
Abstract
Silent information regulator 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase, and the function is linked to cellular metabolism including mitochondrial biogenesis. Hepatic l-serine concentration is decreased significantly in fatty liver disease. We reported that the supplementation of the amino acid ameliorated the alcoholic fatty liver by enhancing l-serine-dependent homocysteine metabolism. In this study, we hypothesized that the metabolic production of NAD+ from l-serine and thus activation of SIRT1 contribute to the action of l-serine. To this end, we evaluated the effects of l-serine on SIRT1 activity and mitochondria biogenesis in C2C12 myotubes. l-Serine increased intracellular NAD+ content and led to the activation of SIRT1 as determined by p53 luciferase assay and western blot analysis of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) acetylation. l-Serine treatment increased the expression of the genes associated with mitochondrial biogenesis and enhanced mitochondrial mass and function. In addition, l-serine reversed cellular insulin resistance determined by insulin-induced phosphorylation of Akt and GLUT4 expression and membrane translocation. l-Serine-induced mitochondrial gene expression, fatty acid oxidation, and insulin sensitization were mediated by enhanced SIRT1 activity, which was verified by selective SIRT1 inhibitor (Ex-527) and siRNA directed to SIRT1. l-Serine effect on cellular NAD+ level is dependent on the l-serine metabolism to pyruvate that is subsequently converted to lactate by lactate dehydrogenase. In summary, these data suggest that l-serine increases cellular NAD+ level and thus SIRT1 activity in C2C12 myotubes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Silent information regulator 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase, and the function is linked to cellular metabolism. Hepatic SIRT1 deficiency in mice impairs mTorc2/Akt or fibroblast growth factor 21 signaling and results in hyperglycemia, insulin resistance, as well as severe hepatic steatosis (Wang et al. 2011; Li et al. 2014). Moreover, reduced SIRT1 expression was reported in muscle biopsies and primary myotubes obtained from patients with type 2 diabetes (Fröjdö et al. 2011). Therefore, SIRT1 activation has attracted attention as a promising strategy to treat diseases associated with metabolic disorder such as type 2 diabetes. Resveratrol or NAD+ precursor nicotinamide riboside improves insulin resistance by inducing mitochondrial biogenesis in genetic or diet induced diabetic animal model (Beaudoin et al. 2013; Cantó et al. 2012). Beneficial effects of resveratrol on systemic insulin sensitivity are also observed when administered to healthy obese men (Timmers et al. 2011).
The essential function of amino acids in most of the eukaryotic cells is to synthesize proteins. However, several amino acids also have non-protein functions such as precursors of the neurotransmitters or signaling molecules involved in many biochemical processes. Some of them have been reported to play an important role in lipid and glucose metabolism. Leucine and arginine prevented mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice (Li et al. 2012; Fu et al. 2005). Glutamine supplementation with high fat diet reduced body weight and attenuated hyperglycemia and hyperinsulinemia (Opara et al. 1996). Recent studies have shown that amino acids also act as modulators of SIRT1 activity. Leucine is the first and the most studied amino acid regulating SIRT1 activity that increased mitochondrial mass by stimulating SIRT1 in C2C12 myocytes and 3T3-L1 adipocytes (Sun and Zemel 2009). Leucine supplementation increased SIRT1 expression and prevented mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Taurine prevents amyloid β-induced mitochondrial dysfunction by activation of SIRT1 (Sun et al. 2014). Tryptophan has a potential to activate SIRT1 activity. Inhibition of the kynurenine pathway, the principle route of tryptophan metabolism leading to the production of NAD+, resulted in the decreased SIRT1 activity (Braidy et al. 2011). However, over-activation of the kynurenine pathway may lead to increased levels of quinolinic acid and other neurotoxic metabolites generated by perivascular macrophages.
Hyperhomocysteinemia is one of the risk factors associated with many metabolic diseases and cancer. Recently, we reported that l-serine ameliorates alcoholic fatty liver by accelerating l-serine-dependent homocysteine metabolism (Sim et al. 2015). The purpose of the present study was to explore molecular mechanisms underlying the beneficial effects of l-serine on hepatic lipid metabolism. l-Serine is metabolized to lactate by lactate dehydrogenase (LDH) and produce NAD+. Therefore, we hypothesized that l-serine activates SIRT1 and induces mitochondria biogenesis in C2C12 myotubes by producing NAD+. In the present study, we found that the incubation of the cells in the presence of l-serine in the media increased cellular NAD+ content. l-Serine increased mitochondrial function of C2C12 myotubes by means of SIRT1 activation. Activation of SIRT1 by l-serine increased fatty acid oxidation and reversed palmitic acid-induced insulin resistance in C2C12 myotubes.
Materials and methods
Cell culture
C2C12 mouse skeletal muscle cells and HepG2 human hepatoma cells were obtained from American Type Culture Collection (Rockville, MD). C2C12 myoblasts and HepG2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) 4.5 g/L glucose supplemented with 10% fetal bovine serum and 50 units/mL penicillin and streptomycin. To initiate differentiation, C2C12 myoblasts were allowed to reach 90% confluence. Then, cells were rinsed twice with DPBS and replenished with fresh warmed differentiation medium (containing 2% horse serum and 50 units/mL penicillin and streptomycin) every 24 h for 6 days. Cells were incubated in a 37 °C incubator in an atmosphere of 5% CO2 in air. For the treatment, fresh media was replaced and l-serine was added at the indicated concentration.
