
Abstract
NMR spectroscopy methods have been used to prove structures of two diastereomers of cyclopropa[a]pyrrolizine spiro-fused with a benzo[4,5]imidazo[1,2-a]indole fragment which were obtained through a three-component 1,3-dipolar cycloaddition reaction and isolated in a ratio of 6:1. Complete signal assignment in 1H and 13C spectra of each compound was made by using some homonuclear (COSY, NOESY) and heteronuclear (HMQC, HMBC) NMR experiments. The spatial structure of the studied diastereomers was proved on the basis of data on interproton through-space interactions obtained from the NOESY spectra at a mixing time of 0.5 s. The comparison of the experimental and calculated values of the vicinal constants and interproton distances indicates that each of the studied diastereomers in solution is characterized by a fast (in the NMR time scale) conformational equilibrium due to the inversion of the nitrogen atom in the cyclopropa[a]pyrrolizine fragment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
One of the important tasks in the synthetic organic chemistry is the formation of azomethine ylide particles, with the help of which various compounds with useful properties can be obtained. In our recent work, we have shown the possibility of generating azomethine ylides from 11H-benzo[4,5]imidazo[1,2-a]indol-11-one and amino acids [1] for the first time. The structure of the compounds considered in the article contains pharmacophore fragments of 3-azabicyclo[3.1.0]hexane and pyrrolysine. They are both often found in many medicinal preparations and represent interesting classes of heterocyclic compounds from the point of view of biological activity [2,3,4]. In the process of work, diastereomers—(4) and (5) were synthesized and individually isolated, which undoubtedly facilitated the task, but did not make it less interesting from the point of view of NMR analysis.
The aim of this article is to prove the structures of two diastereomers (4) and (5), which are produced by the interaction of trimethylsilylcyclopropene 1, 11H-benzo[4,5]imidazo[1,2-a]indol-11-one (2) and L-proline (3) and later were isolated individually by preparative thin-layer chromatography in a ratio of 6:1, respectively (Fig. 1). We do not describe the reaction mechanism, since it has been recently discussed in detail in our separate publication [1], but we present here the most important arguments of the NMR evidence for the structure and intramolecular dynamic of each of the two diastereomers (4) and (5).

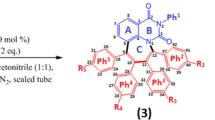
Synthesis of spiro-adducts (4) and (5) via one-pot three-component reactions of cyclopropene (1), ketone (2) and l-proline (3)
One of the main advantages of the NMR method is its high resolution capability, which makes it possible to determine even small changes in the molecular structure [5]. The method is unique, because it makes it possible to study the dynamics of molecular processes, including fast in NMR time scale changes in the conformation of the molecules in solution [6]. Thanks to development of NMR methods and data processing procedures, it has become possible to analyze NMR spectra of very large molecules such as proteins and other biopolymers [6, 7] and also the spectra of complex mixtures of small molecules [8]. However, like any other method, the NMR spectroscopy has its disadvantages. In particular, some proton signals in 1H spectra may be hidden under solvent lines and overlapped on each other. It may lead to so-called strong-coupled effects between scalar connected protons, which may impose some restrictions and create difficulties at spectral analysis. For correct interpretation of NMR data in some cases, it is necessary to obtain additional information about the molecular structures under study and/or their spectral and relaxation data. This can be done on the basis of computer modeling. Such approach, known by the generic acronym CASE (Computer Assisted Structure Elucidation), has been widely and effectively used in recent times [9,10,11].
2 Results and Discussion
2.1 1H and 13C Signal Assignments in NMR Spectra of Compounds (4) and (5)
As the main methods for identifying signals in the 1H NMR spectra of regioisomers (4) and (5), we used the correlation spectra COSY [12] (Figs. S5 and S12, respectively) and NOESY [13,14,15] (Figs. S6 and S13, respectively), in which the cross-peaks between the signals of scalar-coupled and spatially close protons, respectively, provided unambiguous information about their belonging as in aliphatic (2.0–5.0 ppm), and in aromatic (6.0–8.1 ppm) regions of the indicated spectra. In case of both compounds (4) and (5), there is no doubt that the singlet signal (Fig. 2) with relative intensity 9H belongs to the protons of the Si(Me)3 group due to the characteristic chemical shift (− 0.08 ppm for (4) and − 0.15 ppm for (5)) and to the presence of a satellite doublet signal of methyl groups at 29Si atoms with a total relative intensity of 4.7% and 2J(29Si-1H) = 7.5 Hz and presence of a satellite doublet signal of methyl groups at 13C atoms with a total relative intensity of 1.1% and 1J(13C-1H) = 119.9 Hz.

