
Abstract
Amyloidosis is a diverse group of protein conformational disorder which is caused by accumulation and deposition of insoluble protein fibrils in vital tissues or organs, instigating organ dysfunction. Renal amyloidosis is characterized by the acellular Congo red–positive pathologic deposition of amyloid fibrils within glomeruli and/or the interstitium. It is generally composed of serum amyloid A–related protein or an immunoglobulin light chain; other rare forms lysozyme, gelsolin, fibrinogen alpha chain, transthyretin, apolipoproteins AI/AII/AIV/CII/CIII; and the recently identified form ALECT2. This disease typically manifests with heavy proteinuria, nephrotic syndrome, and finally progression to end-stage renal failure. Early diagnosis of renal amyloidosis is arduous as its symptoms appear in later stages with prominent amyloid deposition. The identification of the correct type of amyloidosis is quite troublesome as it can be confused with another related form. Therefore, the exact typing of amyloid is essential for prognosis, treatment, and correct management of renal amyloidosis. The emanation of new techniques of proteomic analysis, for instance, mass spectroscopy/laser microdissection, has provided greater accuracy in amyloid typing. This in-depth review emphasizes on the clinical features, renal pathological findings, and diagnosis of the AL and non-AL forms of renal amyloidosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyloidosis is a group of disorders causing nonphysiologic extracellular depositions of misfolded proteins as amyloid fibrils, subsequently distorting the structure and function of the affected tissues that includes diverse protein conformational disorders such as Alzheimer’s, Parkinson’s, Huntington’s, prion-associated transmissible spongiform encephalitis type 2 diabetes, cystic fibrosis, and systemic amyloidosis. So far, 36 human precursor proteins have been recognized that causes amyloidosis (Benson et al. 2019). Amyloid fibrils bind the widely used dye, Congo red; when the amyloid deposits stained with Congo red dye are viewed under polarized light, they exhibit an apple-green birefringence (Dogan 2017). Many studies have shown that protein conformational diseases are associated to a specific protein that is partially unfolded and aggregated. Misfolding of protein exerts a pathologic effect since because of their conformational alteration, gets accumulated as toxic aggregates aggregated either intracellularly/extracellularly or undergo an accelerated degradation. All protein conformational diseases have their unique, neuropathological, biochemical, and clinical characteristics; nevertheless, their related amyloidogenic precursor proteins have share no sequence homology and distinct functions. Moreover, the hallmark feature of Alzheimer’s disease is the presence of two kinds of intracellular protein aggregates β-amyloid (Aβ) and tau in the brain, while in Parkinson’s disease, alpha-synuclein (αSyn) is a major aggregating protein in brain. Freshly, type 2 diabetes mellitus has been defined as a conformational disease due to islet amyloid polypeptide (IAPP), undergoes a change in tertiary structure followed by self-association and tissue deposition (Lin and Liu 2006). Here, in this review, we will focus on renal amyloidosis.
Renal amyloidosis is a rare and intractable protein misfolding disorder which prompts progressive renal insufficiency. Intractable disease mainly refers to the rare diseases that have resulted from the lack of curable treatment and preventions/ unidentifiable causes. The kidney is one of the most commonly affected organs among AL, AA and several types of hereditary amyloidosis (lysozyme, gelsolin, fibrinogen alpha chain, transthyretin, apolipoproteins AI/ AII/AIV/CII/CIII) (Gurevitz and Stone 2012). The disease shows a number of distribution patterns within the kidney compartment (Hopfer et al. 2011).
Based on immunohistochemistry and immunofluorescence studies, renal amyloidosis can be classified into amyloid A amyloidosis (AA) and light chain amyloidosis (AL) types. Lately, amyloidogenic leukocyte chemotactic factor 2 (ALECT2) has recently been recognized as the most common type of renal amyloidosis (Larsen et al. 2016). The other type of amyloidosis is hereditary amyloidosis, and some variants of this type of amyloidosis may affect multiple organ system of the body. Renal amyloidosis cases are predominantly ALECT2 and AL amyloidosis, but sometimes rare hereditary or familial forms of amyloidosis are also found to be involved in the kidney pathogenesis. Amyloid typing is essential for accurate diagnosis, treatment, and genetic counseling as the treatment modalities of different types of amyloid differ accordingly (Sethi et al. 2010).
This review provides a decisive outline of the pathology of AL, AA, ALECT2, and rare forms of hereditary renal amyloidosis. Numerous approaches have been anticipated for amyloid typing together with a short overview of proteomic approaches for the assessment of the circulating amyloidogenic proteins. The pitfalls of conventional immunohistochemistry-based techniques have urged the need for an alternate solution for direct amyloid typing. The enormous potential of the proteomic approach in describing the composition of amyloid deposits and in studying the molecular features of the amyloidogenic precursors is no longer obscure. The introduction of LMD and MS-based proteomics in clinical practice and SAP scintigraphy has transformed the field of amyloid typing (Lavatelli and Vrana 2011). It is expected that recent studies on renal amyloidosis will help in understanding the molecular mechanisms related to the disease and increase the diagnostic potential of the disease (Ikeda 2008).
Diagnosis
Unambiguous diagnosis is the first step for any medical treatment; here, the demonstration of amyloid deposition in tissues and the accurate typing are imperative for genetic counseling, prognosis, and treatment (Sethi et al. 2012a). The primal method is the histological examination of biopsy specimens which when stained with Congo red dye demonstrates characteristic apple-green birefringence when viewed under polarized light. Congo red can detect with 80–90% sensitivity and 100% specificity, yet pathologists stumble upon optimal results due to poor smears, inadequate sample size, and the misinterpretation caused by autofluorescence (yellow-green) of collagen. Fluorescence, thioflavin S and T, methyl violet, and sulfonated alcian blue are also used, but they are less sensitive and less specific when compared with the former (Picken 2010).
However recently, the abdominal fat aspirate is used for the diagnosis of amyloidosis as it is easy to perform, cost-effective, rapid, and a sensitivity (54 to 82%) (Ong and Rajasingam 2007). Since, biopsy is the invasive method which could lead to several complications and sometimes even internal bleeding; therefore, a less invasive strategy of using subcutaneous fat aspirate is also used. In 1970s, Westermark and Stenkvist exhibited that amyloid can be detected in subcutaneous fat extracted by the fine needle. The aspirate is then poured on the slide and mechanically formed in single cell consistency, air dried, and stained with Congo red. The test samples having amyloids show the apple-green birefringence under polarized light. Using biopsy of fat tissue, amyloid typing can also possible by performing immunofluorescence, western blotting, electron microscopy, mass spectrometry, and various other molecular techniques. As nothing is perfect, even this can give false positive; hence, the results should be confirmed further (Picken 2010). Subsequently, correct amyloid typing of deposited protein is cross-examined by various other methods (Fig. 1). Additionally, an algorithm for diagnosis and typing of renal amyloidosis is shown in Fig. 2.

Schematic representation of various methods used for a pivotal milestone in diagnosis, i.e., amyloid typing

Algorithm for the diagnosis and typing of renal amyloidosis
Since the past two decades, the immunohistochemical method has been used extensively for the classification of amyloid entities using immunoperoxidase staining. This technique uses labeled specific antibodies for locating the amyloid protein in cells or in parts of the cells, where peroxidase enzyme undergoes catalysis and forms the colored complex. However, this technique can present constraints in detecting the complete array of amyloidosis, such as availability of antibodies that are capable of detecting the native precursor and prior selection of antibody panel. Mutations in proteins render them misfolded or truncated which can even cause loss of epitopes thus, antibody usage in vain. By using fluorescein isothiocyanate (FITC) labeled antibodies, AH or AL amyloid can be detected by immunofluorescence. Generally, this technique is executed only on the fresh-frozen tissue. While in the absence of frozen tissue, pronase-digested paraffin-embedded tissue can be used (Leung et al. 2012).
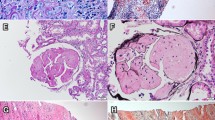
Alternatively, immunogold microscopy which amalgamates IHC with electron microscopy using gold-labeled antibodies can be performed and location of amyloid in tissue can be detected. Typical pictures of hematoxylin and eosin (H&E) stain (Fig. 3), Congo red staining (Fig. 4), immunohistochemistry, and electron microscopy are shown in Fig. 5.

H&E stain shows deposition of abnormal eosinophilic extracellular material within mesangial, peritubular, and interstitial. (Adapted from Kar Wah Fu et al. (2018) BMC Nephrology)

a Reddish-orange Congophilic amyloid deposits observed in glomeruli and surrounding interstitium from renal biopsy. b Congophilic deposits visualized under polarized light demonstrating apple-green birefringence. (Adapted from Kristina Bowen et al. (2012), Case reports in nephrology)

Histology of renal amyloidosis. a H&E stain shows extensive effacement of the glomerular architecture by amorphous amyloid. b The involvement of glomeruli by AA amyloid are revealed by immunohistochemistry using antibody specific for amyloid A protein. c Electron micrograph shows random alignment of amyloid fibrils in the subepithelial zone of a glomerular capillary. (Adapted from Muhammed Hassan et al. (2019), BMC Nephrology)
In the case of familial amyloidosis, an additional step of detection for the DNA mutation is complementary which provides unequivocal identification of the amyloid protein. Techniques such as RFLP, SNP array analysis, and DNA microarray aid the typing process. Furthermore, a definitive analysis of protein is mandatory (Picken 2010).
Proteomics is the new dawn of diagnosis, which includes the entire complement of proteins and eradicates the need to do specific tests for an individual protein of interest. Proteomic technique includes the trailing steps: sample preparation (tissue or serum), protein concentration, protein fragmentation and separation, MS analysis, and finally protein identification by help of informatics. Mass spectrometry (MS) has the potential for high efficiency identification of protein of interest in the whole proteome being analyzed either by the biopsy tissue or by serum samples. Prior to MS analysis, proteins need to be separated, which can be accomplished by liquid chromatography and gel electrophoresis (Leung et al. 2012). The classic approach of 1D-PAGE, 2D-PAGE and then MS analysis of in-gel digested proteins is done for typing. One-dimensional gel electrophoresis separates proteins by size which is a robust method for protein separation. 2D-PAGE (two-dimensional gel electrophoresis) permits higher resolution of a complex protein mixture. In the first dimension, mixtures of proteins are separated on the basis of their charge by isoelectric point focusing (IEF), and in the second dimension, proteins are separated on the basis of mass. These techniques require manual sample handling and consume a lot of time (Brambilla et al. 2013). Only the mass of the charged peptide fragments can be measured by MS, and usually amyloid proteins are not charged. Therefore, for maximum efficiency, modern proteomics combines reverse-phase liquid chromatography with tandem electrospray mass spectrometer for proteins in liquid phase and MALDI/MS for proteins in solid phase. Shotgun proteomics and targeted proteomics are widely used; the former globally identifies the proteins from the mixture, whereas the latter targets the specific protein. The results are demonstrated as the spectra of abundant proteins found, which are then analyzed by correlating the identified to that of the known masses in the databases such as UniProt/SwissPRot (Picken 2015).