Cell viability assay
C2C12 myoblasts were seeded into 96 clear well plates and changed into myotubes according to the differentiation procedure above. After differentiation, cells were treated with resveratrol or l-serine and cell viability was analyzed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MTT solution (1 mg/mL) was added to each well and incubated for 2 h. And then, solution was discarded and the formazan crystals were dissolved in DMSO, and absorbance was measured at 540 nm.
NAD+/NADH measurements
NAD+ and NADH nucleotides were directly measured using NAD+/NADH Quantitation Colorimetric Kit (Biovision, Milpitas, CA) following the manufacturer’s protocols. In brief, two 100π dishes of C2C12 myotubes were freshly homogenized in 200 μL acid extraction buffer or alkali extraction buffer to measure the NAD+ or NADH concentrations. Then, homogenates were filtered with 10 kDa molecular weight cut off filters (Abcam) neutralized using the opposite buffer and the concentration of nucleotides was determined after an enzymatic cycling reaction using 10-μL sample. Values for both nucleotides were normalized by protein concentration.
IP
Immunoprecipitation was performed using specific antibodies against acetyl-lysine (Cell signaling Technology, Beverly, MA) for overnight and subjected to bind protein G agarose bead (Thermo Fisher Scientific, Waltham, MA). Immunoprecipitated bead was washed with IP buffer three times and mixed with sample buffer. After centrifugation, supernatant was subjected to western blotting.
Western blotting
Extracted proteins were separated by SDS-PAGE and then transferred to a polyvinylidene difluoride membrane (Millipore, Darmstadt, Germany). Antibodies against the following proteins were used, typically at 1:2000 dilution; peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), glucose transporter type 4 (GLUT4) (Santa Cruz Biotechnology, Santa Cruz, CA), ATPase (Abcam, Cambridge, MA), pAKT, AKT, Sirtuin 1 (SIRT1), and GAPDH from Cell Signaling Technology. HRP-conjugated secondary anti-rabbit antibody was purchased from Cell Signaling Technology and used at 1:2500 dilution.
qRT-PCR
For qRT-PCR, RNA was prepared from C2C12 myotubes using Easy-Blue™ Total RNA Extraction Kit (Intron Biotechnology, Seoul, Korea). cDNA was synthesized using QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). The resulting cDNA was amplified by qRT-PCR using iTaq™ Universal SYBR® Green Supermix Kit (Bio-Rad, Hercules, CA). The sequences of the gene specific primers are listed in Supplemental Table 1.
MitoTracker red staining
C2C12 myoblasts were differentiated in 96 black well plates and stained by MitoTracker® dye according to the manufacturer’s protocols. In brief, cells were added prewarmed (37 °C) staining solution containing MitoTracker® Red CMXRos probe (Invitrogen) for 30 min. After staining, cells were washed in fresh growth media and replaced with 4% formaldehyde containing Hoechst 33258 (Invitrogen) at 37 °C for 10 min. After fixation, cells were rinsed twice in Dulbecco’s phosphate buffered saline (DPBS) and analyzed by fluorescence detection system (Molecular Devices, Sunnyvale, CA).
mtDNA quantification
Genomic DNA was extracted by QIAamp® DNA Mini Kit (Qiagen) from C2C12 myotubes according to the manufacturer’s protocol. Briefly, cells were harvested and cell pellet was lysed with proteinase K. After lysis, samples were poured on QIAmp Mini spin column with silica membrane and washed twice with washing buffer. Purified DNA was eluted from QIAamp Mini spin column with elution buffer. To quantify mtDNA, Mt-Cyb (forward) (5′-CCA CTT CAT GTT ACC ATT TA-3′) and Mt-Cyb (reverse) (5′-ATC TGC ATC TGA GTT TAA TC-3′) primers were used. The Rn18s (forward) (5′-GGG AGC CTG AGA AAC GGC-3′) and Rn18s (reverse) (5′-GGG TCG GGA GTG GGT AAT TT-3′) primers were used to quantify nuclear DNA. Real-time PCR was carried out by using iTaq™ Universal SYBR® Green Supermix Kit (Bio-Rad, Hercules, CA), and mitochondrial DNA copy number was calculated from the ratio Mt-Cyb/Rn18s.
ATP measurements
Intracellular ATP was measured using CellTiter-Glo® (Promega) according to the manufacturer’s protocol. In brief, C2C12 myoblasts were seeded in 96-well plate and differentiated. After treatment, the plate was equilibrated to room temperature for 30 min and added CellTiter-Glo® reagent in an equal volume (100 μL per well) to all wells. After 2 min mixing contents, luminescence was detected by Centro LB960 luminometer (Berthold Technologies, Oak Ridge, TN).
