-0404-9
Hwang SY, Kang YJ, Sung B, Kim M, Kim DH, Lee Y, Yoo MA, Kim CM, Chung HY, Kim ND (2015) Folic acid promotes the myogenic differentiation of C2C12 murine myoblasts through the Akt signaling pathway. Int J Mol Med 36(4):1073–1080. https://doi.org/10.3892/ijmm.2015.2311
Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK (2012) Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336(6084):1040–1044. https://doi.org/10.1126/science.1218595
Ji L, Tang Y, Pang X, Zhang Y (2019) Increased expression of serine hydroxymethyltransferase 2 (SHMT2) is a negative prognostic marker in patients with hepatocellular carcinoma and is associated with proliferation of HepG2 cells. Med Sci Monit 25:5823–5832. https://doi.org/10.12659/msm.915754
Kim D, Fiske BP, Birsoy K, Freinkman E, Kami K, Possemato RL, Chudnovsky Y, Pacold ME, Chen WW, Cantor JR, Shelton LM, Gui DY, Kwon M, Ramkissoon SH, Ligon KL, Kang SW, Snuderl M, Vander Heiden MG, Sabatini DM (2015) SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 520(7547):363–367. https://doi.org/10.1038/nature14363
Li Z, Xu G, Qin Y, Zhang C, Tang H, Yin Y, Xiang X, Li Y, Zhao J, Mulholland M, Zhang W (2014) Ghrelin promotes hepatic lipogenesis by activation of mTOR-PPARγ signaling pathway. Proc Natl Acad Sci USA 111(36):13163–13168. https://doi.org/10.1073/pnas.1411571111
Liu GY, Sabatini DM (2020) mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 21(4):183–203. https://doi.org/10.1038/s41580-019-0199-y
Locasale JW (2013) Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer 13(8):572–583. https://doi.org/10.1038/nrc3557
Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B Jr, Reitman ML, Gonzalez FJ (2003) Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Investig 111(5):737–747. https://doi.org/10.1172/jci17223
Mejias M, Gallego J, Naranjo-Suarez S, Ramirez M, Pell N, Manzano A, Suñer C, Bartrons R, Mendez R, Fernandez M (2020) CPEB4 increases expression of PFKFB3 to induce glycolysis and activate mouse and human hepatic stellate cells, promoting liver fibrosis. Gastroenterology 159(1):273–288. https://doi.org/10.1053/j.gastro.2020.03.008
Micevic G, Muthusamy V, Damsky W, Theodosakis N, Liu X, Meeth K, Wingrove E, Santhanakrishnan M, Bosenberg M (2016) DNMT3b modulates melanoma growth by controlling levels of mTORC2 component RICTOR. Cell Rep 14(9):2180–2192. https://doi.org/10.1016/j.celrep.2016.02.010
Minton DR, Nam M, McLaughlin DJ, Shin J, Bayraktar EC, Alvarez SW, Sviderskiy VO, Papagiannakopoulos T, Sabatini DM, Birsoy K, Possemato R (2018) Serine catabolism by SHMT2 is required for proper mitochondrial translation initiation and maintenance of formylmethionyl-tRNAs. Mol Cell 69(4):610-621.e615. https://doi.org/10.1016/j.molcel.2018.01.024
Morán-Salvador E, López-Parra M, García-Alonso V, Titos E, Martínez-Clemente M, González-Périz A, López-Vicario C, Barak Y, Arroyo V, Clària J (2011) Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J 25(8):2538–2550. https://doi.org/10.1096/fj.10-173716
Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE, Goldstein JL, Brown MS (2012) Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci USA 109(40):16184–16189. https://doi.org/10.1073/pnas.1213343109
Parsa S, Ortega-Molina A, Ying HY, Jiang M, Teater M, Wang J, Zhao C, Reznik E, Pasion JP, Kuo D, Mohan P, Wang S, Camarillo JM, Thomas PM, Jain N, Garcia-Bermudez J, Cho BK, Tam W, Kelleher NL, Socci N, Dogan A, De Stanchina E, Ciriello G, Green MR, Li S, Birsoy K, Melnick AM, Wendel HG (2020) The serine hydroxymethyltransferase-2 (SHMT2) initiates lymphoma development through epigenetic tumor suppressor silencing. Nat Cancer 1:653–664. https://doi.org/10.1038/s43018-020-0080-0
Pettinelli P, Videla LA (2011) Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab 96(5):1424–1430. https://doi.org/10.1210/jc.2010-2129
Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung Y-L, Schulze A (2008) SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 8(3):224–236. https://doi.org/10.1016/j.cmet.2008.07.007
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11):2281–2308. https://doi.org/10.1038/nprot.2013.143
Rom O, Liu Y, Liu Z, Zhao Y, Wu J, Ghrayeb A, Villacorta L, Fan Y, Chang L, Wang L, Liu C, Yang D, Song J, Rech JC, Guo Y, Wang H, Zhao G, Liang W, Koike Y, Lu H, Koike T, Hayek T, Pennathur S, Xi C, Wen B, Sun D, Garcia-Barrio MT, Aviram M, Gottlieb E, Mor I, Liu W, Zhang J, Chen YE (2020) Glycine-based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci Transl Med 12(572):eaaz841. https://doi.org/10.1126/scitranslmed.aaz2841
Sim WC, Yin HQ, Choi HS, Choi YJ, Kwak HC, Kim SK, Lee BH (2015) l-Serine supplementation attenuates alcoholic fatty liver by enhancing homocysteine metabolism in mice and rats. J Nutr 145(2):260–267. https://doi.org/10.3945/jn.114.199711