The touchstone of proteomics is laser microdissection and tandem mass spectrometry (LMD-MS). LMD-MS has higher specificity than immunohistochemistry; single test with LMD/MS can determine the amyloid protein in place of testing the sample several times by using different antibodies, and it can be done on small renal tissue samples. Positive areas stained with Congo red are dissected under a laser dissecting microscopy supplemented with tryptic digestion. These digested peptides are scrutinized through liquid chromatography-electrospray tandem mass spectrometry. As it directly analyses the real peptide, it can distinguish a protein with or without known mutations. It can be performed on all the tissues including fat aspirates and paraffin-embedded material. (Leung et al. 2012).
LMD/MS and genetic testing are also convenient in differentiating fibrillary glomerulonephritis from amyloidosis. The former is positive for IgG antibody detection and ambiguous for Congo red, therefore making it arduous to differentiate from AH amyloidosis. Here, LMD/MS is quintessential which detects the spectra of SAP and heavy and light chains, thus authenticating the disease (Sethi et al. 2012a). Despite entitling it to be gold class, it is not flawless. Its equipment requires large storage space, and its running is exorbitant (Gilbertson et al. 2015). Genetic mutations can cause loss of epitope in the amyloid protein. Hence, protein conformational changes often lead to hereditary renal amyloidosis. For instance, Zehnyu et al. demonstrated a single base-pair transition from T to C (TGG/ CGG) and T to A of codon 82, leading to the replacement of tryptophan by arginine in the mature protein (p.Trp82Arg) which was confirmed by DNA sequencing, immunohistochemistry, and mass spectrometry on renal biopsy and gene testing as well. Immunohistochemistry and mass spectrometry of renal tissue confirmed the lysozyme nature of the amyloid (Li et al. 2019a).
LMD/MS-based proteomic analysis showed heterozygous mutation c.220 T>C in ApoA-I gene which leads to the replacement of tryptophan by arginine at residue 74 (p. Trp74Arg). Additionally, in fibrinogen alpha chain amyloidosis, the mutation in the fibrinogen gene c.1639delA results in an amyloidogenic p. Arg547GlyfsTer21 variant of fibrinogen (Li et al. 2019b). Using proteomics, several researchers performed germline DNA sequencing of APOC2 gene and found a heterozygous c.206A→T alteration in exon 3, which results in a glutamate to valine substitution at codon 69 (E69V) (Nasr et al. 2017a). Other than this, in transthyretin amyloidosis, the presence of a TTR variant can be recognized by IEF of the serum. Variant forms and wild type of the protein will have typical migration patterns, and the specific TTR gene mutation can be determined subsequently by DNA sequencing (Yamamoto et al. 2018). The pathogenesis related to gelsolin amyloidosis indicated a mutation in Gly167 to Arg were G at 580 positions is converted into A, respectively, causing a dramatic reorganization of the polypeptide chain, forming the domain-swapped dimer resulting in kidney-localized deposition of the Gly167Arg aggregates which was identified by laser microdissection followed by mass spectrometry analysis (Bonì et al. 2018).
Another robust technique is MudPIT (multidimensional protein identification technology), which involves enzymatic digestion of protein mixture, the separation by strong cationic exchange (SCX), followed by C18 reverse phase chromatography using acetonitrile (ACN) gradient. It allows efficient analysis of proteins with elevated hydrophobicity, high molecular weight, and extreme pI (Brambilla et al. 2013).
Two imaging modalities exist, namely magnetic resonance imaging (MRI) and SAP scintigraphy. Diffusion-weighted MRI is a noninvasive diagnostic method that is easily applicable. It also differentiates between amyloidosis from that of CKDs (Rona et al. 2016).
I-123-labeled SAP scintigraphy uses a radiolabeled SAP component to track down amyloid deposits in the body. Since SAP binds to all amyloid proteins, it can be used for each form of amyloidosis (Leung et al. 2012).
Prospectively, proteomics will improve treatment for amyloidosis as it dispenses an understanding of underlying mechanisms at multiple aspects. It is reshaping our knowledge with respect to both pathogenesis and epidemiology whereas no other tool can provide such a plethora of information (Lavatelli and Merlini 2017).
AL amyloidosis
Light chain (AL) amyloidosis is the most common multisystemic amyloidosis, described as monoclonal gammopathies of renal significance, producing abnormal plasma cells. It is derived from immunoglobulin kappa and lambda chain or their fragment that are secreted into circulation, where the light chains misfold, aggregate as amyloid fibrils in target organs, and cause organ dysfunction, organ failure, and death. Fibrils of AL amyloid are collection of both constant and variable domains or only the variable domain of light chains. Mutations in the V λ VI subgroup and of the N-terminus may result in increased amyloidogenic properties (Abraham et al. 2003). Histologically, deposition of AL was seen in all renal compartments, though glomerular deposition frequently involved. The incidence of this disorder is approx 9.7 to 14 persons per million per year (Quock et al. 2018). Clinical manifestations include albuminuria, nephrotic-range proteinuria and ultimately lead to end-stage renal failure if diagnosed late. The median survival of patients associated with AL amyloidosis exceeds 3 years after initiation of dialysis (Milani et al. 2018). The kidney is the most commonly affected organ in AL amyloidosis, which occurs in around 10–15% of patients with multiple myeloma. In the largest case series of 407 renal amyloidosis patients, arterial and arteriolar deposits (56%), interstitial deposits (58%), glomerular deposits (97%), and tubular basement membrane deposits (8%) were observed (Said et al. 2013).
The study done on AL amyloidosis patients demonstrated that the immunofluorescence performed on biopsy tissue of kidney has low specificity, whereas trustworthy techniques, such as mass spectrometry, immune-electron microscopy, or immunohistochemistry, revealed that bone marrow biopsy was consistent with atypical kappa predominant plasma cell neoplasm (50–60%). AL amyloidosis is often treated with melphalan, an alkylating agent, lenalidomide and bortezomib in combination with dexamethasone. (Logan et al. 2016). Melphalan, an antineoplastic chemotherapeutic drug, enters the plasma cells (source of amyloidogenic light chains) and alkylate the guanine in the DNA causing linkages between the strands of DNA, thus inhibiting DNA/RNA synthesis. Additionally, lenalidomide induces the cell apoptosis in metabolically active plasma cells by inhibiting the support from bone marrow stromal cells. Whereas dexamethasone, a corticosteroid being antiinflammatory, counteracts the side effects of chemotherapy (drug bank). Bortezomib is mainly used to treat multiple myeloma. Bortezomib is boron containing molecule and a chemotherapeutic agent present in eukaryotic cells, inhibits the 26S proteasome, an enzyme complex which plays a crucial role in the cell by controlling protein degradation. Proteasome degrades misfolded, abnormal, and ubiquitinylated proteins, thus regulates the protein functions and expression. One central mechanism of bortezomib is the inhibition of the breakdown of inhibitory kappa B (IκB) and consequently stabilization of the nuclear factor kappa B complex which leads to the aggregation and accumulation of misfolded proteins and ultimately plasma cell apoptosis (Field-Smith et al. 2006). Supportive care and treatment play a major role to maintain organ function as chemotherapy needs a certain time to take effect.
Renal non-AL amyloidosis
Non-AL amyloidosis is present with slowly declining renal function without significant proteinuria. The AA and ALECT2 amyloidosis are the most common, and the enduring amyloidosis types associated with lysozyme, gelsolin, fibrinogen, and apolipoproteins AI/AII/AIV/CII/CIII are very rare entities. It is requisite to be familiar with the unique pathologic features of the varied types of non-AL amyloidosis (Sethi and Theis 2018). The amyloid precursor proteins and their clinical manifestations are described in Table 1.
AA amyloidosis
AA amyloidosis is a systemic, fatal, and rare complication; a major cause of this disorder are familial Mediterranean fever or chronic inflammatory disorders (juvenile rheumatoid arthritis) or chronic inflammatory bowel disease (bronchiectasis, osteomyelitis) and tuberculosis (Lachmann et al. 2007). AA amyloidosis derived from the deposition acute phase reactant serum amyloid A (SAA) protein, highly conserved protein which is synthesized as a precursor by the hepatocytes. It is a SAA 1 gene encoded extracellular, soluble, high-density apolipoprotein. N-terminal fragments of SAA can form insoluble fibrils that accumulate in “secondary” amyloid associated diseases. (Erdogmus et al. 2018). It accounts for approx 15% of amyloid deposits. Clinical manifestations of this disease include heavy proteinuria (95%) and nephrotic syndrome (75%) (de Asúa et al. 2014). The median survival of AA amyloidosis associated proteins is 6–9 years. Kidney biopsy revealed predominant deposition of amyloid fibrils in the mesangium of glomeruli, blood vessels walls, and basement membrane of tubules. The median age of diagnosis is rising from 50 to 70 years, which reflects the underlying cause of disease and assesses effective therapies (Papa and Lachmann 2018). AA amyloidosis accounts for 56% in the largest case series of renal amyloidosis patients (de Asúa et al. 2014). Moreover, in epidemiological studies on AA renal amyloidosis from Turkey, familial Mediterranean fever (FMF) is the major cause of this disease. This disease mainly occurs in male 40–50 years of age (Erdogmus et al. 2018). The distribution of AA amyloidosis varies around the world according to the geographical region (Lane et al. 2017).
In addition, AA amyloidosis is very common in the European and developing countries. The life expectancy of this disease is 13 years after diagnosis. The overall prognosis of this disease is poor and associated with the degree of renal involvement (Ansari 2016).
The current gold standard to diagnose AA amyloidosis is the kidney biopsy, and SAP scintigraphy may be helpful for monitoring the type of amyloid deposits. Though SAP scintigraphy has shown amyloid distribution which can be patchy, explanatory false-negative renal biopsy results, and it may vary between different forms of amyloidosis. The primary treatment of AA amyloidosis is the suppression of SAA protein and supportive care. To detect AA amyloid protein, immunostaining has been documented highly sensitive technique (de Asúa et al. 2014; Papa and Lachmann 2018).
Eprodisate is a low molecular weight, sulfonated, and negatively charged molecule which binds to glycosaminoglycan-binding sites on SAA, inhibits fibril polymerization, and decreases the risk of the main endpoint, a composite of aggravating renal function by 42%, and this reduced risk was mostly independent of SAA concentration or baseline renal function but it did not affect the proteinuria. As a result of more effective and less chemotoxic therapy, treatment of AL amyloidosis has significantly improved during the past decade (Dember et al. 2007).
ALECT2 amyloidosis
The leukocyte chemotactic factor 2 (LECT2) gene encodes a secretory 16-kDa protein which acts as a chemokine for the neutrophils and stimulates the growth of chondrocytes and osteoblasts. The LECT2 is expressed by cerebral neurons, epithelial cells of sweat and sebaceous glands, parathyroid glands, monocytes, liver, adipocytes, and vasculature tissue. As a matter of fact, the source of this protein to be in circulatory system is liver, hence even a hepatokine. The LECT2 gene has been mapped to chromosome 5q31.1–32 and contains four exons and three introns. The four exons code for a 151 amino acid protein, which is 133 amino acids in its secreted form (Genecard).