OCR determination
Oxygen consumption rate (OCR) was determined using Extracellular Flux Analyzers XFp (Agilent Technologies, Santa Clara, CA) and Sea. C2C12 cells were differentiated in XFp cell culture miniplates in a CO2 incubator at 37 °C. After treatment, cells were washed twice with assay medium (XF Base Medium) and were incubated in a CO2-free incubator at 37 °C for 30 min prior to the assay. Then, cells were treated consecutively with oligomycin (an ATP synthase inhibitor, 1 μM), FCCP (carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; uncoupling agent, 1 μM), and antimycin A and rotenone (a complex III and I inhibitor, respectively, 0.5 μM) every 20 min.
siRNA transfection
The SignalSilence® SirT1 siRNA was purchased at Cell signaling Technology, and LDH-A siRNA was purchased at Santa Cruz Biotechnology. The control siRNA (sc-37,007) was purchased at Santa Cruz Biotechnology. The target sequence of the siRNA for SIRT1 was forward 5′-GAA GUU GAC CUC CUC AUU GU-3′ and reverse 5′-ACA AUG AGG AGG UCA ACU UC-3′. The target sequence of the siRNA for LDH-A was forward 5′-GUU CAU CAU UCC CAA CAU Utt-3′ and reverse 5′-AAU GUU GGG AAU GAU GAA Ctt-3′. The siRNA was transfected into C2C12 myotubes using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol. Fifty nanomolars of SirT1 and LDH-A siRNA was selected finally for cells and was transfected into the cells for 48 h.
Nile red assay
C2C12 myoblasts were seeded in 96 black well plates and differentiated into myotubes. After differentiation, cells were incubated with or without oleic acid, l-serine, and Ex-527. On the following day, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich). Cells were washed twice with DPBS and stained with 100 ng/mL Nile red (Sigma-Aldrich, St. Louis, MO) and Hoechst 33258 dye in DPBS. After staining, cells were washed twice with DPBS and then analyzed fluorometrically by a microplate fluorescence detection system (Molecular Devices). For quantification, Nile red fluorescence was normalized by Hoechst 33258 fluorescence.
Confocal microscopy
C2C12 myoblasts were seeded in 12-well plates covered with microscope cover glass (Paul Marienfeld GmbH & Co.KG, Lauda-Königshofen, Germany) which were coated with poly-d-lysine (Sigma-Aldrich). Myoblasts were changed into myotubes according to the differentiation procedure above, and the cells were incubated with or without oleic acid, l-serine, and Ex-527. After treatment, cells were stained with 100 ng/mL Nile red dye and Hoechst 33258 (Sigma-Aldrich) in DPBS and fixed with 4% paraformaldehyde and moved onto Microscope slides (Thermo Fisher Scientific) with ProLong® Gold antifade reagent (Invitrogen). And microscopic images were acquired using a confocal microscope with a Nikon Plan Apochromat × 60/0.75 objective (Nikon Eclipse Ti; Nikon, Japan).
Membrane fraction
To detect GLUT4 expression, subcellular fractionation was performed according to Li et al. (2015). Briefly, cells were washed with ice-cold DPBS twice and then suspended in cold sample preparation buffer, sonicated four to five times for 10 s each, and centrifuged at 100,000×g for 1 h at 4 °C. The resulting pellet was resuspended in 0.5% Triton X-100 added homogenization buffer and incubated on ice for 1 h. The resultant supernatant was kept as the plasma membrane fraction and validated by identifying ATPase protein.
Lactate measurement
Intracellular lactate concentration was measured by AVANCE nuclear magnetic resonance (NMR; 600 MHz; Bruker, Billerica, MA) equipped with a cryoprobe (NCIRF, Seoul, Korea). The pulse program was noesypr1d, a classical one-dimensional method. Lactate was identified and quantified using Chenomx spectral database (Edmonton, Alberta, Canada) by fitting the experimental spectra to those in the database.
Statistical analysis
Multiple comparisons were evaluated by one-way analysis of variance followed by Dunnett’s post-test or Tukey’s multiple comparison procedure with p < 0.05 considered significant by using GraphPad Prism version 5.0 (San Diego, CA).
Results
l-Serine increases SIRT1 activity and mitochondrial function
First, we tested the effects of l-serine on SIRT1 activity in C2C12 mouse myotubes. Treatment of the cells with l-serine increased the cellular NAD+ level without affecting that of NADH at a concentration of 5 mg/mL (Fig. 1a). We measured the SIRT1 activity using reporter gene assay in HepG2 cells transfected with p53 luciferase and western blot analysis of PGC-1α acetylation. Resveratrol was treated as a positive control for PGC-1α deacetylation (Price et al. 2012) (Supplemental Figure 1a and b). As shown in Fig. 1b and Supplemental Figure 1c, p53 luciferase activity and acetyl PGC-1α level were decreased significantly by the treatment with l-serine. We also investigated the effect of pyruvate and other amino acids that can increase pyruvate levels on SIRT1 activity. Among these compounds, pyruvate was the only one that increased SIRT1 activity significantly (Supplemental Figure 2). SIRT1 activity can also be affected by its expression; we measured the SIRT1 and PGC-1α expression. The protein levels of SIRT1 and PGC-1α were not affected by l-serine treatment (Fig. 1c).