Fragment of NMR 1H spectrum of compound (4) with methyl signal of Si(Me)3 group. Scalar coupling of these protons with isotopes 29Si and 13C are shown by arrows
In case of NOESY experiments [16], it was necessary to select the appropriate conditions for registration and process data. We used a relatively short mixing time (τm = 0.5 s) and so-called ISPA (isolated spin-pare approximation) approach [17,18,19,20] and PANIC (peak amplitude normalization for improved cross-relaxation) method [21,22,23] to eliminate relaxation effects: the values of the cross-peak integral intensities were reduced to the diagonal ones. Moreover, we used the so-called spherical (or isotropic) model of distance estimation on base NOE data and calibrate method for calculation of unknown distances [15] because of the relatively low calculated value of the diffusion coefficients relation around the main axes: (D║/D┴)calc. ≈ 2. It was assumed that there was no (or insignificant contribution) of the anisotropy of the total rotational diffusion in the solution of the studied molecules (4) and (5) [24, 25].
When analyzing the distances between protons in the rigid part of the molecule (H6a′ or H1′) and degenerate ortho-protons (H12,16 or H18,22) of mobile aromatic rings at 1a′- or 6b′-positions, only the distance to the nearest ortho-proton was taken into account [26,27,28], because the distance to the farthest ortho-proton was significantly (by 1.5–2.0 Å) longer than that of the nearest one. As a result the contribution of distant ortho-proton to the total NOE was negligibly small: no more than 2–3% of the measured value [29, 30].
Comparison of the 1H NMR spectra of compounds (4) and (5) shows (Fig. 3) [1] significant differences in the signal positions of both aliphatic and aromatic protons. In the aliphatic region almost all the protons of compound (5) have higher chemical shifts than of compound (4). There is an exception in the form of proton H1′, the singlet signal of which has a slightly higher chemical shift in the case of compound (4). The greatest changes in the chemical shift are observed for the H4′α proton, which increases by 1.0 ppm. for compound (5) compared to its value of (4). In the case of the aromatic region, we can observe that for compound (5), the chemical shift decreases for the doublet signals of ortho-protons of aromatic rings at positions 1a′ and 6b′ by 0.25 and 0.14 ppm, respectively, and for proton H1 by 0.24 ppm. These differences in chemical shifts in compounds (4) and (5) indicate a significant difference in the chemical structure of these molecules, associated with the change in the mutual orientation of the fragments at the C2′ atom.

(From [1] by permission of IJMS)
a Regioisomers (4) and (5) with atom numbering. b Fragments of NMR 1H spectra of these compounds. The most significant differences in the proton chemical shifts in aromatic and aliphatic regions are shown by lines
When comparing the 1H spectra of compounds (4) and (5), in addition to the above spectral differences, a significant broadening of the lines of proton signals belonging to the C4′H2–C5′H2–C6′H2–C6a′H fragment of the second of them was found, compared to the corresponding multiplet signals of compound (4). At the same time, the signals in the aromatic region were almost identical in width, except the signal of the H1 proton at 7.61 ppm: in the 1H NMR spectrum of compound (5), the line width of the doublet signal of the H1 proton at half height is about 6.0 Hz. This is 2–3 times higher than for other aromatic proton signals. Thus, the detected broadening of proton signals in the spectrum of compound (5) may indicate the presence of a fast (in the NMR time scale) exchange process.
The signal assignments of carbon atoms in NMR 13C spectra for compounds (4) and (5) were made on base HMQC [31] and HMBC [32] spectra (Figs. S7, S8 and S14, S15, respectively) and all results are presented in Table S1.
In 13C spectrum of compound (5), as well as in its proton spectrum, a broadening of the part of both aliphatic and aromatic carbon signals is observed (Fig. 4) [1]. This fact also suggests the existence of a fast (in NMR time scale) dynamic equilibrium between two or more conformers in a five-membered ring –N3′–C4′H2–C5′H2–C6′H2–C6a′H. Probably, a similar dynamic process occurs in compound (4). It is indirectly confirmed by broadening (up to 8.5 Hz) of some carbon signals in the aliphatic region of the 13C NMR spectrum (Fig. S4a), as well as by the similar multiplet structures of the proton signals H4′α (2.86 ppm) and H4′β (2.38 ppm), which due to time-averaging of scalar constants have the same (doublet of triplets) multiplet structure (Fig. S4b) and almost the same sum of the scalar constants: 20.2 and 20.8 Hz, respectively.