Amyloidogenic leukocyte chemotactic factor 2 (ALECT2) has emerged as the latest addition linked to renal amyloidosis, with unknown etiology and pathogenesis (Dember et al. 2007; Picken 2014). This type of renal amyloid is associated with a gradual decline of glomerular filtration rate (GFR), diagnosed in renal histology (Rezk et al. 2016). It typically manifests as progressive renal insufficiency with or without proteinuria and bland urine sediment (Kulkarni et al. 2015; Said et al. 2014).
The first case of ALECT2 was detected in the year 2008 in a 61-year-old female who was presented with glomerular amyloid deposition and nephritic syndrome (Benson et al. 2008). The disease mainly affected elderly Hispanics originated from Mexican descent. The median time for renal survival is 62 months for this disease (Said et al. 2014).
In ALECT2 cases, amyloid deposition was found predominantly in the renal cortical interstitium while minimal deposition was observed in the glomerular, vascular compartment and bone marrow. The pattern of amyloid deposition exhibits a distinctive globular appearance (Rezk et al. 2016; Larsen et al. 2014).
The group of researchers have reported that ALECT2 amyloid fibrils was predominantly found in the lungs, liver, kidneys, spleen, small intestine, gallbladder, bilateral ovaries, and uterus of a Hispanics (Tariq and Sharkey 2018). Glomerular amyloids were found around 70% of kidney biopsies (Rezk et al. 2016).
In renal ALECT2 amyloidosis, substantial cardiac involvement was absent, and patient survival was excellent (Said et al. 2014). Therefore, the prognosis is much better in a patient with ALECT2 amyloidosis than AA and AL amyloidosis patients (Rezk et al. 2016). Mass spectrometry (MS) and immunological staining are helpful tool to confirm the category of amyloid ALECT2 (Tariq and Sharkey 2018). ALECT2 amyloidosis is not likely to be demented with AL amyloidosis by immunofluorescence but can be misdiagnosed as heavy chain amyloidosis (Said et al. 2014).
Currently, there are no efficacious treatment guidelines available because the precursor protein is nonmutated but support measures including renal transplantation have been established (Picken 2014; Nasr et al. 2015; Jimenez-Zepeda and Leung 2014). It is essential that ALECT2 amyloidosis is not mistaken for the AL amyloidosis so that no harmful treatment is prescribed.
Hereditary renal amyloidosis
Around 2% of the renal amyloidosis is rare hereditary forms. Hereditary amyloidosis may affect a variety of organs, primarily depending on the mutant protein and represent a wide spectrum of clinical manifestations (Nasr et al. 2017b). The rare hereditary forms of renal amyloidosis are fibrinogen A alpha-chain, apolipoprotein AI, apolipoprotein AII, apolipoprotein AIV, apolipoprotein CII, apolipoprotein CIII, transthyretin, gelsolin, and lysozyme (Said et al. 2014; Nasr et al. 2017b).
Fibrinogen A alpha-chain amyloidosis
Fibrinogen A alpha-chain (FGA) is the constituent of the coagulation factor fibrinogen which is then cleaved by thrombin to form fibrin following vascular injury. FGA expression is restricted to liver only. Fibrinogen A alpha-chain amyloidosis (Afib) was the first time demonstrated in 1993, a 66-kDa molecular weight protein which is composed of 610 amino acids (Weisel 2005; Weisel and Litvinov 2013).
Afib is an autosomal dominant disease that is typically characterized by proteinuria, hypertension, azotemia, unique histological appearance, lower limb edema, and renal failure (Uemichi et al. 1994; Gillmore et al. 2009). Afib amyloidosis is associated with the deposition of mutant FGA amyloid in kidneys and is predominantly familial. Afib is found to be massively deposited in glomerular compartment of kidney (Sivalingam and Patel 2016).
The median time of the patients with kidney impairment is 4.6 years, from proteinuria progression to end-stage renal disease (ESRD) (Rowczenio et al. 2017). The majority of patients with Afib amyloidosis were found in the USA and European countries while this disease was rarely reported in Asian countries. The most prominent clinical manifestation in Asian patients with A α-chain amyloidosis is progressive nephropathy caused by extreme deposition of amyloids in the glomeruli (Yazaki et al. 2018). Though, no patient with nonneuropathic hereditary renal amyloidosis had been identified in Japan (Yazaki et al. 2015).
Until now, 16 amyloidogenic FGA variants have been found, out of which 9 are deletion insertion-deletion mutations and 7 are missense mutations. In the year 2015, a sporadic case of a 40-year-old woman was reported with promptly depreciating nephropathy with A α-chain amyloidosis having a novel frameshift variant (4899_4902delAGTG) (Yazaki et al. 2015).
In another study, researchers reported 6 novel FGA mutations, out of which five were linked with renal Afib amyloidosis and one was found to be concomitant to AL amyloid deposit present in a patient with plasma cell dyscrasia. Out of six, four were single base substitutions R554H, G555F, E526K, and E524K and two were frameshift mutations G519Efs*30 F521Sfs*27. They found extensive amyloid deposition within the glomeruli of 5 subjects (Rowczenio et al. 2017). A study done in Portugal found a high frequency of the FGA p. Glu545Val variant. Recently, the group of researchers have identified the first patient as homozygous for nephropathic amyloidosis, involving the fibrinogen variant correlated with the fibrinogen alpha-chain p.Glu545Val (E526V) mutation (Tavares et al. 2018). E526V forms are positioned in the C-terminal region (517–555 amino acid residues) of mature A α-chain (Yazaki et al. 2018).
This disorder might be an underrecognized reason for kidney disease; the diagnosis of fibrinogen A alpha-chain amyloidosis should have a high index of doubt mainly for the populations in which hypertension is common owing to the nonspecific nature of its primary clinical features (Tavares et al. 2018).
To circumvent misdiagnosis, especially when a family history of amyloidosis is lacking, accurate typing using mass spectrometry, laser microdissection, coalesced immunohistochemistry, and genetic analysis is valuable (Rowczenio et al. 2017). Laser microdissection and mass spectrometry can also be used to attain information about the entire sequence of the amyloid fibril protein utilizing paraffin-embedded and formalin-fixed renal tissue samples obtained from the patient (Yazaki et al. 2018).
The treatment of Afib amyloidosis encircles supportive measures and kidney transplantation for ESRD patients (Rowczenio et al. 2017).
Lysozyme amyloidosis
Lysozyme is also known as muramidase or N-acetylmuramide glycan hydrolase, discovered by Alexander Fleming in 1922. It is an antimicrobial enzyme which catalyzes the hydrolysis of 1,4-β linkages between the N-acetylmuramic acid and N-acetyl-D-glucosamine residues in peptidoglycan. Lysozyme is mainly found in secretions like tears, saliva, human milk, and mucus. It is also present in cytoplasmic granules of the macrophages and the polymorphonuclear neutrophils. In humans, bacteriolytic enzyme is synthesized by macrophages, gastrointestinal system, and hepatocytes, and hence, lysozyme amyloid deposits typically prevail in the liver, gastrointestinal tract, kidney, and spleen (Reitamo et al. 1978).
Lysozyme amyloidosis (Alys) is an extremely rare, autosomal dominant, and a hereditary systemic nonneuropathic form of amyloidosis (Scafi et al. 2018). Alys typically manifests with mild proteinuria, development of the nephrotic syndrome, gastrointestinal (GI) symptoms, and sicca syndrome while cardiac involvement is very rare (Granel et al. 2006). Lysozyme was first identified in 1993, and Alys is peculiarized by the precipitation of lysozyme within the body, leading to multiorgan failure. In general, lysozyme amyloidosis is considered to be a slowly progressive disease with a median survival rate of 17.9 years after the diagnosis of the disorder. Christopher and group first time reported the amyloidogenic component of lysozyme amyloidosis via mass spectrometry and laser microdissection from a bone marrow biopsy (Pleyer et al. 2015). Foremost, lysozyme C was reported as a specific amyloidogenic protein by the same group in the bone marrow.
Lysozyme (OMIM ID 153450) amyloidosis has been reported to be caused by many lysozyme variants: I56T (Pepys et al. 1993), D67H (Pepys et al. 1993), W64R (Valleix et al. 2002), D68G (Wooliver et al. 2008), T70N /W112R (Röcken et al. 2006), L84S (Nasr et al. 2017b), and F57I (Yazaki et al. 2003). These mutations perturb the hydrogen bonds between α and β domains, causing partial unfolding, attaining beta-sheet fibrillar structure, and then the interprotein interaction plays the major role to transform the mutated protein into amyloid (Griffin and Bradshaw 2018).
In I56T lysozyme variant, heterozygosity for T->C transition in exon 2 of the lysozyme gene occurs and alters the 56 amino acid from isoleucine to threonine (Pepys et al. 1993). The symptoms include predominantly dermal petechiae, renal involvement, and visceral amyloid deposits (Valleix et al. 2002). Whereas, in D67H, G->C transversion happens in lysozyme gene encoding histidine for aspartic acid at the 67th position in lysozyme (Pepys et al. 1993). The clinical features include an unrushed progression of renal involvement and amyloid deposition in GI tracts which leads to surgical and hemorrhagic complications. Both I65T and D67H were discovered in English family’s kindred in 1993 (Pepys et al. 1993).
A W64R variant of lysozyme amyloidosis was the first time reported in a French family, manifests due to the T->C transition rendering replacement of aromatic amino acid tryptophan to cationic arginine at position 64 of the mature protein. The clinical presentation includes a long history of sicca syndrome, renal diseases, dry mouth, and keratoconjunctivitis. The amyloid deposits in the kidney were found in vessels, arterioles, and glomerulus but not often in intestinal tissue (Valleix et al. 2002).
In another study, variant F57I, in which T->A transversion happened at 57th codon of the lysozyme, prompts replacement to isoleucine from phenylalanine. This was first reported in a Canadian Italian family, its distinctive clinical feature is renal amyloidosis (Yazaki et al. 2003).
Furthermore, hereditary Alys amyloidosis is also engendered by compound heterozygosity in exon-2 T70N and exon-4 W112R mutations of the lysozyme gene with both mutations on the same allele. In T70N, C->A transversion was observed, replacing threonine by asparagine at residue 70 of the mature protein. In addition, the W112R type of mutation undergoes T->C transition and replaces tryptophan to arginine at the 112th residue of the mature protein. This variant was discovered for people of German ancestry. The phenotype is typecasted by the incidence of lysozyme fragments in the form of amyloids, gastrointestinal involvement with recurrent bleeding, and slow-progressing nephropathy. The kidney biopsy revealed amyloid deposits in thickened walls of preglomerular vessels, in the enlarged mesangium and along the walls of glomeruli (Röcken et al. 2006).
Additional studies showed another variant D68G was found for Alys, which occurred due to A->G transition leading to the replacement of glycine to aspartic acid at position 68 of the LYZ protein (Wooliver et al. 2008).