a C2C12 myotubes were treated with l-serine at the indicated concentrations for 24 h. Then, intracellular NAD+ and NADH were measured. b C2C12 myotubes were treated with l-serine at the indicated concentrations for 24 h or 50 μM resveratrol (R) for 6 h. Then, PGC-1α levels were checked on acetyl-lysine immunoprecipitates (IP). Lower panel shows the quantification of PGC-1α acetylation. c C2C12 myotubes were treated with indicated concentrations of l-serine for 24 h. Protein extracts were prepared from cell lysates, and western blot analysis was performed to determine SIRT1 and PGC-1α expression. Right panels show the quantification of SIRT1 and PGC-1α expression. Values are expressed as mean ± SD of three independent experiments. Significance (**p < 0.01, ***p < 0.001) relative to the vehicle treatment using one-way ANOVA with Dunnett’s post-test
We next investigated whether l-serine treatment increases the expression of the genes associated with mitochondrial biogenesis in C2C12 myotubes, including Cycs, Atp5b, and Tfam. Treatment of the cells with 5 mg/mL of l-serine increased the expression of the genes significantly (Fig. 2a). The increase in mitochondrial biogenesis genes was associated with enhanced mitochondrial mass, mtDNA copy number, and ATP level (Fig. 2b–d). Finally, mitochondrial function was determined by measuring the OCR in the absence of fatty acid source. Figure 2e clearly shows that incubation of C2C12 cells in the presence of l-serine increased the OCR, which was comparable to the effects of resveratrol.

C2C12 myotubes were treated with 50 μM resveratrol (R) for 6 h or l-serine at the indicated concentrations for 24 h. a Total RNA was extracted from myotubes and PGC-1α target mRNAs were analyzed by qRT-PCR. b Mitochondrial mass was analyzed by Mitotracker red. c mtDNA copy number was analyzed by measuring the ratio of mitochondrial to nuclear DNA (mtDNA/nDNA). d Mitochondrial function was analyzed by measuring ATP levels. e Cells were treated with 5 mg/mL l-serine for 24 h or 50 μM resveratrol (R) for 6 h prior to assay and oxygen consumption ratio (OCR) was analyzed by Seahorse Bioscience XFp extracellular flux analyzer. The OCR in cells was measured in the presence of oligomycin (Oligo) (1 μM), FCCP (1 μM), and antimycin A and rotenone (AA and rot), respectively, (0.5 μM), and the quantification of area under the curve (AUC) was presented at the right panel. Values are expressed as mean ± SD of three independent experiments. Significance (*p < 0.05, **p < 0.01, ***p < 0.001) relative to the vehicle treatment using one-way ANOVA with Dunnett’s post-test
Effects of l-serine on the mitochondrial function is mediated by SIRT1
To investigate whether the effects of l-serine on the mitochondrial function are dependent on the SIRT1 activity, cells were incubated with l-serine in the absence or presence of the selective SIRT1 inhibitor, Ex-527, or siRNA directed to SIRT1. The cell viability was not affected by Ex-527 treatment (Supplemental Figure 3a). The decreased acetylation of PGC-1α and increased expression of Cycs and Atp5b were reversed by the co-treatment with Ex-527 (Fig. 3a, b). Ex-527 or SIRT1 siRNA in the absence of l-serine had no effect on acetylation of PGC-1α and its target gene expression (Supplemental Figure 3b-e). l-Serine-induced increase in the mtDNA copy number and ATP level was reversed by silencing SIRT1 gene (Fig. 3c, d). These data demonstrate that the effects of l-serine on the mitochondrial biogenesis and function are mediated by enhanced SIRT1 activity.

C2C12 myotubes were treated with 5 mg/mL l-serine in the absence or presence of 10 μM Ex-527. After 24 h treatment, a PGC-1α levels were checked on acetyl-lysine immunoprecipitates (IP). Relative quantification of PGC-1α acetylation is shown on the right. b Total RNA was extracted from myotubes and Cycs and Atp5b mRNA level were analyzed by qRT-PCR. c, d C2C12 myotubes were transfected with scrambled or SIRT1 siRNA, and then cells were treated with 5 mg/mL l-serine for 24 h or 50 μM resveratrol (R) for 6 h. ATP levels or mtDNA/nDNA ratio was analyzed. Values are expressed as mean ± SD of three or four (a) independent experiments. Means without a common letter differ; p < 0.05. Significance (*p < 0.05, **p < 0.01, ***p < 0.001) relative to the vehicle treatment; significance (#p < 0.05, ###p < 0.001) relative to the l-serine treatment group; significance ($p < 0.05) relative to the R treatment group using one-way ANOVA with Tukey’s post-test
l-Serine-induced activation of SIRT1 increases fatty acid oxidation and reverses insulin resistance in C2C12 myotubes
In skeletal muscle cells, SIRT1 regulates PGC-1α-mediated mitochondria and fatty acid metabolism (Gerhart-Hines et al. 2007). We investigated whether l-serine increases the expression of genes associated with the β-oxidation of fatty acids including Lcad and Cpt1a in C2C12 myotubes. l-Serine treatment increased the expression of both of the genes that play central role in the fatty acid oxidation and Ex-527 blunted the effect (Fig. 4a). Treatment of C2C12 cells with oleic acid at a concentration of 500 μM increased the neutral lipid accumulation in C2C12 cells, which was suppressed by the co-treatment with l-serine (Fig. 4b) and Ex-527 reversed this effect. Next, we tested the effects of l-serine on fatty acid metabolism in C2C12 cells. Figure 4c shows the OCR and the calculated area under the curve in cells treated with l-serine in the presence of oleic acid with or without Ex-527. There was a significant increase in the fatty acid oxidation by 32% in C2C12 cells and Ex-527 decreased fatty acid oxidation by 22%.

