
Abstract
Corynebacterium glutamicum has a long and successful history in the biotechnological production of l-lysine. Besides the adjustment of metabolic pathways, intracellular and extracellular transport systems are critical for the cellular metabolism of l-lysine or its by-products. Here, three amino acid transmembrane transporters, namely, GluE, BrnE/BrnF, and LysP, which are widely present in C. glutamicum strains, were each investigated by gene knockout. In comparison with that in the wild-type strain, the yield of l-lysine increased by 9.0%, 12.3%, and 10.0% after the deletion of the gluE, brnE/brnF, and lysP genes, respectively, in C. glutamicum 23,604. Moreover, the amount of by-product amino acids decreased significantly when the gluE and brnE/brnF genes were deleted. It was also demonstrated that there was no effect on the growth of the strain when the gluE or lysP gene was deleted, whereas the biomass of C. glutamicum WL1702 (ΔbrnE/ΔbrnF) in the fermentation medium was significantly reduced in comparison with that of the wild type. These results also provide useful information for enhancing the production of l-lysine or other amino acids by C. glutamicum.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
l-Lysine is an essential amino acid for mammals and can promote the development of mammals and strengthen the immune system in humans. l-Lysine has significant value in feed additives (Katheline 2013) and dietary supplements, as well as in cosmetics and pharmaceuticals (Koffas and Stephanopoulos 2005). With the widening of the range of applications of lysine, the demand for lysine is rapidly increasing every year. The fermentative production of lysine by Corynebacterium glutamicum was developed in 1958 by Kyowa Hakko Kogyo Co., Ltd. (Ikeda 2016). Corynebacterium glutamicum is an aerobic nonpathogenic Gram-positive soil bacterium. As an excellent producer, it is utilized for the industrial production of amino acids, including l-glutamate and l-lysine (Shukuo et al. 2004; Kalinowski et al. 2003). Moreover, the main producers of l-lysine are almost all auxotrophic engineered strains of C. glutamicum, particularly in terms of changes in metabolic flux (Wittmann and Becker 2007).
In the fermentative production of lysine, many by-products, such as glutamic acid, methionine, leucine, isoleucine, and valine, are formed. Reducing the accumulation of by-products is an effective approach to reducing production costs, because the downstream purification and recovery process might thereby be simplified (Hou et al. 2012a). The influence of the transport of certain amino acid by-products in the cell will cause the concentration of the by-product in the cell to increase, thereby inhibiting the formation of the by-product (Chen et al. 2015). Furthermore, decreasing the formation of certain by-products may improve the yield of the target product, because the carbon source might thus be utilized more exclusively for the biosynthesis of the target product (Chen et al. 2015). The extracellular export of amino acids from C. glutamicum is very important, and the proteins involved in the export of amino acids have been thoroughly characterized at the biochemical and genetic level (Marin and Krämer 2007). For example, intracellular l-methionine, l-leucine, l-isoleucine, and l-valine can be exported from the cell via a two-component export system consisting of the proteins BrnE and BrnF, which are encoded by the genes brnE and brnF, respectively (Kennerknecht et al. 2002; Lange et al. 2012). Some experiments have indicated that overexpressing the export system BrnE/BrnF could increase the production of l-methionine and l-isoleucine by C. glutamicum (Nicole et al. 2002; Qin et al. 2014). Furthermore, a mutant in which both the above genes were deleted no longer exported l-isoleucine. Moreover, the NCgl1221 gene, which encodes a mechanosensitive channel homolog, is involved in the mechanism of secretion of l-glutamate (Nakamura et al. 2007). The expression, localization, construction, and functions of NCgl1221 have been identified and reported. This gene has also been termed gluE (Yamashita et al. 2013; Yao et al. 2009; Yoshitaka et al. 2012). The amino acid transporter LysE is responsible for the transport of l-lysine and l-arginine in C. glutamicum, and the expression of lysE increases the production of lysine (Vrljic et al. 1997). In this study, the two genes of gluE and brnE/brnF were knocked out by the principle of two homologous single exchanges to inactivate the extracellular transporters GluE and BrnE/BrnF, thereby exploring the effect on the production of l-lysine. Besides, in C. glutamicum the uptake of l-lysine is catalyzed by a lysine/alanine exchange carrier encoded by the lysP gene (Bröer and Krämer 1990). It has also been demonstrated that the excretion of lysine is independent of the lysine uptake system (Seep-Feldhaus et al. 2010).