Another study documented that the transversion from T->A at codon 54, thus encoding asparagine instead of tyrosine in exon-2. It was first reported in the Swedish family. Clinically, the phenotype associated with Y54N features weight loss, chronic abdominal pain, diarrhea, sicca syndrome, and malabsorption. Amyloid deposits were found in bone marrow, abdominal fat, stomach, and duodenum (Girnius et al. 2012).
In a profound study, it was found that Alys variant L84S occurs due to altercation of T->C in exon-3 at codon 102, encoding serine in place of leucine at 84th position in the bacteriolytic enzyme. Clinical implication includes gastrointestinal symptoms, nausea, dysphagia, abdominal bloating, and early satiety. Renal biopsy of the patients with L84S had amyloid deposition in arterial walls, basement membranes of the medullary collecting duct, vasa recta, and glomerular mesangium. Moderate tubular atrophy, arteriosclerosis, and intestinal fibrosis were also observed (Nasr et al. 2017b).
Even though there is no distinct therapy is available for Alys, it is supervised symptomatically. The median age of incurring the disease is 25 (9–70 years). The median age of developing ESRD from a presentation of the renal disease is reported to be 11 years. Renal transplantation (RTx) for ESRD and orthotropic liver transplantation for liver rupture cases have been proven successful with commendable medium-term graft function and patient’s survival (Sattianayagam et al. 2012). In all the cases, the amyloid deposition was checked by staining with Congo red dye, and lysozyme was confirmed as amyloid fibril protein by immunoblot analysis using antibodies against the lysozyme. Peculiar changes in the nucleotides were discerned with the help of DNA sequencing.
The diagnosis of Alys remains challenging as it can be simply misguided for primary amyloidosis, which also presents with the same symptoms. Immunohistochemical staining of renal tissue sample, laser microdissection and mass spectrometry from a bone marrow sample for specific amyloidogenic proteins allows for an exact diagnosis and should be performed in all amyloidosis patients so as to additional patients from potentially futile or harmful therapy. The well-known systemic involvement of lysozyme amyloidosis, at present, provides limited options for treatment, even though liver and kidney transplantation appears to be promising palliative treatments (Pleyer et al. 2015).
Gelsolin renal amyloidosis
Gelsolin is a prototype of large superfamily of protein, widely distributed actin binding protein that regulates actin filament length. Human gelsolin is expressed in both an intracellular (81 kDa) and an extracellular/secreted protein (plasma gelsolin) (83 kDa) form that is derived from a single gene by alternative splicing. Gelsolin plays a key role in several physiological processes for instance muscle contraction, organelle trafficking, cell division, and cell motility (Wen et al. 1996). Mutation in plasma gelsolin leads to the deposition of insoluble protein aggregate deposition in various organ and tissues.
Hereditary gelsolin amyloidosis (Agel) is also known as familial amyloidosis or Finnish type (FAF). It is an autosomal dominant polyneuropathy syndrome, mainly involves the peripheral and cranial nerves and culminating in distal sensorimotor neuropathy, corneal lattice dystrophy, and progressive cranial neuropathy (Rowczenio et al. 2014; Solomon et al. 2012). It was first described in the Finnish population by Jouko Meretoja in 1969 (Solomon et al. 2012). Although it was initially reported in Finland, now the cases have been identified globally (Rowczenio et al. 2014). It is typically characterized by nephrotic syndrome and slow-progressing renal failure with existing comorbidities including diabetes and hypertension. Histologically, gelsolin amyloid deposition was predominantly found in the renal glomeruli. Agel amyloidosis was cited to be most common in the Caucasian population yet was quoted in Asians, American, and Hispanic patients (Sethi et al. 2017). This type of renal amyloidosis has been delineated in the kidney, skin, and heart, though the involvement of the kidney is uncommon and its penetrance is 100% (Solomon et al. 2012; Sethi et al. 2017). Patients with renal Agel amyloidosis are diagnosed at the mean age of 63.8 years; the disease has mostly been documented in older patients (Sethi et al. 2017).
Till now, 4 pathogenic mutations of gelsolin gene have been identified; p.D214Y (G654T), p.D214N (G654A), p.G194R (G580A), and p.N211K (C633A) (Srivastava et al. 2018). Clinical manifestations of p.D214N/Y include cranial neuropathy, distal sensorimotor neuropathy, corneal lattice dystrophy, and changes in the skin. Fascinatingly, patients with p.N211K and p.G194R cause renal impairment and proteinuria. A mutation in the GSN gene at positions G654A/T is eminently prevalent. Mutant gelsolin (D187N/Y) loses calcium regulation, which leads to improper folding of the G2 domain and renders furin cleavage site exposed which is concealed in wild-type structure. Furin is the member of proprotein convertase family which recycles between the trans golgi network, endosomes, and the cell surface. The secreted gelsolin variant encounters furin in trans golgi network and undergoes proteolysis producing 68-kDa C-terminal fragment (amino acids 173–755, C68). The C68 fragment is further cleaved upon secretion from the cell by MT1-MMP (membrane type 1 matrix metalloprotease), generating the 8 and 5-kDa fragments (amino acids 173–242 and 173–225, respectively). These fragments subsequently form amyloid fibrils by nucleated polymerization mechanism (Van Overbeke et al. 2015). In a discrete analysis, it was found that 3 out of 4 patients having homozygosity for the G654A mutation had a severe case of nephrotic syndrome and had to undergo a renal transplant. In homozygous mutant individuals, early onset of nephrotic syndrome happens in the 1920s followed by speedy progression to ESRD in the 1930s. Meanwhile, the heterozygous patients frequently had transient proteinuria, and none of them had nephrotic syndrome. Nonetheless, heterozygous patients too developed the amyloid deposits in the glomerulus. The reason for this difference in clinical manifestations is still not known (Yamanaka et al. 2013). In recent years, p. Asn211Lys gelsolin mutation was detected on MS appeared to be a renal restricted form.
Renal biopsy showed light eosinophilic Congo red–positive amyloid deposits in large amounts in patients associated with renal Agel amyloidosis. Light microscopy divulged mesangial and capillary wall expansion with Congo red stain positive regions restricted to glomeruli except for the single case in which focal interstitial amyloid was also seen. Furthermore, electron microscopy results depicted amyloid deposition was observed all along the capillary walls and in the mesangium. Moreover, vascular or medullary amyloid deposition and spicule formation were absent. In a few areas, the amyloid fibrils were arranged as parallel bundles and sheets that had a storiform or wave-like pattern and, the thickness of the fibrils ranged from 7 to 20 nm.
Immunofluorescence studies were not positive for immunoglobulins in some cases; the rest showed equivocal glomerular staining for IgG. Immunohistochemistry showed positive glomerular staining for gelsolin (Sethi et al. 2017). Furthermore, for timely typing of amyloidosis, laser microdissection followed by mass spectrometry must be done on paraffin-embedded renal biopsy specimens, as mass spectrometry enables detection of the mutant (gelsolin, apolipoproteins, AIV, and serum amyloid P (SAP) component) peptide; this underlying cause of the Agel amyloidosis can be further confirmed by Sanger sequencing. SAP scintigraphy is a painless, safe, and nonintrusive technique used for timely diagnosis of patients with Agel amyloidosis, predominantly those who presented with an abnormal clinical phenotype, or lack a family history (Rowczenio et al. 2014).
Transthyretin amyloidosis
Transthyretin (TTR) is a homotetrameric plasma protein, having four identical 127-amino acid β-sheet monomers restrained as a pair of dimers. It is produced primarily not only by the liver but also by the retinal pigment epithelium and choroid plexus. The homodimer functions prominently as a transport protein for retinol and for about 15% of circulating plasma thyroxine (Lobato and Rocha 2012a). Biopsy of the kidney revealed that massive deposition of TTR was found in glomeruli and medullary interstitium.
Hereditary transthyretin amyloidosis (ATTR) is an autosomal dominant disease, caused by the deposition in ATTR, associated with over 100 different mutations. The mutated form is synthesized in liver and undergoes proteolytic remodeling into monomers and C-terminal fragments, which works as a framework for aggregation. These alterations cause the protein to deposit as amyloid in the heart, gastrointestinal tract, eyes, kidneys, peripheral and autonomic nerves, and connective tissue of transversal carpal ligament. This leads to progressive organ failure, and the median survival rate is 10 years (Conceição et al. 2016). A single amino acid substitution of methionine instead of valine at position 30 is widely observed in ATTR patients. Those patients undergoing hemodialysis have a survival span of 21 months after initiation, with septicemia being the prime cause of death (Lobato and Rocha 2012a). The incidence rate of hereditary ATTR is 10%. The usual clinical manifestations are autonomic neuropathy, peripheral axonal polyneuropathy, carpal tunnel syndrome, cardiomyopathy, and/or vitreous opacities. Loss of kidney function can straightforwardly be caused by amyloid, although not directly as a result of the neurogenic bladder caused by the neuropathy.
Diagnostic screening by Congo red staining of fat tissue has less sensitivity; thus, effective diagnostic techniques with higher sensitivity are used such as immunohistochemistry, SAP scintigraphy, and genome sequencing.
Whereas orthotropic liver transplantation (OLT) and Fx-1006A (tafamidis meglumine) are sole options for the treatment. Fx-1006A is a small molecule pharmacological chaperone that binds and stabilizes the tetramer of TTR thus prevents degradation, misfolding, and their subsequent deposition Fx-1006A stabilizes the tetramer of TTR thus, prevents misfolding (Lobato and Rocha 2012a). On the other hand, OLT was the rationale therapy popularized in 1990s, for abrogating the mutant TTR production; OLT has its limitations as it is not viable for cardiac amyloidosis or renal failure. Therefore, kidney and liver transplantations have been performed together with success (Haagsma et al. 2004a). Various potential novel therapies for ATTR are under clinical trials such as diflunisal (binds to TTR via a T4-binding site, stabilizing TTR tetramer), antisense oligonucleotide (repair genes), and single-stranded oligonucleotides (suppress TTR mRNA levels) and TTR small interfering RNA (inhibits TTR expression) (Lobato and Rocha 2012a). Of late a new potential candidate namely Patisiran, a small interfering RNA, targets the conserved domain in all the TTR variants and mitigates the production of mutated as well as the wild type hepatic TT (Kristen et al. 2019). Besides, several studies demonstrated that Mab ATTR can be a potential candidate for antibody therapy and help in suppressing the amyloid formation (Su et al. 2012). Various ATTR variants are described in Table 2.