a C2C12 myotubes were treated with 5 mg/mL of l-serine with or without 10 μM Ex-527 for 24 h and total RNA was extracted from cells. Cpt1α and Lcad mRNA levels were analyzed by qRT-PCR. b C2C12 myotubes were with 5 mg/mL of l-serine or 10 μM Ex-527 and oleic acid (OLA) (500 μM) for 24 h, and intracellular lipid droplets were quantified fluorometrically with excitation and emission wavelengths of 486 and 528 nm. Right panel shows representative image of lipid droplets by Nile red and nucleus by Hoechst 33258 staining. c C2C12 myotubes were assessed for oleic acid-induced oxygen consumption ratio (OCR) as a measure of fatty acid oxidation using the Seahorse Bioscience XFp extracellular flux analyzer. Cells were treated with oleic acid in the presence or absence of 5 mg/mL l-serine with or without Ex-527 for 24 h prior to assay. The OCR in cells was measured in the presence of oligomycin (Oligo) (1 μM), FCCP (1 μM), and antimycin A and rotenone (AA and rot), respectively (0.5 μM). Right panel shows the quantification of area under the curve (AUC). Values are expressed as mean ± SD of three independent experiments. Significance (***p < 0.001) relative to the vehicle treatment; significance (##p < 0.01, ###p < 0.001) relative to the serine treatment group; significance ($p < 0.05, $$$p < 0.001) relative to the oleic acid treatment group by using one-way ANOVA with Tukey’s post-test
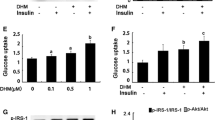
Then, we evaluated whether SIRT1 activation by l-serine contribute to the reversion of insulin resistance induced by palmitic acid. Insulin resistance was induced by the incubation of C2C12 cells with palmitic acid for 24 h, which was confirmed by the reduced Akt phosphorylation and Slc2a4 expression in response to the insulin stimulus (Fig. 5a, b). Treatment of the cells with l-serine increased the level of phospho-Akt and the expression of Slc2a4. However, co-treatment with Ex-527 blunted the effect (Fig. 5a, b). l-Serine also reversed GLUT4 membrane translocation reduced by palmitic acid and Ex-527 diminished the effect (Fig. 5c). These data indicate that l-serine increases fatty acid oxidation and reversed insulin resistance, which is mediated by SIRT1 activity.

C2C12 myotubes were treated with 5 mg/mL l-serine in the absence or presence of 10 μM Ex-527 with palmitic acid (Pal) (250 μM) for 24 h and then insulin (100 nM) was treated for 20 min. a Protein extracts were prepared from cell lysates and western blot analysis was performed to determine the levels of Akt phosphorylation. b Total RNA was extracted from cells, and Slc2a4 mRNA level was determined using qRT-PCR. c Total cell lysates were collected and subjected to subcellular fractionation. GLUT4, ATPase, and GAPDH levels were determined from membrane fraction by western blot analysis. Values are expressed as mean ± SD of three independent experiments. Significance (***p < 0.001) relative to the insulin and palmitic acid treated group; significance (##p < 0.01) relative to the insulin, palmitic acid, and l-serine treated group by using one-way ANOVA with Tukey’s post-test
l-Serine metabolism to pyruvate increases NAD+ production and SIRT1 activity
l-Serine is converted to pyruvate by serine dehydratase that is reduced to lactate by LDH using NADH (Burgner and Ray 1984). We hypothesized that the SIRT1 activating effect of l-serine ascribe to the production of NAD+ in the course of pyruvate metabolism. To verify the hypothesis, we measured the cellular lactate levels in l-serine treated cells and found lactate was increased by l-serine treatment (Fig. 6a). We treated the cells with l-serine in the absence or presence of specific LDH inhibitor or siRNA for LDH. Figure 6b shows that l-serine increased the intracellular NAD+ level, which was not observed in the case of sodium oxamate co-treatment. Intracellular NADH was not changed. Similarly, decreased PGC-1α acetylation and increased expression of Cycs were reversed by the inhibition of LDH (Fig. 6d, f). The same was true when we transfected the cells with siRNA directed for LDH (Fig. 6c, e, g). Taken together, l-serine effect on the SIRT1 activity is mediated by the production of NAD+ in the course of l-serine metabolism to lactate.