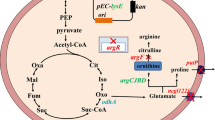
The functions and transport mechanisms of the three abovementioned amino acid transporters have been extensively studied. The effects of inactivating these three transporters on the respective target amino acids have also been reported (Marin and Krämer 2007). The transfer of intracellular glutamate to the outside of the cell is controlled by the NCgl1221 protein encoded by the gluE gene, which is a mechanically sensitive channel homologue and an important glutamate transporter (Yao et al. 2009). The intracellular methionine and branched amino acids are transported to the outside of the cell through a transport system composed of BrnE and BrnF (Qin et al. 2014). In addition, reducing the accumulation of by-products may increase the yield of the target product, because the carbon source will be more concentrated for the synthesis of the target product (Chen et al. 2015). However, there has thus far been no report on the effect of inactivating these three transporters in C. glutamicum on the yield of l-lysine. Because the transport systems of bacterial cells are usually related to the excretion as well as the reuptake of products, which makes the engineering of transporters a useful strategy for the improvement of strains, we investigated the effect of inactivating the three abovementioned transmembrane transporters on the production of extracellular l-lysine (Fig. 1). We hypothesized that deleting the gluE gene would not affect the growth of the strain and, because the concentration of extracellular l-glutamate was not significantly decreased, the carbon source could not be employed to accumulate intracellular l-glutamate and hence could not be utilized more exclusively for the production of l-lysine.

Pathway engineering for production of l-lysine in recombinant C. glutamicum
Materials and methods
Strains, plasmids, primers, and culture conditions
All the strains, plasmids, and primers used in this study are listed in Table 1. The nonreplicable integration plasmid pK19mobsacB was used to construct C. glutamicum deletion vectors. Escherichia coli DH5 cells were used as hosts for gene cloning and were routinely grown in Luria–Bertani (LB) medium at 37 °C. Corynebacterium glutamicum 23604 was used as the parent strain for generating mutants and was cultivated aerobically at 30 °C with LBG medium (LB medium supplemented with 5 g L−1 glucose). The fermentation medium used for production of l-lysine by C. glutamicum consisted of (per liter): 20 g glucose, 30 g corn pulp, 20 g (NH4)2SO4, 10 g CH3COONa, 5 g urea, 1.34 g l-alanine, 2 g KH2PO4, 1.35 g MgSO4·7H2O, 0.02 g FeSO4·7H2O, 0.04 g MnSO4·H2O, 0.008 g nicotinamide, 0.001 g biotin, 0.04 g leucine. The basic medium is used for evaluate the synthetic ability of all amino acids by C. glutamicum consisted of (per liter): 5 g glucose, 1 g (NH4)2SO4, 1 g sodium citrate, 0.2 g MgSO4·7H2O, 4 g K2HPO4, 6 g KH2PO4 and adjusted to pH 7.2 with NaOH. When appropriate, kanamycin was added at a final concentration of 50 mg L−1 (E. coli) or 25 mg L−1 (C. glutamicum).
Construction of deletion vectors
The genome was extracted from C. glutamicum 23604 using an Ezup Column Bacteria Genomic DNA Purification Kit (Sangon Biotech, Shanghai, China) according to the manufacturer’s instructions. The polymerase chain reaction (PCR) primers used in this study are listed in Supplementary Table 1. In the first round of PCR, the chromosomal DNA of C. glutamicum 23604 was used as the template, and the primer pairs A1/A2, B1/B2, and C1/C2 were used to amplify the upstream regions of the gluE, brnE/brnF, and lysP genes, respectively. The primer pairs A3/A4, B3/B4, and C3/C4 were used to amplify the downstream regions of the respective genes. For the above six fragments, the conditions used for PCR amplification were as follows: pre-degeneration at 95 ℃ for 5 min, then 30 cycles of degeneration at 94 ℃ for 30 s, annealing at 57 ℃ for 30 s, and pre-extension at 72 ℃ for 90 s, followed by extension at 72 ℃ for 10 min and holding at 4 ℃. Then, gel purification was used to purify the PCR products using a gel extraction kit (Sangon Biotech, Shanghai, China). In the second round of overlap PCR, the products of the first round of PCR were used as templates in a molar ratio of 1:1, and A1/A4, B1/B4, and C1/C4 were used as primer pairs. The conditions used for the second PCR amplification were as follows: (1) pre-degeneration at 95 ℃ for 5 min, then five cycles of degeneration at 94 ℃ for 30 s, annealing at 57 ℃ for 30 s, and pre-extension at 72 ℃ for 90 s, followed by extension at 72 ℃ for 2 min; and (2) pre-degeneration at 95 ℃ for 5 min, then 30 cycles of degeneration at 94 ℃ for 30 s, annealing at 56 ℃ for 30 s, and pre-extension at 72 ℃ for 2.5 min, followed by extension at 72 ℃ for 10 min and holding at 4 ℃. Fragments of a fusion gene, namely, a 1205 bp fragment of gluE, a 1130 bp fragment of brnE/brnF, and a 1134 bp fragment of lysP, were thus obtained. The plasmid pK19mobsacB was digested using the restriction enzyme EcoRI (Thermo Fisher Scientific, United States). Via seamless cloning using a One Step Cloning Kit (Vazyme Biotech, Nanjing, China), the fragments obtained by overlap PCR were inserted into the plasmid pK19mobsacB, and then the recombinant plasmids were transfected into competent E. coli cells. Finally, colony PCR was used to screen for positive clones, and the identity of the fusion fragments was confirmed by sequencing.