Apolipoprotein A1
Apolipoprotein A1 amyloidosis is an autosomal dominant, rare, late-onset condition, a multisystemic disease not to mention easily misdiagnosed caused by a mutation in the APOA1 gene (located on chromosome 11q23-q24) that encodes a group of plasma proteins (Gregorini et al. 2005; Lu et al. 2017). It is the major lipoprotein of high-density lipoprotein that promotes efflux of cholesterol from cells and a cofactor of lecithin cholesterol-acyl transferase. This apolipoprotein is expressed primarily in liver, small intestine, and duodenum. It is nonglycosylated protein (molecular weight, 28 kDa) composed of 243 amino acids, and its N-terminal fragment (83–93 residues) has been known as the major components of AApoA-I amyloid fibrils (Pleyer et al. 2015; Gregorini et al. 2005). The disease is mainly observed in elder patients with a mean age of 58 years (Pleyer et al. 2015). It was histologically confirmed that AApoA-I amyloidosis is primarily tubulointerstitial nephritis, typically characterized by nephrotic syndrome, slow-progressing renal dysfunction, associated with secondary focal glomerular sclerosis. Hepatic, cardiac, and renal involvement is the major feature of this disease (Gregorini et al. 2005). Mutations in coding regions (50–93) are most probably to cause renal and hepatic involvement. To date, more than 20 AApoA-I mutations have been reported that are related to the deposition of amyloid fibrils primarily in the kidney, heart, and liver (Lu et al. 2017). Leu75Pro mutation is the most common associated with renal AApoA-I amyloidosis, providing a significant perception of the outcome and natural history of the disease (Pleyer et al. 2015). The group of researchers reported AApoA-I-related renal amyloidosis in Chinese Han population demonstrated with hereditary and nonhereditary forms. Glu34Lys mutation in the APOA1 gene is rare, presented in which a case resulted in major renal, conjunctival, and testicular involvement by the process of amyloid deposition; lipid accumulation was found inside renal amyloid deposits (Andeen et al. 2014). Renal AApoA-I amyloidosis shows the contribution of the medulla with tubulointerstitial nephritis. Glomeruli involvement was absent (Gregorini et al. 2005).
Diagnosis of the renal tubulointerstitial disease is challenging because it is generally not tested in amyloid typing. Immunofluorescence and immunohistochemistry can identify the amyloid subtype but can be confused by background staining caused by loss of antigenic determinants in the fibrillar conformation and serum contamination. LMD and MS-based proteomic analysis of kidney biopsy specimens revealed that ApoA-1 was the causative agent of amyloidosis. MS-based and LMD proteomic analysis can provide 98 to 100% accuracy in the diagnosis of the subtypes of amyloidosis. At present, there is no successful drug for AApoA-I amyloidosis; organ transplantation and supportive treatment are the major therapeutic approaches (Lu et al. 2017).
Apolipoprotein AII
Apolipoprotein A-II amyloidosis (AApoA-II) is an extremely rare and nonneuropathic form of familial amyloidosis. It has been characterized by elevated creatinine and nephrotic range proteinuria with progression to end-stage renal disease.
It is a 17-kDa protein composed of 77 amino acids, expressed in the liver. Each circulating wild-type ApoA-II is bound to plasma HDL by the curiously large apolar faces of its amphipathic α-helices. ApoA-II which is bound with lipid achieves an α-helical structure on HDL and binds strongly to HDL which makes wild-type ApoA-II basically nonexchangeable, protecting from misfolding in vivo. On the other hand, in the absence of bound lipids in vitro, ApoA-II shall become labile to proteolysis and misfolding (Prokaeva et al. 2017). Worldwide, only three AApoA-II mutations were found. A nucleotide replacement at the stop codon of APOA-II brought forth an ApoA-II variant with a 21-residue C-terminal expansion 78Serext21, 78Glyext21, and 78Argext21 [59]. Very recently a group of researchers reported a novel AApoAII variant, 78Leuext21 in a female patient of Filipino-Portuguese descent associated with hereditary renal amyloidosis. Kidney biopsy shows extensive amyloid deposition in glomeruli and modest deposition observed in small vessels. Although, no clinical evidence of interstitium or blood vessel involvement was demonstrated. immunohistochemistry of renal biopsy sample showed positive glomerular staining (Prokaeva et al. 2017).
Apolipoprotein A-IV
Apolipoprotein A-IV (ApoA-IV) is a lately described, uncommon type of systemic amyloidosis characterized by progressive chronic kidney disease and exhibits extensive medullary involvement with sparing of the cortex (Dasari et al. 2016). This is a 46-kDa glycoprotein, synthesized in the small intestine and plays a crucial role in the absorption, metabolism, and transport of dietary lipids (Dasari et al. 2016; Sethi et al. 2012b).
ApoA-IV is the biggest known exchangeable apolipoprotein, circulating in plasma both as in high-density lipoprotein bound and a lipid-free status in the same distribution, sparkly modest lipid-binding affinity. The kidney is the main organ of HDL catabolism: probably locally high concentrations of ApoA-IV and other, still undecided, factors present in the renal medullary location might generate local amyloid fibrils formation and deposition (Obici et al. 2016).
Amyloid deposition was predominantly seen in the glomeruli, vascular, and inner medulla, where the extensive peritubular and interstitial deposition exists whereas it was absent in the cortex region (Dasari et al. 2016; Sethi et al. 2012b). Sethi and group first time reported a unique case of renal amyloidosis that showed ApoA-IV deposition and renal involvement was limited to the medulla. Immunofluorescence microscopy showed that glomeruli were negative for kappa and lambda light chain deposition (Sethi et al. 2012b).
Laser microdissection (LMD) and mass spectrometry (MS/MS) are an insightful tool used for subsequent protein analysis that exposed the amyloid deposition to be made up of chiefly ApoA-IV (Sethi et al. 2012b). Therefore, a significant degree of suspicion and awareness is needed for proper diagnosis (Dasari et al. 2016).
Apolipoprotein C II
Apolipoprotein CII (AApoCII) amyloidosis is another newly described rare form of renal amyloidosis related with apolipoproteins. ApoCII is synthesized in liver and is a major component of chylomicrons, HDL particles, and VLDL particles which plays a key role in the lipid transport in the circulatory system. However, in the absence of lipid, at the physiological pH and ionic strength, ApoCII exhibit propensity to form ribbon-type amyloid fibrils that are acquiescent to a range of structural and biophysical analysis. ApoCII amyloidosis was found higher in men than in women subjects (Kei et al. 2012).
ApoCII was mainly seen in older patients and presented with proteinuria, gradual loss of kidney function, and chronic kidney disease. It was recently found that heterozygous c.206A → T alteration in exon 3 of ApoCII gene (glutamate to valine substitution at codon 69, E69V). The E69V mutation located in the linker region alters the conformation of the apoAII protein, which mediates amyloid fibrillogenesis (Nasr et al. 2017c).
Freshly, it was established that APOC2 p. Lys41Thr mutant is a frequent form related to renal AApoCII amyloidosis. MS findings of the kidney biopsy specimen identified the APOC2 p. Lys41Thr protein. Therefore, Sanger sequencing studies of the ApoCII gene established the p. Lys41Thr (c.122A → C transition) substitution in the protein at the DNA level. This form of amyloidosis is characterized by enormously full-sized mesangial amyloid nodules, frequently obliterating the capillary lumen. The nodules have the relatively peppery and flaky look, are periodic acid Schiff negative and weakly eosinophilic. Even though mesangial nodules are enormous, a few of the glomeruli reveals only segmental involvement. p.Lys41Thr mutation is located in the lipid-binding domain, changing lipid-binding capability of the protein, making the ApoCII protein unstable (Sethi et al. 2018).
Apolipoprotein CII amyloidosis showed predominantly medullary interstitium and glomerular involvement. The glomerular involvement can result in the formation of larger mesangial nodules and can be mystified with Afib amyloidosis; the presence of medullary interstitial and cortical deposits in AApoCII amyloidosis is useful in ruling out Afib amyloidosis. Vascular and cortical involvement is minimal or absent. As assessed by immunofluorescence, amyloid deposits were negative for IgA (kappa and lambda) and IgG. Electron microscopy of renal biopsy specimens revealed the randomly oriented large aggregate of amyloid fibrils (8–14 nm) beside the mesangium and the capillary walls. Immunohistochemistry was performed on paraffin sections of kidney biopsy specimens found to be strongly positive in ApoCII cases (Nasr et al. 2017a).
Apolipoprotein C-III
Apolipoprotein C-III is glycosylated 9-kDa protein composed of 79 amino acids, fabricated in the liver and to some extent in the intestine. It constituent the major structural part of VLDL, although it is also part of chylomicron and HDL. The protein inhibits the hepatic uptake of chylomicrons.
Apolipoprotein C-III (ApoCIII) is a recently described novel form of renal amyloidosis, deficiency of ApoCIII providing cardiovascular protection even in the unfavorable milieu of renal failure. To the best of our knowledge, very recently, a novel D25V ApoCIII variant was demonstrated in French kindred with renal amyloidosis and hypotriglyceridemia. In the lipid-free state, D25V protein is highly fibrillogenic and forms amyloid fibrils under physiological conditions in vitro. It provides clear-sightedness into the genetics of ApoC-III and into HDL and VLDL metabolism in health and disease, illustrating the growing importance of ApoC-III in different biological processes.
Clinical renal manifestations include chronic tubulointerstitial nephritis, sicca syndrome at the age of 20 years, and progressive renal insufficiency. Amyloid deposition was found in the renal cortex, glomeruli with major mesangial distribution, within the peritubular basement membranes and interstitium with tubular atrophy constrained to the inner medulla or nephrotic syndrome. Deposition of amyloid fibrils mostly affected the vascular compartment, along with the walls of renal arterioles leading to lumen obliteration. Immunoelectron microscopic examined that these renal amyloid fibrils were labeled by gold-conjugated anti-apoC-III antibody. In addition to, LMD MS-based proteomics was performed on various tissues: heart, kidney, digestive tract, and salivary glands with several amyloid deposits microdissected per tissue. The variant apoC-III containing Valine 25 was consistently found in the various amyloid deposits together with apoE and SAP in all the proteomes. Thus, 123I-SAP scintigraphy, immunohistochemical staining, immunogold technology, and LMD and MS-based proteomics are the best ways for accurate typing of ApoCIII Amyloidosis (Valleix et al. 2016).
Conclusion
This review article focuses on the recent advancement of renal amyloidosis especially in the area of pathogenesis and diagnosis. In conclusion, renal amyloidosis is a disease that continues to pose challenges to all the stakeholders including patients, physicians, and researchers. Constructive treatment requires accurate diagnosis and molecular typing of the amyloid protein. The histological tests must be confirmed with immunohistochemistry analysis. Furthermore, proteomic analysis can be efficacious in elucidating various facets of renal amyloidosis, such as identification of misfolded protein precursor and discovery of new prognostic and diagnostic biomarkers. MS-based proteomics is currently the gold standard for amyloid typing, due to its high efficiency, and it is capable to overcome drawbacks of antibody-based techniques. LMD/MS enables evaluation of global protein expression patterns; identification of amyloidogenic precursor proteins in a single test is also often possible such as gelsolin, fibrinogen-α, and apolipoproteins and is helpful in differentiating the fibrillary deposits from that of amyloidosis. Apparently, proteomics will enhance our understanding of disease pathogenesis and epidemiology. It provides a comparable amount of information in a single analysis when used with bioinformatic aids, which will allow us to dig deeper into the complexities. Currently, successful treatment for renal amyloidosis is yet to be achieved; there is a sensible need for further investigation on this rare entity to the clearer characterization of the natural course and pathophysiology of this disease to develop new potential diagnosis and treatment strategies.