(From [1] by permission of IJMS)
a Aromatic and b aliphatic fragments of 13C NMR spectrum of compound (5). Peaks, affected by dynamic effects are depicted by asterisk
Thus, all the above observations of dynamic effects in the 1H and 13C NMR spectra of compounds (4) and (5) indicate the existence of a fast conformational equilibrium, apparently associated with the fast rearrangement of the five-membered ring –N3′–C4′H2–C5′H2–C6′H2–C6a′H– envelope transfer type and possible nitrogen N3′ inversion process.
2.2 The Usage of Vicinal Constants 3 J H-H for Analysis of Compounds (4) and (5)
In order to confirm the assumption of conformational exchange in compounds (4) and (5) independently, an increase in temperature by 30 degrees (up to 50 °C) was used to shift the conformational equilibrium toward a thermodynamically less stable state. This, in its turn, should lead to a corresponding change in the exchange-averaged vicinal constants in accordance with the relation for the two-position exchange: \(\langle\)3JH–H\(\rangle\) = PA3JH-H(A) + PB3JH-H(B), where PA and PB are the populations of the (A) and (B) states (PA + PB = 1), while 3JH-H(A) and 3JH-H(B) are the values of the constant in states (A) and (B). Usually, as 3JH-H(A) and 3JH-H(B), their calculated values 3JH-H(calc.) based on the Karplus dependence 3JH-H = f(θ) [33,34,35,36] can be used for known (or calculated by molecular mechanic method MM2) dihedral angles. Unfortunately, this approach can be used only for compound (4), which 1H spectrum exhibits well-resolved multiplet signals of the C4′H2–C5′H2–C6′H2–C6a′H fragment. However, due to the overlap of H5′α, H5′β, and H6′β signals, only the proton signals H6a′ (dd, 4.82 ppm), H4′α (dt, 2.84 ppm) and H4′β (dt, 2.36 ppm.) are suitable for accurate measurement of vicinal constants.
For compound (4), the temperature increasing across 5 degrees from 25 to 50 °C led to a slight (on 0.22 Hz) increase in the averaged value of constant 3J6a′–6′α from 9.05 to 9.27 Hz (Fig. 5a). At the same time the value of the constant 3J6a′–6′β remained on identical level about 5.14 ± 0.02 Hz. To get such high accuracy (digital resolution less 0.02 Hz) 512K data points, LG-transformation (LB = − 4.0 Hz, GB = 2.0 Hz) and straightforward linear prediction (LP) procedures were used during data processing.

Experimental dependence of averaged a constant \(\langle\)3J H6a′–H6′α\(\rangle\) and b sum of constants \(\langle\)3JH6a′-H6′α + 3JH6a′-H6′β\(\rangle\) on temperature
It should be noted that in the case of compound (4), the so-called strong-coupling effects are quite possible on the signals of H4′α, H4′β and H6a′ protons due to the overlap of the signals of H5′α, H5′β and H6′β protons in the region 2.18‒1.95 ppm. These effects can lead to distortion of the values of scalar constants or even to the appearance of additional so-called combination lines. Since the effects of strong coupling in spin systems of the ABX type, which in this case are the protons H4′α, H4′β and H6a′, do not change the value of the sum of constants on the signal of the nucleus X, the real (and not imaginary due to the effects of strong coupling) temperature effect on the constant 3J6a′–6′α can be established if the sum of the constants (i.e. 3J6a′–6′α + 3J6a′–6′β) changes simultaneously, which is the full width of the doublet-doublet signal of the H6a′ proton at 4.82 (or doublet-triplet signals of protons H4′α at 2.84 ppm and H4′β at 2.36 ppm). Therefore, in Fig. 5b, it is additionally shown the experimental dependence of the sum of constants (3J6a′–6′α + 3J6a′–6′β) on temperature, which synchronously increases by approximately 0.2 Hz. Similar verification of the absence of strong-coupling effects was also carried out in the analysis of the temperature dependence of the scalar interactions of H4′α and H4′β protons.
The calculated values of dihedral angles θij, which were obtained by MM2 optimizing the geometry of three selected conformers of compound (4), are presented in Table 1. These selected conformers (A), (B) and (C), which differ in the mutual spatial orientation of protons in the five-membered ring N3–C4′H2–C5′H2–C6′H2–C6a′H are shown in Fig. 6 [1].

(From [1] by permission of IJMS)
Conformational exchange of the compound (4) between 5′β-envelope (A), 5′α-envelope (B) and 6a′β-envelope (C) forms
The lowest total energy was found for conformer (A) and conformers (B) and (C) are most preferable for interpretation of some detected spectral and cross-relaxation discrepancies. For each of the conformers, using three modifications of the Karplus dependence [33,34,35], six vicinal constants were calculated and compared with their experimental values.