Transformation and isolation of engineered strains
Competent C. glutamicum 23604 cells were prepared in advance. A single colony of C. glutamicum 23604 was inoculated into liquid LBG medium and cultured overnight. Then a 2% inoculant was inoculated into neutral complex medium (NCM) containing Tween 80 (0.1%) and glycine (3%) to give a value of the optical density at 600 nm (OD600) of 0.3. When the OD600 value reached 0.9, the cells were chilled on ice for 15 min and harvested by centrifugation at 4 ℃ and 4000 g for 10 min. Then the cell pellets were washed four times with 20 mL ice-cold 10% glycerol. The cells were resuspended in 0.2 mL 10% glycerol, and 80 μL competent cells were selected. Next, the three different recombinant plasmids were extracted and concentrated. An 8 μL aliquot of DNA was added to the competent cells, which were transferred to a 1 mm electroporation cuvette (Bio-Rad Laboratories, Hercules, California, United States), of which the parameters were set at 1.8 kV and 5 ms. In comparison with the study by Ruan et al. (2015), there was a slight difference in the composition of NCM. After electroporation was carried out, a heat shock was applied immediately for 6 min.
Two rounds of positive selection for homologous recombination were performed (Fig. 2). Kanamycin resistance was first used to select for the integration of the plasmid into the chromosome. Resistant clones were selected and tested by PCR analysis with the primer pairs A1/A4, B1/B4, or C1/C4 using the chromosomal DNA as a template. DNA fragments with lengths of 1205 and 2264 bp were obtained by the deletion of the gluE gene. Deletion of the brnE/brnF gene yielded fragments with lengths of 1130 and 2060 bp, and deletion of the lysP gene yielded fragments with lengths of 1128 and 1377 bp. Subsequently, clones that survived and grew in the presence of sucrose were selected, because it was assumed that they had lost the pK19mobsacB vector (Hou et al. 2012a). The second recombination resulted in either the desired deletion or the restoration of the wild-type characteristics. Clones selected using sucrose were tested by PCR analysis using A1/A4, B1/B4, or C1/C4 as primer pairs to identify clones that carried the desired deletion or had undergone allelic exchange. In the case of knockout of the gluE gene, only the 1205 bp PCR fragment was obtained. The 1130 and 1128 bp DNA fragments were amplified in the cases of knockout of the brnE/brnF and lysP genes, respectively.

Flowchart of construction of C. glutamicum engineered strains
Culture conditions used for l-lysine production
Three colonies of the engineered and wild-type strains were inoculated into liquid LBG medium and cultured overnight at 200 rpm and 30 ℃ in 200 mL flasks. After 12 h, 5% of each overnight culture was transferred to 100 mL fermentation medium. The remaining portions of the C. glutamicum WL1701 and C. glutamicum WL1702 strains, in which the gluE and brnE/brnF genes, respectively, were knocked out, were centrifuged and inoculated into the basic medium to give the same initial OD600 value. During the fermentation process, samples were taken from the fermentation medium and basic medium at intervals of 4 h to measure the OD600 value and concentrations of glucose and l-lysine. In the case of the C. glutamicum WL1701 strain, the yield of l-glutamate was measured. Each experiment was repeated three times, and the average values of the measurements were taken as the experimental results.
Analysis of growth, glucose consumption, and l-lysine production by engineered strains
The biomass concentration in a 100 μL aliquot of bacterial culture was measured by a photometer at 600 nm after an appropriate dilution or by gravimetric analysis, as described previously (Hou et al. 2012b). The supernatant of the culture broth obtained by centrifugation was used for the determination of the concentrations of glucose, l-lysine and l-glutamate, which were measured enzymatically using a biosensor (SBA-40C, Biology Institute of Shandong Academy of Sciences, Jinan, China; https://www.bio-sensor.org) with a standard process described in the instruction manual. An automatic amino acid analyzer (L-8900, Hitachi, Tokyo, Japan) was used to determine the compositions and contents of various amino acids in the fermentation broth and basic broth of the three strains of recombinant bacteria with a standard pipeline method described in the instruction manual.