a C2C12 myotubes were treated with 5 mg/mL l-serine and cellular lactate was measured. b, c To inhibit LDH activity, 10 μM sodium oxamate (Na+ oxamate) or LDH siRNA was used. After 24-h l-serine treatment, intracellular NAD+ and NADH were measured. d, e PGC-1α levels were checked on acetyl-lysine immunoprecipitates (IP). f, g Total RNA was extracted from myotubes, and Cycs mRNA level was analyzed by qRT-PCR. Values are expressed as mean ± SD of three independent experiments. Means without a common letter differ; p < 0.05. Significance (*p < 0.05, **p < 0.01, ***p < 0.001) relative to the vehicle treatment; significance (#p < 0.05, ###p < 0.001) relative to the l-serine treated group using one-way ANOVA with Tukey’s post-test
Discussion
High protein intake exerts beneficial metabolic effects on the body weight, glucose homeostasis and intrahepatic lipid metabolism (Tremblay et al. 2007; Bortolotti et al. 2009). The mechanisms by which the proteins show such beneficial effects are not limited to their role as a nutrient for de novo protein synthesis but may be related to the function of several amino acids as signaling molecules regulating various metabolic pathways. For example, we reported that l-serine replenishment in acute and chronic ethanol-fed mice and rats decreased hepatic lipid accumulation reversed fatty liver (Sim et al. 2015). For the mechanism of anti-steatotic effects of l-serine, we reported that l-serine reduced intracellular homocysteine concentrations by inhibiting cellular homocysteine uptake and by accelerating l-serine-dependent homocysteine metabolism and thus suppressed homocysteine-induced endoplasmic reticulum (ER) stress in our previous study (Sim et al. 2016). Resolution of ER stress is required but not sufficient for the attenuation of fatty liver (Kandel-Kfir et al. 2015). Moreover, exposure of cells to ethanol alone had little effect on the expression of markers for ER stress but enhanced the expression of ER stress markers when induced by tunicamycin or palmitic acid (Chen et al. 2008; Yi et al. 2015). Therefore, we hypothesized that there would be additional mechanisms of l-serine for the positive effects in the abnormal lipid metabolism. We investigated whether the beneficial effects of l-serine on several pathological conditions of metabolic syndrome are related to the SIRT1 activity. We found that l-serine increased cellular NAD+ content and thus activated SIRT1 in C2C12 myotubes. l-Serine-induced SIRT1 activation led to the increase in the expression of the genes associated with mitochondrial biogenesis and of the mitochondrial mass accompanied with increased ATP production, OCR, and fatty acid oxidation, while SIRT1 inhibition completely reversed the effect of l-serine, but did not affect mitochondrial biogenesis or ATP levels. In general, the basal level of its acetylation status or its transcriptional activity is not changed even when SIRT1 is blocked (Gerhart-Hines et al. 2011; Cantó et al. 2009; Singh et al. 2013). Because of sustained transcriptional activity of PGC-1α, basal levels of mitochondrial biogenesis can be occurred and these results were reported by Bai et al. (2011) and Iwabu et al. (2010). Moreover, l-serine reversed the insulin resistance induced by palmitic acid in C2C12 cells.
Our hypothesis was based on the idea that supplementation of l-serine could increase intracellular NAD+ level due to its unique metabolism. First, l-serine is an amino acid participating in the biosynthesis of nucleotides, amino acids and sphingolipids. It is also catabolized into pyruvate producing glucose by gluconeogenesis or acetyl-CoA which enters TCA cycle to produce ATP or lactate by LDH. In the course of pyruvate reduction to lactate, NADH is oxidized to NAD+. Second, l-serine is a nonessential amino acid and synthesized in the body. There are two major routes of l-serine biosynthesis. The one is the conversion from glycine by serine hydroxymethyltransferase. The other one starts with the oxidation of 3-phosphoglycerate, an intermediate metabolite of glycolysis, by phosphoglycerate dehydrogenase (PHGDH) using NAD+ as a cofactor. l-Serine is then synthesized by subsequent transamination and hydrolysis. PHGDH is allosterically regulated by its product, l-serine (Mattaini et al. 2016). Therefore, addition of l-serine diverts the metabolic flow of 3-phosphoglycerate to pyruvate rather than to l-serine preventing cellular NAD+ pool from exhaustion. In muscle fibers, pyruvate is reduced to lactate to satisfy cellular energy demand. Our cell studies were conducted in aerobic condition, and it is true that anaerobic condition ensues lactate production and accumulation. However, a well-oxygenated cell also can produce lactate. Gladden (2004) reported that net lactate production and efflux from cells can occur under aerobic conditions. The equilibrium constant of lactate dehydrogenase reaction is strongly in favor of lactate (1.62 × 1011 M−1) (Suchankova et al. 2009). Thus, l-serine can be converted to lactate by LDH in C2C12 cells and NAD+ is produced in this step, which is essential for SIRT1 activity. l-Serine exerts very diverse effects in a physiological setting. For example, we reported that l-serine decreases systemic and hepatic homocysteine level that has adverse health effects with regard to the metabolic syndrome in our previous study (Sim et al. 2015). l-Serine also prevented high-fat diet-induced oxidative stress by activating AMP-activated protein kinase (AMPK) and epigenetically modulating the expression of glutathione synthesis-related genes (Zhou et al. 2018). Collectively, these data support our hypothesis that l-serine supplementation in a tolerable dose could exert beneficial health effects.
Theoretically, any amino acid that is metabolized to pyruvate could activate SIRT1. However, incubation of C2C12 cells with alanine, glycine, tryptophan, or cysteine failed to induce SIRT1 activation. Unlike l-serine and alanine, other three amino acids are metabolized to pyruvate through more complex steps. More importantly, serine is the only amino acid that activates pyruvate kinase M2 (PKM2) responsible for the synthesis of pyruvate in glycolysis (Chaneton et al. 2012). Therefore, we can conclude that l-serine specifically activates SIRT1 in our experimental conditions.
Wilson et al. (2007) reported that pyruvate induces mitochondrial biogenesis in a PGC-1α-independent manner, and Philp et al. (2010) showed the suppressing effect of pyruvate on PGC-1α expression. But there are some critical differences between two studies and our paper in terms of the experimental conditions. The two studies treat the cells with pyruvate for a longer period (72 h) while we used l-serine for a relatively shorter period (24 h). The time factor may important considering the paper that only acute treatment of pyruvate activated SIRT1 (Suchankova et al. 2009).