From the analysis of vicinal constant data, it is quite obvious that none of the conformers can give the satisfactory agreement between their calculated and experimental values. Therefore, this discrepancy is the direct evidence of the existence of a fast (on the NMR time scale) exchange between two or more conformations of compound (4), which leads to averaging of the observed characteristics in accordance with their populations in solution. It should be noted that the detected quantitative differences between the calculated and experimental constants go beyond the usual accuracy of their calculation using the well-known modifications of the Karplus type empirical relations 3JH-H = f(θ).
The significant difference between the calculated and experimental values of the vicinal constants is especially noticeable when their sums are compared. For example, given in Table 1 data clearly show that for the sum of constants (3JHa–H6′α + 3JHa–H6′β) their calculated values for conformers (A) and (C) exceed their experimental value of 14.19 Hz, and for conformer (B), on the contrary, this amount is underestimated. A similar strong discrepancy is observed for the sums of the constants (3J4′α–5′α + 3J4′α–5′β) and (3J4′β–5′α + 3J4′β–5′β), which were measured on the signals H4′α and H4′β, respectively, and had, for example, almost the same values for the conformer (A): 12.8 and 12.6 Hz, respectively. However, their calculated values obtained using the HLA modification of the Karplus equation [33] turned out to differ by more than 10 Hz and equal to 18.02 and 7.23 Hz, respectively. At the same time, for conformer (B), the calculated values of the same sums of constants obtained by the same method (HLA) turned out to be completely opposite: 8.07 and 18.23 Hz, respectively. For conformer (C), the difference in the sums of constants decreases by almost 5 Hz: 12.8 and 17.43 Hz, respectively. Thus, a good agreement between the experimental and calculated values of the sums of constants can be obtained only under the conditions of the presence of two (or more) conformers in the solution, which rapidly (in the NMR time scale) exchange with each other.
It should be added that for compound (5) an increase in temperature to 50 °C led only to a slight narrowing of the broadened signals of C4′H2–C5′H2–C6′H2–C6a′H fragment, but not to the appearance of their multiplet splitting, which is observed for the corresponding proton signals of compound (4).
2.3 The Usage of NOESY Data in Conformational Analysis of Compounds (4) and (5)
The comparison of NOESY spectra of products (4) and (5) is the easiest way to obtain the proof of their structural and conformational feature. In Fig. 7, red asterisks indicate the most important proton–proton spatial interactions for compound (4). The normalized integral intensities of the corresponding cross-peaks are shown by the figures near integral rectangles. The comparison of their values with the others allows us to estimate proton–proton distances or their relations quickly and accurately enough. It should be noted that in this case only one NOESY spectrum is used, obtained at a mixing time of 0.5 s, its processing was carried out as described in [22]. For this approach, we used graphic dependence of normalized cross-peak intensity (Sij/Sii) on distance rij at reference distance value 1.78 Å for r(ref.) = r4′α–4′β and (Sij/Sii)(ref.) = 18.6 (Fig. S16 in SI). The obtained experimental distances rij(exp.) or their ratios rij(exp.)/rik(exp.) were compared with the corresponding calculated values, which were defined after optimizing of the molecular geometry by the molecular mechanics method MM2.

(From [1] by permission of IJMS)
The spatial structures of the conformers and the spectrum NOESY of the compound (4). Double arrows and rectangles on the 3D model of the molecule indicate the interproton distances the most important for proving the structures and the corresponding cross-peaks, respectively. The calculated (MM2) distance values are given by figures in angstroms (Å), and the volume integrals are given in % relatively to the intensity of the diagonal signal of the proton H1′
For compounds (4) and (5), the most important difference in their spatial structures is the spatial proximity of the H1 proton with neighbor protons in the α-region and β-region of these molecules. Compound (4) is characterized by proximity to H1′ (2.4 Å) and H4α (2.2 Å) protons, the reduced integral cross-peak intensities of which are 7.1 and 2.0%, respectively (see Fig. 7) [1]. In the NOESY spectrum of compound (5), these cross-peaks are simply absent (Fig. 8), but next intense spatial interactions are observed in the β-region of this molecule: 1/6a′ (2.7 Å), and 1′/18 (2.5 Å), which cross-peak intensities are 5.7 and 3.0%, respectively.