UHPLC-QE-MS non-target metabolomics detection
100 μL of samples (the supernatant of the basic medium culture broth at 96 h) were transferred to an EP tube. After the addition of 400 μL of extract solution (acetonitrile: methanol = 1: 1, containing isotopically-labeled internal standard mixture), the samples were vortexed for 30 s, sonicated for 10 min in ice-water bath, and incubated for 1 h at – 40 ℃ to precipitate proteins. Then the sample was centrifuged at 12,000 rpm for 15 min at 4 ℃. The resulting supernatant was transferred to a fresh glass vial for LC/MS analysis.
LC–MS/MS analyses were performed using an UHPLC system (Vanquish, Thermo Fisher Scientific) with a UPLC BEH Amide column (2.1 mm × 100 mm, 1.7 μm) coupled to Q Exactive HFX mass spectrometer (Orbitrap MS, Thermo). The mobile phase consisted of 25 mmol/L ammonium acetate and 25 ammonia hydroxide in water (pH 9.75) (A) and acetonitrile (B). The analysis was carried with elution gradient as follows: 0–0.5 min, 95% B; 0.5–7.0 min, 95%—65% B; 7.0–8.0 min, 65%—40% B; 8.0–9.0 min, 40% B; 9.0–9.1 min, 40%—95% B; 9.1–12.0 min, 95% B. The column temperature was 30 ℃. The auto-sampler temperature was 4 ℃, and the injection volume was 2 μL.
The QE HFX mass spectrometer was used for its ability to acquire MS/MS spectra on information-dependent acquisition (IDA) mode in the control of the acquisition software (Xcalibur, Thermo). In this mode, the acquisition software continuously evaluates the full scan MS spectrum. The ESI source conditions were set as following: sheath gas flow rate as 50 Arb, Aux gas flow rate as 10 Arb, capillary temperature 320 ℃, full MS resolution as 60,000, MS/MS resolution as 7500, collision energy as 10/30/60 in NCE mode, spray Voltage as 3.5 kV (positive) or − 3.2 kV (negative), respectively.
The raw data were converted to the mzXML format using ProteoWizard and processed with an in-house program, which was developed using R and based on XCMS, for peak detection, extraction, alignment, and integration by BiotreeCo., Ltd. Then an in-house MS2 database (BiotreeDB) was applied in metabolite annotation. The cutoff for annotation was set at 0.3.
Statistical analysis
Statistical analyses were performed using GraphPad Prism4 software. A two-tailed Student’s t test was used to test statistical significance. The symbol (*) denotes a p value of at least < 0.05. The symbol (**) denotes a p value of at least < 0.01. All data were obtained from three independent experiments performed in triplicate, and the results are presented as mean and standard error of the mean.
Results and discussion
Effects of gluE gene knockout on l-lysine production
Using the pK19mobsacB plasmid to construct knockout vectors, we generated strains derived from the wild-type strain C. glutamicum 23604 with three different genic deletions in their genomes. The resulting recombinant strains were denoted as C. glutamicum WL1701, C. glutamicum WL1702, and C. glutamicum WL1703, respectively. To construct the strain C. glutamicum WL1701, we knocked out gluE by homologous recombination between the chromosomal gluE gene and a truncated gluE gene. The plasmid pK19mobsacB-ΔgluE, which incorporated the upstream and downstream regions of gluE, was introduced into wild-type C. glutamicum 23604. After two rounds of positive selection for homologous recombination and testing by PCR analysis, we obtained the gluE gene knockout strain denoted as C. glutamicum WL1701 (Supplementary Fig. 1). In this mutant, the gluE gene was completely inactivated by selecting the position of homologous arms in the genome. In comparison with wild-type C. glutamicum 23604, the yield of l-lysine increased by 9.0% after the deletion of the gluE gene (Fig. 3a, p < 0.01, Supplementary Fig. 6). However, whereas wild-type C. glutamicum 23604 secreted 0.907 g L−1 l-glutamate after fermentation for 96 h, the yield of l-glutamate secreted by C. glutamicum WL1701 was 0.775 g L−1 and was thus reduced by only 17% (Fig. 3b, p < 0.01 Supplementary Fig. 3). Moreover, after fermentation on the basic medium, it was found that the glucose consumption curves of the recombinant bacteria were basically identical to the control bacteria (Supplementary Fig. 3). While, it can be seen from Fig. 4 that the yield of l-glutamate from the recombinant bacteria after fermentation was 69.7% lower than that from the control bacteri a (Supplementary Fig. 3, p < 0.01, Supplementary Fig. 3). However, these results show that it is different from the previous research by Nakamura et al. (2007), they observed a significant drop in glutamate production after the entire gluE gene was deleted: the yield was 7.1 times lower than the wild type. A possible reason for our results is that the wild-type strain C. glutamicum 23604 is a glutamate-secreting strain. Even though we deleted the gluE gene completely, the mutant also secreted l-glutamate. Furthermore, from the growth and glucose consumption curves (Fig. 3c, d), we can conclude that the wild-type strain and mutant exhibited almost the same trends in growth and glucose consumption.