Abbreviations
- AA:
-
Amyloid A
- ACN:
-
Acetonitrile
- Alys:
-
Lysozyme amyloidosis
- Agel:
-
Gelsolin amyloidosis
- ApoA1:
-
Apolipoprotein A1
- ApoAII:
-
Apolipoprotein AII
- ApoAIII:
-
Apolipoprotein AIII
- ApoAIV:
-
Apolipoprotein AIV
- ApoACII:
-
Apolipoprotein CII
- ApoACIII:
-
Apolipoprotein CIII
- ALECT2:
-
Amyloidogenic leukocyte chemotactic factor 2
- AL:
-
Light chain amyloidosis
- Afib:
-
Fibrinogen amyloidosis
- AN:
-
Autonomic neuropathy
- ATTR:
-
Transthyretin amyloidosis
- CKD:
-
Chronic kidney disease
- CRF:
-
Chronic renal failure
- CTS:
-
Carpel tunnel syndrome
- 1D-PAGE:
-
One-dimensional gel electrophoresis
- 2D-PAGE:
-
Two-dimensional polyacrylamide gel electrophoresis
- ESRD:
-
End-stage renal diseases
- FITC:
-
Fluorescein isothiocyanate
- FTIR:
-
Fourier transform infrared spectroscopy
- FGA:
-
Fibrinogen alpha chain precursor
- GFR:
-
Glomerular filtration rate
- H:
-
Hereditary
- HDL:
-
High-density lipoprotein
- IgG:
-
Immunoglobulin G
- IHC:
-
Immunohistochemistry
- IEF:
-
Isoelectric focusing
- kDa:
-
Kilodalton
- LMD:
-
Laser microdissection
- LYZ:
-
Lysozyme
- L:
-
Localized
- MS:
-
Mass spectrometry
- MT1-MMP:
-
Membrane type 1 matrix metalloprotease
- MRI:
-
Magnetic resonance imaging
- NAC:
-
National amyloidosis centre
- OLT:
-
Orthotropic liver transplantation
- pI :
-
Isoelectric point
- PCR:
-
Polymerase chain reaction
- PN:
-
Peripheral neuropathy
- RTx:
-
Renal transplantation
- RFLP:
-
Restricted length fragmented polymorphism
- S:
-
Systemic
- SAA:
-
Serum amyloid A
- SAP:
-
Serum amyloid P component
- ssNMR:
-
Solid-state nuclear magnetic resonance
- SNP:
-
Single nucleotide polymorphism
- TTR:
-
Transthyretin
References
Abraham RS, Geyer SM, Price-Troska TL, Allmer C, Kyle RA, Gertz MA, Fonseca R (2003) Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain–associated amyloidosis (AL). Blood 101(10):3801–3807. https://doi.org/10.1182/blood-2002-09-2707
Andeen NK, Lam DY, De Boer IH, Nicosia RF (2014) Renal ApoA-1 amyloidosis with Glu34Lys mutation and intra-amyloid lipid accumulation. J Am Soc Nephrol 25(12):2703–2705. https://doi.org/10.1681/ASN.2013060651
Ansari NJJUNJUN (2016) Longest survival with renal AA amyloidosis: development of end stage renal disease after 25 years of AA amyloidosis diagnosis. 3(1):5. https://doi.org/10.2147/CLEP.S39981
Benson MD, James S, Scott K, Liepnieks JJ, Kluve-Beckerman B (2008) Leukocyte chemotactic factor 2: a novel renal amyloid protein. Kidney Int 74(2):218–222. https://doi.org/10.1038/ki.2008.152
Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, Sipe JD, Westermark P (2019) Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid:1–5. https://doi.org/10.1080/13506129.2018.1549825
Bhatia K, Reilly M, Adams D, Davis MB, Hawkes CH, Thomas PK, Said G, Harding AE (1993) Transthyretin gene mutations in British and French patients with amyloid neuropathy. J Neurol Neurosurg Psychiatry 56(6):694–697. https://doi.org/10.1136/jnnp.56.6.694
Bonì F, Milani M, Barbiroli A, Diomede L, Mastrangelo E, de Rosa M (2018) Gelsolin pathogenic Gly167Arg mutation promotes domain-swap dimerization of the protein. Hum Mol Genet 27(1):53–65. https://doi.org/10.1093/hmg/ddx383
Booth DR, Gillmore JD, Persey MR, Booth SE, Cafferty KD, Tennent GA, Madhoo S, Cochrane SW, Whitehead TC, Pasvol G, Hawkins PN (1998) Transthyretin Ile73Val is associated with familial amyloidotic polyneuropathy in a Bangladeshi family. Mutations in brief no. 158. Online. Hum Mutat 12(2):135. https://doi.org/10.1002/(sici)1098-1004(1998)12:2<135::Aid-humu10>3.0.Co;2-6
Bowen K, Shah N, Lewin M (2012) AL-amyloidosis presenting with negative Congo red staining in the setting of high clinical suspicion: a case report. Case reports in nephrology, 2012. https://doi.org/10.1155/2012/593460
Brambilla F, Lavatelli F, Merlini G, Mauri P (2013) Clinical proteomics for diagnosis and typing of systemic amyloidoses. Proteomics–Clin Appl 7(1–2):136–143. https://doi.org/10.1002/prca.201200097
Conceição I, González-Duarte A, Obici L, Schmidt HHJ, Simoneau D, Ong ML, Amass L (2016) “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst 21(1):5–9. https://doi.org/10.1111/jns.12153
Dasari S, Amin MS, Kurtin PJ, Vrana JA, Theis JD, Grogg KL, Alexander MP, Nasr SH, Fervenza FC, Leung N (2016) Clinical, biopsy, and mass spectrometry characteristics of renal apolipoprotein A-IV amyloidosis. Kidney Int 90(3):658–664. https://doi.org/10.1016/j.kint.2016.04.003
de Asúa DR, Costa R, Galván JM, Filigheddu MT, Trujillo D, Cadiñanos JJC (2014) Systemic AA amyloidosis: epidemiology, diagnosis, and management. 6:369. https://doi.org/10.2147/CLEP.S39981
Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, Livneh A, Lesnyak O, Puéchal X, Lachmann HJJNEJM (2007) Eprodisate for the treatment of renal disease in AA amyloidosis. 356(23):2349–2360. https://doi.org/10.1080/13506129.2018.1474733
Dogan A (2017) Amyloidosis: insights from proteomics. Annu Rev Pathol 12:277–304. https://doi.org/10.1146/annurev-pathol-052016-100
Erdogmus S, Kendi Celebi Z, Akturk S, Kumru G, Duman N, Ates K, Erturk S, Nergizoglu G, Kutlay S, Sengul SJA (2018) Profile of renal AA amyloidosis in older and younger individuals: a single-centre experience. 25(2):115–119. https://doi.org/10.1080/13506129.2018.1474733
Field-Smith A, Morgan GJ, Davies FE (2006) Bortezomib (Velcade™) in the treatment of multiple myeloma. Ther Clin Risk Manag 2(3):271–279. https://doi.org/10.2147/tcrm.2006.2.3.271
Fuah KW, Lim CTS (2018) Renal-limited AL amyloidosis–a diagnostic and management dilemma. BMC nephrology, 19(1):307. https://doi.org/10.1186/s12882-018-1118-8
Gafni J, Fischel B, Reif R, Yaron M, Pras M (1985) Amyloidotic polyneuropathy in a Jewish family. Evidence for the genetic heterogeneity of the lower limb familial amyloidotic neuropathies. Q J Med 55(216):33–44. https://doi.org/10.1093/qjmed.a067851
Gilbertson JA, Theis JD, Vrana JA, Lachmann H, Wechalekar A, Whelan C, Hawkins PN, Dogan A, Gillmore JD (2015, 2014) A comparison of immunohistochemistry and mass spectrometry for determining the amyloid fibril protein from formalin-fixed biopsy tissue. J Clin Pathol:202722. https://doi.org/10.1136/jclinpath-2014-202722
Gillmore JD, Lachmann HJ, Rowczenio D, Gilbertson JA, Zeng C-H, Liu Z-H, Li L-S, Wechalekar A, Hawkins PNJJASN (2009) Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen Aα-chain amyloidosis. 20(2):444–451. https://doi.org/10.1681/ASN.2008060614
Girnius S, Skinner M, Spencer B, Prokaeva T, Bartholomew C, O’Hara C, Seldin DC, Connors LH (2012) A new lysozyme tyr54asn mutation causing amyloidosis in a family of Swedish ancestry with gastrointestinal symptoms. Amyloid 19(4):182–185. https://doi.org/10.3109/13506129.2012.723074
Granel B, Valleix S, Serratrice J, Chérin P, Texeira A, Disdier P, Weiller P-J, Grateau G (2006) Lysozyme amyloidosis: report of 4 cases and a review of the literature. Medicine 85(1):66–73. https://doi.org/10.1097/01.md.0000200467.51816.6d
Gregorini G, Izzi C, Obici L, Tardanico R, Röcken C, Viola BF, Capistrano M, Donadei S, Biasi L, Scalvini T (2005) Renal apolipoprotein AI amyloidosis: a rare and usually ignored cause of hereditary tubulointerstitial nephritis. J Am Soc Nephrol 16(12):3680–3686. https://doi.org/10.1681/ASN.2005040382
Griffin JW, Bradshaw PC (2018) In silico prediction of novel residues involved in amyloid primary nucleation of human I56T and D67H lysozyme. BMC Struct Biol 18(1):9. https://doi.org/10.1186/s12900-018-0088-1
Gurevitz SA, Stone MJ (2012) Amyloidosis: diagnosis and treatment edited by Morie a. Gertz, MD, and S. Vincent Rajkumar, MD. Proc (Baylor Univ Med Cent) 25 (2):166–166 https://doi.org/10.1080/08998280.2012.11928822, 168
Haagsma EB, Hawkins PN, Benson MD, Lachmann HJ, Bybee A, Hazenberg BP (2004a) Familial amyloidotic polyneuropathy with severe renal involvement in association with transthyretin Gly47Glu in Dutch, British and American-Finnish families. Amyloid 11(1):44–49. https://doi.org/10.1080/13506120410001682578
Haagsma EB, Hawkins PN, Benson MD, Lachmann HJ, Bybee A, Hazenberg BP (2004b) Familial amyloidotic polyneuropathy with severe renal involvement in association with transthyretin Gly47Glu in Dutch, British and American-Finnish families. Amyloid 11(1):44–49. https://doi.org/10.1080/13506120410001682578
Hassen M, Bates W, Moosa MR (2019) Pattern of renal amyloidosis in South Africa. BMC nephrology, 20(1):406. https://doi.org/10.1186/s12882-019-1601-x
Hopfer H, Wiech T, Mihatsch MJ (2011) Renal amyloidosis revisited: amyloid distribution, dynamics and biochemical type. Nephrol Dial Transplant 26(9):2877–2884. https://doi.org/10.1093/ndt/gfq83
Ikeda S (2008) Diagnosis and treatment in systemic amyloidosis. Rinsho byori. Jpn J Clin Pathol 56(2):121–129
Jimenez-Zepeda VH, Leung NJRIC (2014) ALECT2 amyloidosis: a new type of systemic amyloid highly prevalent in the Hispanic population 66(3):269–273