(From [1] by permission of IJMS)
The spatial structure and fragments of spectrum NOESY of compound (5). Double arrows and rectangles on the 3D model of the molecule indicate the interproton distances most important for proving structures and the corresponding cross-peaks, respectively. The calculated (MM2) distance values are given by figures in angstroms (Å); the volume integrals are given in % relatively to the intensity of the diagonal signal of the proton H1′
It should be added that when establishing the structure of compounds (4) and (5) using NOESY, not only the differences in the intensities of the cross-peaks were taken into account, but also their coincidence. For example, the spatial β-orientation of the H6a′ proton in compounds (4) and (5) is confirmed by the same weak (0.4%) cross-peak 6a′/1′. This value fully corresponds to the calculated distance r6a′–1′(calc.), equal to 3.9 Å for the preferable conformer (A). However, in the NOESY spectra of compounds (4) and (5), there are obvious quantitative inconsistencies in the ratio of some pairs of distances. For example, in case compound (4), the intensity of the 6a′/4′β cross-peak is significantly overestimated compared to 6a′/6′β. For the most energetically favorable conformation (A) of this compound, the calculated distances r6a′–4′α(calc.) and r6a′–6′β(calc.) are 3.9 and 2.3 Å, respectively. Therefore, the integrated intensity of the 6a′/4′α cross-peak should be almost 24 times less than the intensity of the 6a′/6′β cross-peak. According to the NOESY spectrum, the 6a′/4′α cross-peak is only 2.5 times smaller than the 6a′/6′β cross-peak. A similar discrepancy is also observed between the intensities of the 1′/4α and 1′/6α cross-peaks, for which the corresponding distances in conformation (A) are 2.2 and 2.5 Å. Therefore, the integrated intensity of the first of the cross-peaks should be approximately 2 times higher than the second, but according to the experimental NOE data (Fig. 7), it turns out to be 1.4 times lower (S1′/4′α(exp.) = 2.3%, S1′/6′α(exp.) = 3.3%).
The obvious reason for such significant differences between the experimental and calculated NOE data is the fast conformational exchange on the NMR time scale. It means that in experiments on measuring NOEs its time-averaged values are observed, which leads to a deviation from the calculated values of distances for each of the conformers if they differ significantly from each other [14,15,16]. Therefore, the obtained NOE data quantitatively confirm the previously made assumption about the existence of compounds (4) and (5) in solution in the form of a fast (in time NMR scale) conformational equilibrium. Thus, thanks to NOESY data, we can speak about conformational exchange more confidently than in case of analysis of scalar constants 3JH-H in Sect. 2.2.
In the right fragment of the NOESY spectrum (Fig. 8) [1], the spatial interactions of the proton H1 are indicated by double arrows. The double red arrow shows the interaction between protons H1 (7.61 ppm) and H12 (7.48 ppm), which could be a significant proof of the structure. However, due to the spectral closeness of the corresponding signals (∆δ = 0.13 ppm), the cross-peak at 400 MHz is covered by diagonal signals and is inaccessible for observation. It should be also noted that for product (5) in the NOESY spectrum, there is no significant increase in the integrated intensity of the 6a′/4′β cross-peak associated with dynamic averaging of the observed (i.e., effective) NOE. This may indicate a shift in the conformational equilibrium in product (5) toward the more energetically favorable conformer (A) as compared to the product (4).
The discrepancies found in the NOESY spectra of compounds (4) and (5) in the ratios of the integrated intensities for some pairs of interproton distances compared with their calculated values for the most preferred conformation (A) make it possible to estimate experimentally the population of the second conformer, the presence of which under conditions of fast (in the NMR time scale) conformational exchange with form (A) leads to distortion of the NOE data. The choice of the second conformer depends on the kind of spatial interaction chosen for such quantitative estimates.
There is no doubt that the most convenient for these purposes is the conformation for which geometry calculations using the method of molecular mechanics predict the maximum change in its structure, leading to the decrease in the measured distance (rather than an increase) compared to conformation (A). For this choice, the location of the corresponding pair of signals in the 1H NMR spectrum convenient for NOE detection, as well as the presence of a reference pair with a comparable distance between protons in the measured pair [15, 22] are important factors.