taken from the fermentation medium every 4 h to measure the l-glutamate, l-lysine, glucose and OD600 production of C. glutamicum 23604 and C. glutamicumWL1701, symbols: filled square strain C. glutamicum 23604, hollow triangle strain C. glutamicum WL1701. The standard errors are shown as bars
Production of l-lysine by engineered strains of C. glutamicum WL1701 at fermentation medium. Samples were

taken from the fermentation medium every 4 h to measure the l-glutamate concentration of C. glutamicum 23604 and C. glutamicum WL1702, symbols: filled square strain C. glutamicum 23604, filled triangle strain C. glutamicum WL1702. The standard errors are shown as bars
Production of l-glutamate by engineered strains of C. glutamicum WL1701 at basic medium. Samples were
Effects of brnE/brnF gene knockout on l-lysine production
To obtain the brnE/brnF gene knockout strain C. glutamicum WL1702, we carried out two rounds of screening and testing by PCR analysis (Supplementary Fig. 4). In the fermentation medium, we found that the biomass and glucose consumption of the mutant significantly decreased in comparison with that of the wild-type strain after the same stable growth period (Fig. 5a). However, the yield of l-lysine was 12.3% higher than that from the wild-type C. glutamicum 23604 (Fig. 5b, p < 0.01 Supplementary Fig. 6), and the residual glucose of C. glutamicum WL1702 was close to zero earlier than that of the wild type during the fermentation (Fig. 5c). From this result, we can conclude that after the deletion of brnE/brnF the yield of l-lysine produced by C. glutamicum WL1702 was higher than that produced by the wild type.

taken from the fermentation medium every 4 h to measure the l-lysine, glucose and OD600 of C. glutamicum 23604 and C. glutamicum WL1702, symbols: filled square strain C. glutamicum 23604, filled triangle strain C. glutamicum WL1702. The standard errors are shown as bars
Production of l-lysine by engineered strains of C. glutamicum WL1702 at fermentation medium. Samples were
After fermentation on the basic medium and amino acid analysis, we found that the yield of l-lysine from C. glutamicum WL1702 increased by 14.2% in comparison with that from C. glutamicum 23604 (Fig. 6, p < 0.05, Supplementary Fig. 6). The yields of l-valine, l-leucine, and l-isoleucine from the wild type and C. glutamicum WL1702 were all 0 mg L−1. However, the yield of l-methionine from C. glutamicum WL1702 was 0.331 mg L−1, which represented a decrease of 63.7% in comparison with C. glutamicum 23604. The results showed that after the deletion of brnE/brnF the strain still had the ability to transport l-methionine. This is consistent with the findings of Christian et al. (2005) and indicates the presence of more than one additional l-methionine export system.

taken from the fermentation medium every 4 h to measure the l-lysine concentration of C. glutamicum 23604 and C. glutamicum WL1702, symbols: filled square strain C. glutamicum 23604, filled triangle strain C. glutamicum WL1702. The standard errors are shown as bars
Production of l-lysine by engineered strains of C. glutamicum WL1702 at basic medium. Samples were
Effects of lysP gene knockout on l-lysine production
According to the methods used for screening C. glutamicum WL1701, a colony of C. glutamicum WL1703 was successfully obtained (Supplementary Fig. 5). After fermentation for 96 h, the yield of l-lysine was 4.84 g L−1. In comparison with that from the wild-type C. glutamicum 23604, the yield of l-lysine increased by 10.0% (Fig. 7a, p < 0.01 Supplementary Fig. 6). The growth and glucose consumption curves of the mutant were similar to those of the wild type (Fig. 7b, c). These results demonstrated that lysP knockout had no significant influence on the growth of the strain. From data obtained using the automatic amino acid analyzer (Table 2), we found that, although the yield of l-lysine increased, the yields and types of other amino acids were basically identical during in the fermentation with fermentation medium.