Kei AA, Filippatos TD, Tsimihodimos V, Elisaf MS (2012) A review of the role of apolipoprotein C-II in lipoprotein metabolism and cardiovascular disease. Metabolism 61(7):906–921. https://doi.org/10.1016/j.metabol.2011.12.002
Kristen AV, Ehlermann P, Helmke B, Hund E, Haberkorn U, Linke RP, Katus HA, Winter P, Altland K, Dengler TJ (2007) Transthyretin valine-94-alanine, a novel variant associated with late-onset systemic amyloidosis with cardiac involvement. Amyloid 14(4):283–287. https://doi.org/10.1080/13506120701616383
Kristen AV, Ajroud-Driss S, Conceição I, Gorevic P, Kyriakides T, Obici L (2019) Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegenerat Dis Manag 9(1):5–23
Kulkarni U, Valson A, Korula A, Mathews V (2015) Leukocyte derived chemotaxin 2 (ALECT2) amyloidosis. Mediterr J Hematol Infect Dis 7(1). https://doi.org/10.4084/MJHID.2015.043
Lachmann HJ, Goodman HJ, Gilbertson JA, Gallimore JR, Sabin CA, Gillmore JD, Hawkins PNJNEJM (2007) Natural history and outcome in systemic AA amyloidosis. N Engl J Med 356(23):2361–2371. https://doi.org/10.1056/NEJMoa070265
Lane T, Pinney JH, Gilbertson JA, Hutt DF, Rowczenio DM, Mahmood S, Sachchithanantham S, Fontana M, Youngstein T, Quarta CCJA (2017) Changing epidemiology of AA amyloidosis: clinical observations over 25 years at a single national referral centre. 24(3):162–166. https://doi.org/10.1080/13506129.2017.1342235
Larsen CP, Kossmann RJ, Beggs ML, Solomon A, Walker PD (2014) Clinical, morphologic, and genetic features of renal leukocyte chemotactic factor 2 amyloidosis. Kidney Int 86(2):378–382. https://doi.org/10.1038/ki.2014.11
Larsen CP, Ismail W, Kurtin PJ, Vrana JA, Dasari S, Nasr SH (2016) Leukocyte chemotactic factor 2 amyloidosis (ALECT2) is a common form of renal amyloidosis among Egyptians. Mod Pathol 29(4):416–420. https://doi.org/10.1038/modpathol.2016.29
Lavatelli F, Merlini G (2017) How do we improve treatments for patients with amyloidosis using proteomics? Expert Rev Proteomics 14(7):561–563. https://doi.org/10.1080/14789450.2017.1331737
Lavatelli F, Vrana JA (2011) Proteomic typing of amyloid deposits in systemic amyloidoses. Amyloid 18(4):177–182. https://doi.org/10.3109/13506129.2011.630762
Leung N, Nasr SH, Sethi S (2012) How I diagnose amyloidosis. Blood 2012:2003–413682
Li Z, Xu H, Liu D, Li D, Liu G, Wang S-x (2019a) Hereditary renal amyloidosis with a variant lysozyme p. Trp82Arg in a Chinese family: case report and literature review. BMC Nephrol 20(1):1–6. https://doi.org/10.1186/s12882-019-1496-6
Li D, Liu D, Xu H, Yu X-j, M-h Z, Wang S-x (2019b) Typing of hereditary renal amyloidosis presenting with isolated glomerular amyloid deposition. BMC Nephrol 20(1):1–8. https://doi.org/10.1186/s12882-019-1667-5
Lim A, Prokaeva T, McComb ME, Connors LH, Skinner M, Costello CE (2003) Identification of S-sulfonation and S-thiolation of a novel transthyretin Phe33Cys variant from a patient diagnosed with familial transthyretin amyloidosis. Prot Sci 12(8):1775–1785. https://doi.org/10.1110/ps.0349703
Lin J-C, Liu H-L (2006) Protein conformational diseases: from mechanisms to drug designs. Curr Drug Discov Technol 3(2):145–153. https://doi.org/10.2174/157016306778108866
Liu JY, Guo YJ, Zhou CK, Ye YQ, Feng JQ, Yin F, Jiang XM (2011) Clinical and histopathological features of familial amyloidotic polyneuropathy with transthyretin Val30Ala in a Chinese family. J Neurol Sci 304(1–2):83–86. https://doi.org/10.1016/j.jns.2011.02.005
Lobato L, Rocha A (2012a) Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol 7(8):1337–1346. https://doi.org/10.2215/CJN.08720811
Lobato L, Rocha A (2012b) Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol 7(8):1337–1346. https://doi.org/10.2215/cjn.08720811
Logan A, Zuppan C, Pi A, Zhang Z, Jaipaul N (2016) Rare and unusual clinicopathologic presentation of renal AL amyloidosis. JRSM open 7(5):2054270416640156. https://doi.org/10.1177/2054270416640156
Lu C, Zuo K, Lu Y, Liang S, Huang X, Zeng C, Zhang J, An Y, Wang J (2017) Apolipoprotein A-1-related amyloidosis 2 case reports and review of the literature. Medicine 96(39):e8148. https://doi.org/10.1097/MD.0000000000008148
Milani P, Merlini G, Palladini G (2018) Novel therapies in light chain amyloidosis. Kidney Int Rep 3(3):530–541. https://doi.org/10.1016/j.ekir.2017.11.017
Nasr SH, Dogan A, Larsen CPJCJASN (2015) Leukocyte cell–derived chemotaxin 2–associated amyloidosis: A recently recognized disease with distinct clinicopathologic characteristics. 10(11):2084–2093. https://doi.org/10.2215/CJN.12551214
Nasr SH, Dasari S, Hasadsri L, Theis JD, Vrana JA, Gertz MA, Muppa P, Zimmermann MT, Grogg KL, Dispenzieri A (2017a) Novel type of renal amyloidosis derived from apolipoprotein-CII. J Am Soc Nephrol 28(2):439–445. https://doi.org/10.1681/ASN.2015111228
Nasr SH, Dasari S, Mills JR, Theis JD, Zimmermann MT, Fonseca R, Vrana JA, Lester SJ, McLaughlin BM, Gillespie R (2017b) Hereditary lysozyme amyloidosis variant p. Leu102Ser associates with unique phenotype. J Am Soc Nephrol 28(2):431–438. https://doi.org/10.1681/ASN.2016090951
Nasr SH, Dasari S, Mills JR, Theis JD, Zimmermann MT, Fonseca R, Vrana JA, Lester SJ, McLaughlin BM, Gillespie R, Highsmith WE Jr, Lee JJ, Dispenzieri A, Kurtin PJ (2017c) Hereditary lysozyme amyloidosis variant p.Leu102Ser associates with unique phenotype. J Am Soc Nephrol 28(2):431–438. https://doi.org/10.1681/asn.2016090951
Obici L, Nuvolone M, Merlini G (2016) Expanding the spectrum of systemic amyloid diseases: a new hint from the kidney. Kidney Int 90(3):479–481. https://doi.org/10.1016/j.kint.2016.05.029
Ong S, Rajasingam R (2007) Abdominal subcutaneous fat aspiration—an alternative method to diagnose amyloidosis. Med J Malaysia 62(1):68–69
Papa R, Lachmann HJJRDC (2018) Secondary, AA, amyloidosis. 44(4):585–603. https://doi.org/10.1016/j.rdc.2018.06.004
Pelo E, Da Prato L, Ciaccheri M, Castelli G, Gori F, Pizzi A, Torricelli F, Marconi G (2002) Familial amyloid polyneuropathy with genetic anticipation associated to a gly47glu transthyretin variant in an Italian kindred. Amyloid 9(1):35–41. https://doi.org/10.3109/13506120209072443
Pepys M, Hawkins P, Booth D, Vigushin D, Tennent G, Soutar A, Totty N, Nguyen O, Blake C, Terry C (1993) Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 362(6420):553–557. https://doi.org/10.1038/362553a0
Picken MM (2010) Amyloidosis—where are we now and where are we heading? Arch Pathol Lab Med 134(4):545–551. https://doi.org/10.1043/1543-2165-134.4.545
Picken MMJK (2014) Alect2 amyloidosis: primum non nocere (first, do no harm). 86(2):229–232. https://doi.org/10.1038/ki.2014.45
Picken MM (2015) Proteomics and mass spectrometry in the diagnosis of renal amyloidosis. Clin Kidney J 8(6):665–672. https://doi.org/10.1093/ckj/sfv087
Pleyer C, Flesche J, Saeed F (2015) Lysozyme amyloidosis—a case report and review of the literature. Clin Nephrol Case Stud 3:42. https://doi.org/10.5414/CNCS108538
Prokaeva T, Akar H, Spencer B, Havasi A, Cui H, O’Hara CJ, Gursky O, Leszyk J, Steffen M, Browning S (2017) Hereditary renal amyloidosis associated with a novel apolipoprotein A-II variant. Kidney Int Rep 2(6):1223–1232. https://doi.org/10.1016/j.ekir.2017.07.009
Quock TP, Yan T, Chang E, Guthrie S, Broder MSJB (2018) Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv 2(10):1046–1053. https://doi.org/10.1182/bloodadvances.2018016402
Reitamo S, Klockars M, Adinolfi M, Osserman E (1978) Human lysozyme (origin and distribution in health and disease). La Ricerca in Clinica e in Laboratorio 8(4):211–231
Rezk T, Gilbertson JA, Rowczenio D, Bass P, Lachmann HJ, Wechalekar AD, Fontana M, Mahmood S, Sachchithanantham S, Whelan CJ (2016) Diagnosis, pathogenesis and outcome in leucocyte chemotactic factor 2 (ALECT2) amyloidosis. Nephrol Dial Transplant 33(2):241–247. https://doi.org/10.1093/ndt/gfw375
Riboldi G, Del Bo R, Ranieri M, Magri F, Sciacco M, Moggio M, Bresolin N, Corti S, Comi GP (2011) Tyr78Phe transthyretin mutation with predominant motor neuropathy as the initial presentation. Case Rep Neurol 3(1):62–68. https://doi.org/10.1159/00032492
Röcken C, Becker K, Fändrich M, Schroeckh V, Stix B, Rath T, Kähne T, Dierkes J, Roessner A, Albert FW (2006) ALys amyloidosis caused by compound heterozygosity in exon 2 (Thr70Asn) and exon 4 (Trp112Arg) of the lysozyme gene. Hum Mutat 27(1):119–120. https://doi.org/10.1002/humu.9393
Rona G, Pasaoglu L, Ozkayar N, Ciliz D, Toprak U, Kaya T, Abat G (2016) Functional evaluation of secondary renal amyloidosis with diffusion-weighted MR imaging. Ren Fail 38(2):249–255. https://doi.org/10.3109/0886022X.2015.1128252
Rowczenio D, Tennent GA, Gilbertson J, Lachmann HJ, Hutt DF, Bybee A, Hawkins PN, Gillmore JD (2014) Clinical characteristics and SAP scintigraphic findings in 10 patients with AGel amyloidosis. Amyloid 21(4):276–281. https://doi.org/10.3109/13506129.2014.973105