For compounds (4) and (5), all the above conditions are satisfied by the distance r6a′–4′β, since it varies from 3.91 Å in conformer (A) to 2.59 Å in conformer (C), which has the maximum proximity between separate H6a′ and H4′β protons (see Fig. 6). A convenient reference pair can be H6a′ and H6′β protons, the distance between which in conformations (A) and (C) is almost the same: r6a′–6β(A) = 2.32 Å and r6a′–6β(C) = 2.41 Å. It should be noted that the given values of the calculated distances for compounds (4) and (5) differ from each other within about (± 0.02 Å),
Therefore, taking into account the common experimental error in measuring the integral intensities of cross-peaks in NOESY spectra and the corresponding accuracy of estimating interproton distances based on only one spectrum within ± 0.1 Å [21, 22], we can use the above initial data to plot the dependence of the average distance \(\langle\)rij\(\rangle\) on the population (P) of the conformers (A) or (C). In the case of two-position exchange (A) ↔ (C) this dependence has a simple form, which is shown in Fig. 9 both in the form of an analytical dependence and its non-linear graph \(\langle\)rij\(\rangle\) = f(PC).

The calculated dependence of time-averaged interproton distance \(\langle\)r6a′–4′β\(\rangle\) at two-position exchange between (A) and (C) conformer on population of the second one Pc
To estimate the proportion of the second conformer of compound (4), we used the normalized integral intensities of the 6a′/4β and 6a′/6β cross-peaks, which are 2.0 and 5.0% relative to the diagonal peak 6a′/6a′ (see NOESY spectrum in Fig. 7). The ratio of their intensities is 1:2.5. Therefore, the ratio of the corresponding distances should be equal to (2.5)1/6 = 1.165. Then, assuming that the reference distance r6a′–6′β does not depend on the population of conformers and is equal to its average value \(\langle\)r6a′–6′β\(\rangle\) = 2.36 Å for conformers (A) and (B), we obtain the observed (i.e., exchange-averaged) value distances between H6a′ and H4′β protons: \(\langle\)r6a′–4′β\(\rangle\) = 1.165*\(\langle\)r6a′–6′β(ref.)\(\rangle\) = 2.75 Å. The graphical solution for determining the population of the conformer (C) is shown in Fig. 9. If the experimental error in measuring the average distance \(\langle\)r6′–4′β\(\rangle\) is within 0.05 Å, its share is about 2/3 (67 ± 7) %.
A similar integration and calculation procedure was carried out for compound (5). In the NOESY spectrum of this compound (Fig. 8 and Fig. S16), the integrated intensities of 6a′/4′β and 6a′/6′β cross-peaks are 0.44 and 4.66% relative to the 6a′/6a′ diagonal peak, and their ratio is 1: 10.6. In this case, the ratio of the corresponding distances should be equal to (10.6)1/6 = 1.482. Then, we obtain for compound (5) the observed distance between the H6a′ and H4′β protons: \(\langle\)r6a′–4′β\(\rangle\) = 1.482*\(\langle\)r6a′–6′β(ref.)\(\rangle\) = 3.50 Å. This leads to the value of the PC population equal to (8 ± 3) %, if the experimental error in measuring the average distance \(\langle\)r6′–4′β\(\rangle\) is within 0.1 Å. Thus, the population of conformer (C) in compounds (4) and (5) differs significantly.
The difference in the population of the conformers of the studied diastereomers is probably because of the difference in the dynamic processes occurring in the cyclopropa[a]pyrrolizine tricyclic system of each of them. Among the factors that can affect the equilibrium position, steric interactions seem to play the most significant role, which, due to the difference in the spatial structure of compounds (4) and (5), should differ significantly. A more detailed and reasoned conclusion on this matter can only be made after an additional study of the thermodynamic and kinetic characteristics of these and other similar systems, as well as testing other alternative hypotheses.
3 Conclusion
Thanks to the NMR spectroscopy method, an important problem of determining the spatial structures of two diastereomers (4) and (5) obtained during the three-component reaction of 1,3-dipolar cycloaddition was solved. At the same time, the processes of dynamic equilibrium between the conformers of each of these compounds were considered. A complete identification of signals in the 1H, 13C NMR spectra was performed by analyzing COSY, NOESY, HMQC and HMBC spectra. The spatial structures of compounds (4) and (5) have been proven by the quantitative NOE data, which were compared with their proton–proton distance values calculated by the molecular mechanics method MM2. In some cases, the calculated and the experimental distances were different, that allowed us to suppose the presence of a dynamic equilibrium between two or more conformers which is connected with the fast (in NMR time scale) envelope change of five-membered ring –N3–C4′H2–C5′H2–C6′H2–C6a′H– for both studied compounds (4) and (5). The comparison of NOE data for compounds (4) and (5) indicates a shift in the conformational equilibrium for product (5) toward the more energetically favorable conformer (A) as compared to the product (4).