Several amino acids are known to modulate SIRT1 activity by diverse mechanisms. When an essential amino acid leucine was given to C57BL/6 mice, expression of SIRT1 protein and mRNA was increased. Addition of leucine to high fat diet correlated with increased expression of SIRT1 and nicotinamide phosphoribosyltransferase (NAMPT) (Li et al. 2012). NAMPT is an enzyme catalyzing the rate limiting step in the mammalian salvage pathway of NAD+ synthesis and thus regulating SIRT1 function. NAMPT activity converts nicotinamide into nicotinamide mononucleotide (NMN) which is the ultimate precursor for the synthesis of NAD+. Leucine exerts also a direct effect on SIRT1 kinetics, lowering the activation energy for NAD+, thus mimicking the effects of calorie restriction, and enabling synergic effects of AMPK or SIRT1 activators (Bruckbauer and Zemel 2014). Although other amino acids such as tryptophan and taurine also have been reported to activate SIRT1 activity (Sun et al. 2014; Braidy et al. 2011), little is known about the mechanisms of the effects. In this study, we found for the first time that l-serine is a natural amino acid that activates SIRT1 through a metabolism-dependent increase in intracellular NAD+ content. Increasing cellular NAD+ content is of interest, given that the NAD+ levels decline with aging due to the reduced activity of NAMPT (Yoshino et al. 2011). Therefore, induction of NAMPT activity by exercise (Costford et al. 2010) or the use of NAD+ intermediates such as nicotinamide riboside or NMN would be efficient in enhancing NAD+ biosynthesis, and thus SIRT1 activity, in aged individuals (Yoshino et al. 2011; Cantó et al. 2012). Other reasons for the NAD+ decline in elderly are chronic inflammation and defects in central circadian function. Tumor necrosis factor-α and oxidative stress reduce NAMPT activity and NAD+ level in hepatocytes (Yoshino et al. 2011). Moreover, one of the key target genes of the circadian transcription factor BMAL and CLOCK is Nampt encoding NAMPT. As the activity of the circadian machinery becomes defective with aging, negative regulation of NAMPT expression and NAD+ level is evident (Chang and Guarente 2013). For these reasons, the use of l-serine can be beneficial not only for the treatment of metabolic syndrome but also to the elderly persons for the purpose of increasing SIRT1 activity.
In summary, we proved that l-serine significantly increased SIRT1 activity in C2C12 myotubes by increasing intracellular NAD+ content. Our data interestingly revealed that the effects of l-serine on SIRT1 activity augmented mitochondrial mass and function as well as cellular insulin resistance induced by fatty acid overloading. l-Serine effect on cellular NAD+ increase is dependent on the l-serine metabolism to pyruvate that is subsequently converted to lactate by LDH. The data from our study support the idea that the natural amino acid l-serine can be safely used to increase cellular NAD+ content and thus SIRT1 activity in cases where the NAD+ biosynthetic pathway is compromised.
Change history
20 May 2019
Abstract
The original version of this article unfortunately contained a mistake in the article title.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DPBS:
-
Dulbecco’s phosphate buffered saline
- ER:
-
Endoplasmic reticulum
- GLUT4:
-
Glucose transporter type 4
- IP:
-
Immunoprecipitation
- LDH:
-
Lactate dehydrogenase
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NMN:
-
Nicotinamide mononucleotide
- NAMPT:
-
Nicotinamide phosphoribosyltransferase
- OCR:
-
Oxygen consumption rate
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PHGDH:
-
Phosphoglycerate dehydrogenase
- PKM2:
-
Pyruvate kinase M2
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- SIRT1:
-
Silent information regulator 1
References
Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–8.
Beaudoin MS, Snook LA, Arkell AM, Simpson JA, Holloway GP, Wright DC. Resveratrol supplementation improves white adipose tissue function in a depot-specific manner in Zucker diabetic fatty rats. Am J Phys Regul Integr Comp Phys. 2013;305:R542–51.
Bortolotti M, Kreis R, Debard C, Cariou B, Faeh D, Chetiveaux M, et al. High protein intake reduces intrahepatocellular lipid deposition in humans. Am J Clin Nutr. 2009;90:1002–10.
Braidy N, Guillemin GJ, Grant R. Effects of kynurenine pathway inhibition on NAD metabolism and cell viability in human primary astrocytes and neurons. Int J Tryptophan Res. 2011;4:29–37.
Bruckbauer A, Zemel MB. Synergistic effects of polyphenols and methylxanthines with leucine on AMPK/Sirtuin-mediated metabolism in muscle cells and adipocytes. PLoS One. 2014;9:e89166.
Burgner JW 2nd, Ray WJ Jr. On the origin of the lactate dehydrogenase induced rate effect. Biochemistry. 1984;23:3636–48.
Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–60.
Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–47.
Chaneton B, Hillmann P, Zheng L, Martin ACL, Maddocks ODK, Chokkathukalam A, et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012;491:458–62.
Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013;153:1448–60.
Chen G, Ma C, Bower KA, Shi X, Ke Z, Luo J. Ethanol promotes endoplasmic reticulum stress-induced neuronal death: involvement of oxidative stress. J Neurosci Res. 2008;86:937–46.
Costford SR, Bajpeyi S, Pasarica M, Albarado DC, Thomas SC, Xie H, et al. Skeletal muscle NAMPT is induced by exercise in humans. Am J Physiol Endocrinol Metab. 2010;298:E117–26.
Fröjdö S, Durand C, Molin L, Carey AL, El-Osta A, et al. Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by SIRT1. Mol Cell Endocrinol. 2011;335:166–76.