taken from the fermentation medium every 4 h to measure the l-lysine, glucose and OD600 production of C. glutamicum 23604 and C. glutamicum WL1703, symbols: hollow square strain C. glutamicum 23604, filled triangle strain C. glutamicum WL1703. The standard errors are shown as bars
Production of l-lysine by engineered strains of C. glutamicum WL1703 at fermentation medium. During the fermentation process, samples were
Overview of the C. glutamicum metabolomics data after gene knockout
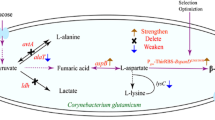
In our metabolomics results, 28 core metabolites were identified and mapped to metabolic pathways in C. glutamicum (Fig. 8). These metabolites included amino acids, organic acids, carbohydrates, lipids and lipid-like molecules, purines/pyrimidines and other metabolites. In the fermentation medium, we found that some core metabolites of the mutant changed in comparison with that of the wild-type strain after fermentation for 96 h with the basic medium. The content of l-aspartic acid, a precursor of l-lysine, were increased after the deletion of the gluE, brnE/brnF and lysP genes, respectively, which may lead to the increase in l-lysine production. After the deletion of the gluE gene, the content of citrate and isocitrate (the precursors of l-lysine) were increased, it also led to the increase of some amino acids (such as l-tyrosine, l-alanine, l-isoleucine and l-methionine) production in other metabolic pathways. Meanwhile, the deletion of brnE/brnF and lysP genes may also affect TCA cycle. More dramatically, the deletion of the gluE, brnE/brnF and lysP genes also affects fatty acids and nucleotides metabolism, especially the increase of oleic acid, capric acid and stearic acid production.

Metabolic pathway analysis results in C. glutamicum after gene knocke out. Overview of the metabolic changes in C. glutamicum after gluE, lysP or brnE/brnF genes were knocked out. The left, middle and right boxes respectively indicate that the levels of metabolites after knocking out gluE, lysP or brnE/brnF genes have changed compared with the original strain
Effects of double and triple knockout on l-lysine production
According to the methods used for screening C. glutamicum WL1701, a colony of C. glutamicum WL1704, C. glutamicum WL1706 was successfully obtained (Supplementary Fig. 7a–d). After the plasmid pK19mobsacB-lysP was electroporated into C. glutamicum WL1704, according to the method used to screen C. glutamicum WL1701, a colony of C. glutamicum WL1705 was successfully obtained (Supplementary Fig. 7e, f). The growth status and glucose consumption of double deletion bacteria C. glutamicum WL1704 and C. glutamicum WL1706 were basically the same as those of the control bacteria. The l-lysine production of the two double deletion bacteria was increased by 3% and 2% respectively compared with the wild-type C. glutamicum 23604 (Supplementary Fig. 8). From the above results, it can be concluded that the l-lysine production of double-deleted bacteria is not as high as that of single-deleted bacteria. The simultaneous deletion of two genes in one strain did not increase the production of l-lysine. The three-deleted bacteria C. glutamicum WL1705 grew more slowly than the control bacteria, and the biomass in the stable phase was not as high as that of the control bacteria (Supplementary Fig. 9). The glucose consumption curves of the triple deletion bacteria and the control bacteria are basically the same (Supplementary Fig. 9). The lysine production of the triple deletion bacteria was 10.8% lower than that of the wild-type C. glutamicum 23604 (Supplementary Fig. 9). After the two membrane transporters were simultaneously inactivated, the production of l-lysine was not much different from the control bacteria, but there is a certain gap with the single deletion bacteria. The study also found that the simultaneous deletion of three genes will further reduce the production of lysine, the specific reasons need to be further analyzed in the future study.
In conclusion, we have studied the development of higher-yielding strains for the production of l-lysine and demonstrated the importance of amino acid transporters involved in l-lysine-related carbon flux pathways for increasing the yield of l-lysine. By knocking out the gluE, brnE/brnF, and lysP genes, we found that the three corresponding amino acid transporters have different effects on the production of l-lysine. The yield of l-lysine in the fermentation medium increased by 9.0%, 12.3%, and 10.0% after the deletion of gluE, brnE/brnF, and lysP, respectively, in C. glutamicum 23604. However, after the inactivation of gluE and brnE/brnF, the strain retained the ability to transport l-glutamate and l-methionine, which means that other l-glutamate and l-methionine transporters are present in C. glutamicum.