4 Experimental Details
1D and 2D homonuclear NMR spectra (COSY, NOESY) of compounds (4) and (5) were acquired on Bruker Avance III HD (400 MHz for 1H nuclei) NMR spectrometer. JEOL ECX400A NMR spectrometer (400 MHz for 1H nuclei and 100 MHz for 13C nuclei) with inverse probe was used for heteronuclear experiments HMQC and HMBC. Chemical shifts of 1H and 13C signals are referenced using residual signal of CDCl3/CHCl3 (7.26 ppm) and triplet signal of CDCI3 (76.9 ppm), respectively. All measurements were carried out without sample spinning at 295K. Initial time-domain NMR signals (FID) were processed in MestReNova program [37]. Routine 1D data were apodized with 0.5 Hz exponential functions while 2D-data were treated with 90-degree sine squared (t1) and 90-degree sine bell (t2) functions prior to Fourier Transform.
Experimental vicinal constants 3JH-H(exp.) for compound (4) were determined directly from NMR 1H spectrum (SW = 4 kHz), which was obtained as a result of the following additional processing of the free induction decay signal (32K data points, digital resolution 0.122 Hz/pt): Lorentz–Gauss transformation (LB = − 4 Hz, GB = 2.0 Hz), zero filling up to 256K and direct linear prediction (LP) using 24 coefficients. These procedures provided a digital resolution of 0.015 Hz/pt and an accuracy of the vicinal constants determining no worse than ± 0.02 Hz. The values of the calculated scalar constants 3JH-H(calc.) were determined using the graphical program MestRe-J [38]. The values of the calculated dihedral angles θH-H(calc.) were obtained by optimizing the geometry of the studied molecule (4) by the molecular mechanics method MM2 in the HyperChem program [39].
Data Availability
Part of the spectral drawings (Figs. 3, 4, 6, 7, 8 in manuscript, and Fig. S4, S5 in Suppl. Inf.) were borrowed (partially or completely) from the Supplementary Information (Figs. S53–S59) of the previous our article [1] and permission from International Journal of Molecular Science was obtained for their use in this article.
References
A.S. Filatov, Yu.A. Pronina, S.I. Selivanov, S.V. Shmakov, A.A. Uspenski, V.M. Boitsov, A.V. Stepakov, Int. J. Mol. Sci. 23(21), 13202 (2022)
L.A. McAllister, C.R. Butler, S. Mente, S.V. O’Neil, K.R. Fonseca, J.R. Piro, J.A. Cianfrogna, T.L. Foley, A.M. Gilbert, A.R. Harris et al., J. Med. Chem. 61(7), 3008–3026 (2018)
J. Qiao, Y.-S. Li, R. Zeng, F.-L. Liu, R.-H. Luo, C. Huang, Y.-F. Wang, J. Zhang, B. Quan, C. Shen et al., Science 371(6536), 1374–1378 (2021)
E. Schweizer, A. Hoffmann-Röder, J.A. Olsen, P. Seiler, U. Obst-Sander, B. Wagner, M. Kansy, D.W. Banner, F. Diederich, Org. Biomol. Chem. 4(12), 2364–2375 (2006)
N.S. Bhacca, D.H. Williams, Application of NMR Spectroscopy in Organic Chemistry. Illustrations from the Steroid Field (Holden-Day, San Francisco, 1964)
R.R. Ernst, G. Bodenhausen, A. Wokaun, Principles of Nuclear Magnetic Resonance in One and Two Dimensions (Oxford University Press, Oxford, 1987)
K. Pervushin, R. Riek, G. Wider, K. Wüthrich, Proc. Natl. Acad. Sci. USA 94(23), 12366–12371 (1997)
J. Wist, Magn. Reson. Chem. 55(1), 22–28 (2017)
M. Jaspars, Nat. Prod. Rep. 16, 241–248 (1999)
M. Elyashberg, D. Argyropoulos, Magn. Reson. Chem. 59(7), 667–668 (2021)
A. Navarro-Vázquez, Magn. Reson. Chem. 55(1), 29–32 (2017)
W.P. Aue, E. Bartholdi, R.R. Ernst, J. Chem. Phys. 64(5), 2229–2246 (1976)
D. Neuhaus, M.P. Williamson, The Nuclear Overhauser Effect in Structural and Conformational Analysis, 2nd edn. (VCH Publishers Inc., New York, 2000)
R.A. Bell, J.K. Saunders, Can. J. Chem. 48(7), 1114–1122 (1970)
N.H. Andersen, H.L. Eaton, X. Lai, Magn. Reson. Chem. 27(6), 515–528 (1989)
A. Kumar, Magn. Reson. Chem. 41(Special Issue), S26–S32 (2003)
I. Solomon, Phys. Rev. 99, 559–565 (1955)
V.A. Likić, Concepts Magn. Reson. 8(6), 423–436 (1996)