Sim WC, Kim DG, Lee W, Sim H, Choi YJ, Lee BH (2019) Activation of SIRT1 by L-serine increases fatty acid oxidation and reverses insulin resistance in C2C12 myotubes. Cell Biol Toxicol 35(5):457–470. https://doi.org/10.1007/s10565-019-09463-x
Sim WC, Lee W, Sim H, Lee KY, Jung SH, Choi YJ, Kim HY, Kang KW, Lee JY, Choi YJ, Kim SK, Jun DW, Kim W, Lee BH (2020) Downregulation of PHGDH expression and hepatic serine level contribute to the development of fatty liver disease. Metabolism 102:154000. https://doi.org/10.1016/j.metabol.2019.154000
Skat-Rørdam J, Højland Ipsen D, Lykkesfeldt J, Tveden-Nyborg P (2019) A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic Clin Pharmacol Toxicol 124(5):528–537. https://doi.org/10.1111/bcpt.13190
Sun K, Wu Z, Ji Y, Wu G (2016) Glycine regulates protein turnover by activating protein kinase B/mammalian target of rapamycin and by inhibiting MuRF1 and atrogin-1 gene expression in C2C12 myoblasts. J Nutr 146(12):2461–2467. https://doi.org/10.3945/jn.116.231266
Tani H, Mito T, Velagapudi V, Ishikawa K, Umehara M, Nakada K, Suomalainen A, Hayashi J-I (2019) Disruption of the mouse Shmt2 gene confers embryonic anaemia via foetal liver-specific metabolomic disorders. Sci Rep 9(1):16054. https://doi.org/10.1038/s41598-019-52372-6
Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, Hue L, Andreelli F (2006) Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J Physiol 574(Pt 1):41–53. https://doi.org/10.1113/jphysiol.2006.108506
Wang W, Wu Z, Dai Z, Yang Y, Wang J, Wu G (2013) Glycine metabolism in animals and humans: implications for nutrition and health. Amino Acids 45(3):463–477. https://doi.org/10.1007/s00726-013-1493-1
Wang W, Wu Z, Lin G, Hu S, Wang B, Dai Z, Wu G (2014) Glycine stimulates protein synthesis and inhibits oxidative stress in pig small intestinal epithelial cells. J Nutr 144(10):1540–1548. https://doi.org/10.3945/jn.114.194001
Wang M, Yuan F, Bai H, Zhang J, Wu H, Zheng K, Zhang W, Miao M, Gong J (2019) SHMT2 promotes liver regeneration through glycine-activated Akt/mTOR pathway. Transplantation 103(7):e188–e197. https://doi.org/10.1097/tp.0000000000002747
Woo CC, Chen WC, Teo XQ, Radda GK, Lee PTH (2016) Downregulating serine hydroxymethyltransferase 2 (SHMT2) suppresses tumorigenesis in human hepatocellular carcinoma. Oncotarget 7(33):53005–53017. https://doi.org/10.18632/oncotarget.10415
Yamashita T, Honda M, Takatori H, Nishino R, Minato H, Takamura H, Ohta T, Kaneko S (2009) Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J Hepatol 50(1):100–110. https://doi.org/10.1016/j.jhep.2008.07.036
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M (2016) Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64(1):73–84. https://doi.org/10.1002/hep.28431
Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, Reddy JK (2003) Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J Biol Chem 278(1):498–505. https://doi.org/10.1074/jbc.M210062200
Yuan M, Pino E, Wu L, Kacergis M, Soukas AA (2012) Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem 287(35):29579–29588. https://doi.org/10.1074/jbc.M112.386854
Zhang YL, Hernandez-Ono A, Siri P, Weisberg S, Conlon D, Graham MJ, Crooke RM, Huang LS, Ginsberg HN (2006) Aberrant hepatic expression of PPARgamma2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J Biol Chem 281(49):37603–37615. https://doi.org/10.1074/jbc.M604709200
Zhou X, He L, Zuo S, Zhang Y, Wan D, Long C, Huang P, Wu X, Wu C, Liu G, Liu Y (2018) Yin Y Serine prevented high-fat diet-induced oxidative stress by activating AMPK and epigenetically modulating the expression of glutathione synthesis-related genes. Biochim Biophys Acta Mol Basis Dis 2:488–498. https://doi.org/10.1016/j.bbadis.2017.11.009
Funding
This work was supported by the the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1A2B5B01001920 and 2021R1C1C2004529).
Author information
Authors and Affiliations
Contributions
Conceptualization: Y-JC and B-HL; methodology, Y-JC, GL, SY, and JY; formal analysis and investigation, Y-JC, GL, SY, WL and JY; writing—original draft preparation, Y-JC and B-HL; writing—review and editing, Y-JC, SKK and B-HL; funding acquisition, Y-JC and B-HL; supervision, B-HL.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Ethical approval
The study was conducted according to the guidelines of the Institutional Animal Care and Use Committee of Seoul National University.
Additional information
Handling editor: J. M Phang.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Choi, YJ., Lee, G., Yun, S.H. et al. The role of SHMT2 in modulating lipid metabolism in hepatocytes via glycine-mediated mTOR activation. Amino Acids 54, 823–834 (2022). https://doi.org/10.1007/s00726-022-03141-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-022-03141-9