Abbreviations
- AcCoA:
-
Acetyl coenzyme A
- Ala:
-
l-Alanine
- Asn:
-
l-Asparagine
- Asp:
-
l-Aspartate
- AspSa:
-
Aspartate semialdehyde
- Chor:
-
Chorismate
- Cit:
-
Citrate
- Fum:
-
Fumaric acid
- G6P:
-
Glucose 6-phosphate
- GDL:
-
Gluconolactone
- Glc:
-
Glucose
- Gln:
-
l-Glutamine
- Glu:
-
l-Glutamate
- Gluc:
-
d-Gluconic acid
- Gluc6P:
-
6-Phospho-d-gluconate
- Glx:
-
Glyoxylate
- Gly:
-
l-Glycine
- Hcys:
-
Homocysteine
- His:
-
l-Histidine
- Hser:
-
Homoserine
- ICit:
-
Isocitrate
- Ile:
-
l-Isoleucine
- αKG:
-
α-Ketoglutarate
- Kval:
-
2-Ketovaline
- Leu:
-
l-Leucine
- Lys:
-
l-Lysine
- Mal:
-
Malic acid
- OA:
-
Oxaloacetic acid
- Met:
-
l-Methionine
- Orn:
-
Ornithine
- PEP:
-
Phosphoenolpyruvate
- PG3:
-
Glycerate 3-phosphate
- Phe:
-
l-Phenylalanine
- Pro:
-
l-Proline
- PRPP:
-
5-Phospho-d-ribosylpyrophosphate
- Pyr:
-
Pyruvate
- R5P:
-
Ribulose 5-phosphate
- Ser:
-
l-Serine
- Suc:
-
Succinate
- SucCoA:
-
Succinate coenzyme A
- Thr:
-
l-Threonine
- Trp:
-
l-Tryptophan
- Tyr:
-
l-Tyrosine
- Val:
-
l-Valine
References
Bröer S, Krämer R (1990) Lysine uptake and exchange in Corynebacterium glutamicum. J Bacteriol 172:7241–7248. https://doi.org/10.1128/jb.172.12.7241-7248.1990
Chen C, Li Y, Hu J, Dong X, Wang X (2015) Metabolic engineering of Corynebacterium glutamicum ATCC13869 for l-valine production. Metab Eng 29:66–75. https://doi.org/10.1016/j.ymben.2015.03.004
Christian TT, Dietrich D, Brigitte B, Andreas B, Reinhard KM (2005) Characterization of methionine export in Corynebacterium glutamicum. J Bacteriol 187:3786. https://doi.org/10.1128/JB.187.11.3786-3794.2005
Hou X, Ge X, Wu D, Qian H, Zhang W (2012a) Improvement of l-valine production at high temperature in Brevibacterium flavum by overexpressing ilvEBNrC genes. J Ind Microbiol Biotechnol 39:63–72. https://doi.org/10.1007/s10295-011-1000-1
Hou X, Zhang Y, Qian H, Zhang W (2012b) l-Valine production with minimization of by-products’ synthesis in Corynebacterium glutamicum and Brevibacterium flavum. Amino Acids 43:2301–2311. https://doi.org/10.1007/s00726-012-1308-9
Ikeda M (2017) Lysine fermentation: history and genome breeding. Adv Biochem Eng Biotechnol 159:73–102. https://doi.org/10.1007/10_2016_27
Jun N, Seiko H, Hisao I, Masaaki W (2007) Mutations of the Corynebacterium glutamicum NCgl1221 gene, encoding a mechanosensitive channel homolog, induce l-glutamic acid production. Appl Environ Microbiol 73:4491–4498. https://doi.org/10.1128/AEM.02446-06
Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A et al (2003) The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J Biotechnol 104(1–3):5–25. https://doi.org/10.1016/s0168-1656(03)00154-8
Katheline H (2013) Investigating the appropriate mode of expressing lysine requirement of fish through non-linear mixed model analysis and multilevel analysis. Br J Nutr 109:1013–1021. https://doi.org/10.1017/S0007114512002863
Kennerknecht N, Sahm H, Yen MR, Pátek M, Eggeling L (2002) Export of l-isoleucine from Corynebacterium glutamicum: a two-gene-encoded member of a new translocator family. J Bacteriol 184:3947–3956. https://doi.org/10.1128/JB.184.14.3947-3956.2002
Koffas M, Stephanopoulos G (2005) Strain improvement by metabolic engineering: lysine production as a case study for systems biology. Curr Opin Biotechnol 16:361–366. https://doi.org/10.1016/j.copbio.2005.04.010
Lange C, Mustafi N, Frunzke J, Kennerknecht N, Wessel M, Bott M, Wendisch VF (2012) Lrp of Corynebacterium glutamicum controls expression of the brnEF operon encoding the export system for l-methionine and branched-chain amino acids. J Biotechnol 158:231–241. https://doi.org/10.1016/j.jbiotec.2011.06.003
Marin K, Krämer R (2007) Amino acid transport systems in biotechnologically relevant bacteria. Amino Acid 5:289–325. https://doi.org/10.1007/7171_2006_069