S. Boros, Z. Gáspári, G. Batta, Annu. Rep. NMR Spectrosc. 94, 1–38 (2018)
B. Vögeli, Prog. Nucl. Magn. Reson. Spectrosc. 78, 1–46 (2014)
S. Macura, B.T. Farmer II., L.R. Brown, J. Magn. Reson. 70(3), 493–499 (1986)
G. Esposito, A. Pastore, J. Magn. Reson. 76(2), 331–336 (1988)
H. Hu, K. Krishnamurthy, J. Magn. Reson. 182(1), 173–177 (2006)
D.E. Woessner, J. Chem. Phys. 36(1), 1–4 (1962)
S.B. Landy, B.D.N. Rao, J. Magn. Reson. 83(1), 29–43 (1989)
H. Liu, P.D. Thomas, T.L. James, J. Magn. Reson. 98(1), 163–175 (1992)
W. Lee, N. Krishna, J. Magn. Reson. 98(1), 36–48 (1992)
J. Tropp, J. Chem. Phys. 72(11), 6035–6043 (1980)
C.P. Butts, C.R. Jones, J.N. Harvey, Chem. Commun. 47(4), 1193–1195 (2011)
I.A. Khodov, S.V. Efimov, V.V. Klochkov, L.A.E. Batista de Carvalho, M.G. Kiselev, J. Mol. Struct. 1106, 373–381 (2016)
A. Bax, R.G. Griffey, B.L. Hawkins, J. Am. Chem. Soc. 105(24), 7188–7190 (1983)
A. Bax, M. Summers, J. Am. Chem. Soc. 108(8), 2093–2094 (1986)
C.A.G. Haasnoot, F.A.A.M. Leeuw, C. Altona, Tetrahedron 36(19), 2783–2795 (1980)
W.B. Smith, M. Barfield, Magn. Reson. Chem. 31(7), 696–697 (1992)
E. Diez, J. San Fabian, J. Guilleme, C. Altona, L.A. Donders, Mol. Phys. 68(1), 49–63 (1989)
S.I. Selivanov, Appl. Magn. Reson. 49(6), 563–578 (2018)
J.C. Cobas, S. Domínguez, N. Larin, I. Iglesias, C. Geada, F. Seoane, M. Sordo, MestReNova, Version 6.0.2-5475. Mestrelab Research S.L. (2009)
A. Navarro-Vazguez, J.C. Cobas, F.J. Sardina, J. Casanueva, E. Diez, J. Chem. Inf. Comput. Sci. 44, 1680–1685 (2004)
HyperChem TM Release 5.1 Windows Molecular Modeling System. HyperCube, Waterloo (1997)
Acknowledgements
NMR investigations have been done in the Resource Centers “Magnetic Resonance Research Center” of the Saint-Petersburg State University and in the Center of collective use at the Faculty of Chemistry of Herzen State Pedagogical University “Instrumental methods for the study of nitro compounds, coordination, biologically active substances and nanostructured substances”. This work was supported by the Ministry of Science and Higher Education of the Russian Federation (state assignment FSEN-2023-0002). V.M.B. is grateful to the Ministry of Education and Science of the Russian Federation (project FSRM-2023-0005) for financial support. S.I.S. acknowledges Saint-Petersburg State University for a research grant 104236506.
Funding
The work of Yu.A.P. and A.V.S. was funded by the Ministry of Science and Higher Education of the Russian Federation (state assignment FSEN-2023-0002); the work of V.M.B. was funded by the Ministry of Education and Science of the Russian Federation (project FSRM-2023-0005); the work of S.I.S. was funded by Saint-Petersburg State University (research grant 104236506).
Author information
Authors and Affiliations
Contributions
YuAP, AVS and VMB synthesized compounds (4) and (5) and together with EAP wrote and proofread the text of the article. RIB recorded all NMR spectra and SIS planned NMR experiments and together with YuAP analyzed their results to get complete signal assignments in 1H and 13C spectra of studied compounds. SIS also wrote text of Sects. 2.2 and 2.3 including the preparing spectral figures for these sections and for Supplementary Information. All authors discussed results and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no competing interests as defined by Springer, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Ethical Approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pronina, Y.A., Stepakov, A.V., Popova, E.A. et al. Diastereomers of Cyclopropa[a]pyrrolizine Spiro-fused with a Benzo[4,5]imidazo[1,2-a]indole Fragment: Structure Determinations Using NMR Methods. Appl Magn Reson 54, 999–1014 (2023). https://doi.org/10.1007/s00723-023-01608-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-023-01608-w