Fu WJ, Haynes TE, Kohli R, Hu J, Shi W, Spencer TE, et al. Dietary L-arginine supplementation reduces fat mass in Zucker diabetic fatty rats. J Nutr. 2005;135:714–21.
Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–23.
Gerhart-Hines Z, Dominy JE Jr, Blättler SM, Jedrychowski MP, Banks AS, Lim JH, et al. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol Cell. 2011;44:851–63.
Gladden LB. Lactate metabolism during exercise. In: Poortmans JR, editor. Principles of exercise biochemistry. 3rd ed. Basel: Karger Press; 2004. p. 152–96.
Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464:1313–9.
Kandel-Kfir M, Almog T, Shaish A, Shlomai G, Anafi L, Avivi C, et al. Interleukin-1α deficiency attenuates endoplasmic reticulum stress-induced liver damage and CHOP expression in mice. J Hepatol. 2015;63:926–33.
Li H, Xu M, Lee J, He C, Xie Z. Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Am J Physiol Endocrinol Metab. 2012;303:E1234–44.
Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology. 2014;146:539–49.
Li HB, Yang YRY, Mo ZJ, Ding Y, Jiang WJ. Silibinin improves palmitate-induced insulin resistance in C2C12 myotubes by attenuating IRS-1/PI3K/Akt pathway inhibition. Braz J Med Biol Res. 2015;48:440–6.
Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. J Cell Biol. 2016;214:249–57.
Opara EC, Petro A, Tevrizian A, Feinglosk MN, Surwit RS. L-glutamine supplementation of a high fat diet reduces body weight and attenuates hyperglycemia and hyperinsulinemia in C57BL/6J mice. J Nutr. 1996;126:273–9.
Philp A, Perez-Schindler J, Green C, Hamilton DL, Baar K. Pyruvate suppresses PGC1alpha expression and substrate utilization despite increased respiratory chain content in C2C12 myotubes. Am J Phys Cell Phys. 2010;299:C240–50.
Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–90.
Sim WC, Yin HQ, Choi HS, Choi YJ, Kwak HC, Kim SK, et al. L-serine supplementation attenuates alcoholic fatty liver by enhancing homocysteine metabolism in mice and rats. J Nutr. 2015;145:260–7.
Sim WC, Han I, Lee W, Choi YJ, Lee KY, Kim DG, et al. Inhibition of homocysteine-induced endoplasmic reticulum stress and endothelial cell damage by l-serine and glycine. Toxicol in Vitro. 2016;34:138–45.
Singh BK, Sinha RA, Zhou J, Xie SY, You SH, Gauthier K, et al. FoxO1 deacetylation regulates thyroid hormone-induced transcription of key hepatic gluconeogenic genes. J Biol Chem. 2013;288:30365–72.
Suchankova G, Nelson LE, Gerhart-Hines Z, Kelly M, Gauthier MS, Saha AK, et al. Concurrent regulation of AMP-activated protein kinase and SIRT1 in mammalian cells. Biochem Biophys Res Commun. 2009;378:836–41.
Sun X, Zemel MB. Leucine modulation of mitochondrial mass and oxygen consumption in skeletal muscle cells and adipocytes. Nutr Metab (Lond). 2009;6:26. https://doi.org/10.1186/1743-7075-6-26.
Sun Q, Hu H, Wang W, Jin H, Feng G, Jia N. Taurine attenuates amyloid β 1-42-induced mitochondrial dysfunction by activating of SIRT1 in SK-N-SH cells. Biochem Biophys Res Commun. 2014;447:485–9.
Timmers S, Konings E, Bilet L, Houtkooper RH, van de Weijer T, Goossens GH, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14:612–22.
Tremblay F, Lavigne C, Jacques H, Marette A. Role of dietary proteins and amino acids in the pathogenesis of insulin resistance. Annu Rev Nutr. 2007;27:293–310.
Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, et al. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest. 2011;121:4470–90.
Wilson L, Yang Q, Szustakowski JD, Gullicksen PS, Halse R. Pyruvate induces mitochondrial biogenesis by a PGC-1 alpha-independent mechanism. Am J Phys Cell Phys. 2007;292:C1599–605.
Yi HW, Ma YX, Wang XN, Wang CF, Lu J, et al. Ethanol promotes saturated fatty acid-induced hepatoxicity through endoplasmic reticulum (ER) stress response. Chin J Nat Med. 2015;13:250–6.
Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–36.
Zhou X, He L, Zuo S, Zhang Y, Wan D, Long C, et al. Serine prevented high-fat diet-induced oxidative stress by activating AMPK and epigenetically modulating the expression of glutathione synthesis-related genes. Biochim Biophys Acta. 2018;1864:488–98.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2017R1A2B4003179) and a grant (16173MFDS009) from Ministry of Food and Drug Safety in 2017.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplemental Table 1
(DOCX 15 kb)
Supplemental Figure 1
(PPTX 213 kb)
Supplemental Figure 2
(PPTX 126 kb)
Supplemental Figure 3
(PPTX 295 kb)
Rights and permissions
About this article

Cite this article
Sim, WC., Kim, D.G., Lee, W. et al. Activation of SIRT1 by l-serine increases fatty acid oxidation and reverses insulin resistance in C2C12 myotubes (l-serine activates SIRT1 in C2C12 myotubes). Cell Biol Toxicol 35, 457–470 (2019). https://doi.org/10.1007/s10565-019-09463-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-019-09463-x