Nakamura J, Hirano S, Ito H, Wachi M (2007) Mutations of the Corynebacterium glutamicum NCgl1221 gene, encoding a mechanosensitive channel homolog, induce l-glutamic acid production. Appl Environ Microbiol 73:4491–4498. https://doi.org/10.1128/AEM.02446-06
Nicole K, Hermann S, Ming-Ren Y, Miroslav P Jr, Saier MH, Lothar E (2002) Export of l-isoleucine from Corynebacterium glutamicum: a two-gene-encoded member of a new translocator family. J Bacteriol 184:3947–3956. https://doi.org/10.1128/JB.184.14.3947-3956.2002
Nji E, Li D, Doyle DA, Caffrey M (2014) Cloning, expression, purification, crystallization and preliminary X-ray diffraction of a lysine-specific permease from Pseudomonas aeruginosa. Acta Crystallogr A 70:1362–1367. https://doi.org/10.1107/S2053230X14017865
Qin T, Hu X, Hu J, Wang X (2014) Metabolic engineering of Corynebacterium glutamicum strain ATCC13032 to produce l-methionine. Biotechnol Appl Biochem 62(4):563–573. https://doi.org/10.1117/12.2061040
Ruan Y, Zhu L, Qi L (2015) Improving the electro-transformation efficiency of Corynebacterium glutamicum by weakening its cell wall and increasing the cytoplasmic membrane fluidity. Biotechnol Lett 37:2445–2452. https://doi.org/10.1007/s10529-015-1934-x
Seep-Feldhaus AH, Kalinowski J, Pühler A (2010) Molecular analysis of the Corynebacterium glutamicum lysl gene involved in lysine uptake. Mol Microbiol 5:2995–3005. https://doi.org/10.1111/j.1365-2958.1991.tb01859.x
Shukuo K, Shigezo U, Masakazu S (2004) Studies on the amino acid fermentation. Part 1. Production of l-glutamic acid by various microorganisms. J Gen Appl Microbiol 50:331–343. https://doi.org/10.1002/jgm.614
Vrljic M, Sahm H, Eggeling L (1997a) A new type of transporter with a new type of cellular function: l-lysine export from Corynebacterium glutamicum. Mol Microbiol 22:815–826. https://doi.org/10.1046/j.1365-2958.1996.01527.x
Wittmann C, Becker J (2007) The l-lysine story: from metabolic pathways to industrial production. Amino Acid. https://doi.org/10.1007/7171_2006_089
Yamashita C, Hashimoto KI, Kumagai K, Maeda T, Wachi M (2013) l-Glutamate secretion by the N-terminal domain of the Corynebacterium glutamicum NCgl1221 mechanosensitive channel. Biosci Biotechnol Biochem 77:1008–1013. https://doi.org/10.1271/bbb.120988
Yao W, Deng X, Liu M, Zheng P, Sun Z, Zhang Y (2009) Expression and localization of the Corynebacterium glutamicum NCgl1221 protein encoding an l-glutamic acid exporter. Microbiol Res 164:680–687. https://doi.org/10.1016/j.micres.2009.01.001
Yoshitaka N, Kenjiro Y, Hidetoshi I (2012) A gain-of-function mutation in gating of Corynebacterium glutamicum NCgl1221 causes constitutive glutamate secretion. Appl Environ Microbiol 78:5432–5434. https://doi.org/10.1128/AEM.01310-12
Acknowledgements
This work was supported by the National Science Foundation of China (31801527), Focus on Research and Development Plan in Shandong Province (2019JZZY011003, 2018YFJH0401, 2016CYJS07A01), Taishan industry leading talent (tscy20180103), Shandong Provincial Natural Science Foundation (ZR2016CB04), Major Program of National Natural Science Foundation of Shandong (ZR2017ZB0208) and Cultivation Project of Shandong Synthetic Biotechnology Innovation Center (sdsynbio-2018-PY-02).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Additional information
Communicated by S. Stuchlík.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiao, J., Wang, D., Wang, L. et al. Increasing l-lysine production in Corynebacterium glutamicum by engineering amino acid transporters. Amino Acids 52, 1363–1374 (2020). https://doi.org/10.1007/s00726-020-02893-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-020-02893-6