Rowczenio D, Stensland M, de Souza GA, Strøm EH, Gilbertson JA, Taylor G, Rendell N, Minogue S, Efebera YA, Lachmann HJ (2017) Renal amyloidosis associated with 5 novel variants in the fibrinogen a alpha chain protein. Kidney Int Rep 2(3):461–469. https://doi.org/10.1016/j.ekir.2016.11.005
Said SM, Sethi S, Valeri AM, Leung N, Cornell LD, Fidler ME, Hernandez LH, Vrana JA, Theis JD, Quint PSJCJASN (2013) Renal amyloidosis: origin and clinicopathologic correlations of 474 recent cases. CJN:10491012. https://doi.org/10.2215/CJN.10491012
Said SM, Sethi S, Valeri AM, Chang A, Nast CC, Krahl L, Molloy P, Barry M, Fidler ME, Cornell LD (2014) Characterization and outcomes of renal leukocyte chemotactic factor 2-associated amyloidosis. Kidney Int 86(2):370–377. https://doi.org/10.1038/ki.2013.558
Saito F, Nakazato M, Akiyama H, Kitahara Y, Date Y, Iwasaki Y, Harasawa S, Hisaki R, Horie T, Kinukawa N, Watanabe T, Sakamaki T, Yagi H, Hoshii Y, Yutani C, Kanmatsuse K (2001) A case of late onset cardiac amyloidosis with a new transthyretin variant (lysine 92). Hum Pathol 32(2):237–239. https://doi.org/10.1053/hupa.2001.22013
Saraiva MJ, Costa PP, Goodman DS (1985) Biochemical marker in familial amyloidotic polyneuropathy, Portuguese type. Family studies on the transthyretin (prealbumin)-methionine-30 variant. J Clin Invest 76(6):2171–2177. https://doi.org/10.1172/jci112224
Sattianayagam P, Gibbs S, Rowczenio D, Pinney J, Wechalekar A, Gilbertson J, Hawkins P, Lachmann H, Gillmore J (2012) Hereditary lysozyme amyloidosis–phenotypic heterogeneity and the role of solid organ transplantation. J Intern Med 272(1):36–44. https://doi.org/10.1111/j.1365-2796.2011.02470.x
Scafi M, Valleix S, Benyamine A, Jean E, Harle JR, Rossi P, Daniel L, Schleinitz N, Granel B (2018) Lysozyme amyloidosis. Rev Med Interne. https://doi.org/10.1016/j.revmed.2018.08.008
Sethi S, Theis JD (2018) Pathology and diagnosis of renal non-AL amyloidosis. J Nephrol:1–8. https://doi.org/10.1007/s40620-017-0426-6
Sethi S, Theis JD, Leung N, Dispenzieri A, Nasr SH, Fidler ME, Cornell LD, Gamez JD, Vrana JA, Dogan A (2010) Mass spectrometry–based proteomic diagnosis of renal immunoglobulin heavy chain amyloidosis. Clin J Am Soc Nephrol 5(12):2180–2187. https://doi.org/10.2215/CJN.02890310
Sethi S, Vrana JA, Theis JD, Leung N, Sethi A, Nasr SH, Fervenza FC, Cornell LD, Fidler ME, Dogan A (2012a) Laser microdissection and mass spectrometry–based proteomics aids the diagnosis and typing of renal amyloidosis. Kidney Int 82(2):226–234. https://doi.org/10.1038/ki.2012.108
Sethi S, Theis JD, Shiller SM, Nast CC, Harrison D, Rennke HG, Vrana JA, Dogan A (2012b) Medullary amyloidosis associated with apolipoprotein A-IV deposition. Kidney Int 81(2):201–206. https://doi.org/10.1038/ki.2011.316
Sethi S, Dasari S, Amin MS, Vrana JA, Theis JD, Alexander MP, Kurtin PJ (2017) Clinical, biopsy, and mass spectrometry findings of renal gelsolin amyloidosis. Kidney Int 91(4):964–971. https://doi.org/10.1016/j.kint.2016.11.017
Sethi S, Dasari S, Plaisier E, Ronco P, Nasr SH, Brocheriou I, Theis JD, Vrana JA, Zimmermann MT, Quint PS (2018) Apolipoprotein CII amyloidosis associated with p. Lys41Thr mutation. Kidney International Reports https://doi.org/10.1016/j.ekir.2018.04.009
Sivalingam V, Patel BK (2016) Familial mutations in fibrinogen Aα (FGA) chain identified in renal amyloidosis increase in vitro amyloidogenicity of FGA fragment. Biochimie 127:44–49. https://doi.org/10.1016/j.biochi.2016.04.020
Solomon JP, Page LJ, Balch WE, Kelly JW (2012) Gelsolin amyloidosis: genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit Rev Biochem Mol Biol 47(3):282–296. https://doi.org/10.3109/10409238.2012.661401
Srivastava A, Singh J, Singh Yadav SP, Arya P, Kalim F, Rose P, Ashish KB (2018) The gelsolin pathogenic D187N mutant exhibits altered conformational stability and forms amyloidogenic oligomers. Biochemistry 57(16):2359–2372. https://doi.org/10.1021/acs.biochem.8b00039
Su Y, Jono H, Torikai M, Hosoi A, Soejima K, Guo J, Tasaki M, Misumi Y, Ueda M, Shinriki S (2012) Antibody therapy for familial amyloidotic polyneuropathy. Amyloid 19(sup1):45–46. https://doi.org/10.3109/13506129.2012.674075
Tariq H, Sharkey FE (2018) Leukocyte cell-derived chemotaxin-2 amyloidosis (ALECT2) in a patient with lung adenocarcinoma: an autopsy report and literature review. Int J Surg Pathol 26(3):271–275. https://doi.org/10.1177/1066896917735893
Tavares I, Silvano J, Pereira PR, Silva R, Lobato L, Henriques AC, Escada L, Macario F, Sampaio S (2018) Renal transplantation in patients with hereditary fibrinogen amyloidosis. Transplantation 102:S550–S144. https://doi.org/10.3109/13506129.2015.1037389
Uemichi T, Liepnieks JJ, Benson MD (1994) Hereditary renal amyloidosis with a novel variant fibrinogen. J Clin Invest 93(2):731–736. https://doi.org/10.1172/JCI117027
Valleix S, Drunat S, Philit J-B, Adoue D, Piette J-C, Droz D, MacGregor B, Canet D, Delpech M, Grateau G (2002) Hereditary renal amyloidosis caused by a new variant lysozyme W64R in a French family. Kidney Int 61(3):907–912. https://doi.org/10.5414/CNCS10853810.1046/j.1523-1755.2002.00205.x
Valleix S, Verona G, Jourde-Chiche N, Nédelec B, Mangione PP, Bridoux F, Mangé A, Dogan A, Goujon J-M, Lhomme M (2016) D25V apolipoprotein C-III variant causes dominant hereditary systemic amyloidosis and confers cardiovascular protective lipoprotein profile. Nat Commun 7:10353. https://doi.org/10.1038/ncomms10353
Van Overbeke W, Wongsantichon J, Everaert I, Verhelle A, Zwaenepoel O, Loonchanta A, Burtnick LD, De Ganck A, Hochepied T, Haigh J (2015) An ER-directed gelsolin nanobody targets the first step in amyloid formation in a gelsolin amyloidosis mouse model. Hum Mol Genet 24(9):2492–2507. https://doi.org/10.1093/hmg/ddv010
Wallace MR, Dwulet FE, Williams EC, Conneally PM, Benson MD (1988) Identification of a new hereditary amyloidosis prealbumin variant, Tyr-77, and detection of the gene by DNA analysis. J Clin Invest 81(1):189–193. https://doi.org/10.1172/jci113293
Weisel JW (2005) Fibrinogen and fibrin. In: advances in protein chemistry, vol 70. Elsevier, pp 247–299. https://doi.org/10.1016/S0065-3233(05)70008-5
Weisel JW, Litvinov RI (2013, 2012) Mechanisms of fibrin polymerization and clinical implications. Blood:2009–306639. https://doi.org/10.1182/blood-2012-09-306639
Wen D, Corina K, Chow EP, Miller S, Janmey PA, Pepinsky RB (1996) The plasma and cytoplasmic forms of human gelsolin differ in disulfide structure. Biochemistry 35(30):9700–9709. https://doi.org/10.1021/bi960920n
Wooliver C, Coriu D, Murphy C, Kestler D, Wang S, Weiss D, Solomon A Familial amyloidosis associated with a novel mutation (D68G) in the lysozyme gene. In: XIth Int Symp Amyloidosis, 2008. pp 208–210 https://doi.org/10.1201/9781420043358.ch70
Yamamoto H, Hashimoto T, Kawamura S, Hiroe M, Yamashita T, Ando Y, Yokochi T (2018) Hereditary cardiac amyloidosis associated with Pro24Ser transthyretin mutation: a case report. J Med Case Rep 12(1):1–5. https://doi.org/10.1186/s13256-018-1931-5
Yamanaka S, Miyazaki Y, Kasai K, S-i I, Kiuru-Enari S, Hosoya T (2013) Hereditary renal amyloidosis caused by a heterozygous G654A gelsolin mutation: a report of two cases. Clin Kidney J 6(2):189–193. https://doi.org/10.1093/ckj/sft007
Yazaki M, Farrell SA, Benson MD (2003) A novel lysozyme mutation Phe57Ile associated with hereditary renal amyloidosis. Kidney Int 63(5):1652–1657. https://doi.org/10.1046/j.1523-1755.2003.00904.x
Yazaki M, Yoshinaga T, Sekijima Y, Nishio S, Kanizawa Y, Kametani F, Miyashita K, Hachiya N, Higuchi K, S-i I (2015) The first pure form of Ostertag-type amyloidosis in Japan: a sporadic case of hereditary fibrinogen A α-chain amyloidosis associated with a novel frameshift variant. Amyloid 22(2):142–144. https://doi.org/10.3109/13506129.2015.1037389
Yazaki M, Yoshinaga T, Sekijima Y, Kametani F, Okumura N (2018) Hereditary fibrinogen Aα-chain amyloidosis in Asia: clinical and molecular characteristics. Int J Mol Sci 19(1):320. https://doi.org/10.3390/ijms19010320
Acknowledgments
The authors acknowledge the Indian Council of Medical Research (ICMR), Govt. of India for providing Junior Research Fellowship to Nimisha Gupta, Grant No. 5/4/7-8/2012/NCD-II.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Consent for publication
All authors read the finalized manuscript and gave their consent for its publication.
Additional information
Handling Editor: Klaudia Brix
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gupta, N., Kaur, H. & Wajid, S. Renal amyloidosis: an update on diagnosis and pathogenesis. Protoplasma 257, 1259–1276 (2020). https://doi.org/10.1007/s00709-020-01513-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00709-020-01513-0