
Abstract
Amyloidosis is a group of diseases characterized by deposition of amyloid fibrils in soft tissues and organs. These fibrils all have the similar characteristics that distinguish them as amyloid. More than 24 proteins have been identified to form amyloid via several mechanisms such as genetic mutation, accumulation, age associated, and multifactorial. The kidney is one of the most commonly affected organs, but affinity to different organs may be predetermined by the native protein. The diagnosis of amyloidosis required demonstration of amyloid deposits in the tissues. In kidney tissue, amyloid appears as pale amorphous extracellular deposits that are periodic acid Schiff (PAS) and silver negative. Congo red staining will produce a green birefringence when viewed by polarized light. Amyloid fibrils are characteristically 7–12 nm in diameter and are randomly arranged on electron microscopy. Once amyloid is detected, typing must be performed to identify the native protein as treatment is type specific. The most common method of amyloid typing is with immunofluorescence or immunohistochemistry. If antibodies are not available, genetic testing may be helpful in identifying hereditary cases. The most advanced method of amyloid typing is liquid chromatography–tandem mass spectrometry. Once amyloid is identified, serum amyloid P (SAP) scintigraphy can be used to locate amyloid in the body. Treatment of amyloidosis depends on the type. Anti-myeloma therapy is used to treat immunoglobulin light chain amyloidosis (AL), antimicrobials or anti-inflammatory medications are used for serum amyloid A (AA), and organ transplantation may be used for some hereditary amyloidoses.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Amyloidosis represents a group of diseases characterized by the deposition of amyloid fibrils in various tissues [1]. The result is progressive organ failure that can lead to death. These fibrils are 7–12 nm in diameter and are randomly arranged. They have the ability to take up Congo red dye and give off an apple-green birefringence when viewed with polarized light. This is a pathognomonic feature which separates amyloid from other fibrils which cause kidney disease.
To date, over 24 types of protein are known to cause amyloidosis in human [2]. The fibrils form as a result of misfolding which causes the protein to take up a pathologic confirmation that allows for subsequent self-aggregation. Several amyloidogenic mechanisms have been identified [1, 2]. In some, there is a natural propensity in the native protein to misfold. With these proteins, amyloid is formed as a result of accumulation or overproduction. Examples of this include senile amyloidosis (wild type transthyretin) which accumulates as a result of aging, dialysis-related amyloidosis (Aβ[beta]2m) where β[beta]2-microglobulin accumulates as a result of renal failure, and secondary amyloidosis (AA) where serum amyloid A is overproduced as a result of persistent inflammation or infection. Others are the result of a genetic mutation that produces an amyloidogenic protein (Table 21.1). Examples of this are (mutant) transthyretin (ATTR), fibrinogen α[alpha]-chain (AFib), lysozyme (ALys), apolipoprotein AI, and AII (AApoAI and AApoAII). Proteins can also become amyloidogenic after proteolytic modification. This is seen in Alzheimer’s disease with β[beta]-amyloid precursor protein (APP) and immunoglobulin light chain (AL). Prion which causes transmissible spongiform encephalopathies such as Creutzfeldt–Jacob disease, Gerstmann–Sträussler–Scheinker syndrome, fatal familial insomnia, and kuru is also considered an amyloidogenic protein (APrP). In some, multiple mechanisms may be involved. The best example of this is AL amyloidosis in which the monoclonal light chain is overproduced by the plasma-cell clones. These light chains undergo a partial proteolytic digestion, and some have mutations which may enhance the amyloidogenic potential of the monoclonal light chain [1].
Different forms of amyloidosis have different organ tropism [2]. Some are quite specific such as the case of cystatin c (ACys) which causes the Icelandic form hereditary cerebral amyloid angiopathy resulting in cerebral hemorrhage, stroke, and dementia. Similarly, Aβ[beta] protein precursor (Aβ[beta]) is responsible for Alzheimer’s disease and is localized to the central nervous system. In others, skin is a preferential organ of deposition. Lichenoid or macular amyloidosis is the result of keratin (AD) deposition possibly as a result of trauma. AIns (insulin) can be found in diabetic patients near their insulin injection site. Nodular localized cutaneous amyloidosis is a form of localized AL that is often produced by polyclonal plasma cells probably in response to a localized inflammatory reaction. Systemic amyloidosis occurs when visceral organs are involved. These forms are often fatal as the persistent deposition of amyloid results in progressive organ failure (Table 21.2).
Renal involvement is common in many forms of amyloidosis. Proteinuria frequently occurs and can be massive (>10 g/day) [3]. Renal insufficiency is variable and often depends on the severity of disease. Renal insufficiency with little proteinuria has also been described in a small subset of patients in which the amyloid preferentially deposits in walls of blood vessels instead of glomerular basement membranes or interstitium [4, 5]. Types of amyloid that involve the kidney include AL, AH (immunoglobulin heavy chain), AA, AFib, AApoAII, and ALECT2 (leukocyte cell-derived chemotaxin-2) [2, 6]. Renal involvement is uncommon in patients with ATTR which usually present with cardiomyopathy and neuropathy, but rare reports of renal involvement including end-stage renal disease have been reported. Finally, Aβ[beta]2m occurs only in dialysis-dependent patients due to a lack of clearance by dialysis [7]. Its main manifestations are soft tissue deposition (carpal tunnel) and arthropathy. However, autopsy studies have discovered systemic deposition usually in vascular beds including those of the kidneys. However, since these patients already had end-stage renal disease, the renal amyloidosis is never manifested.
Pathogenesis
The precursor amyloid proteins all share a common characteristic to misfold into β[beta]-sheet fibrillar protein [1]. This propensity can be natural or as a result of partial digestion as in the case of AL. The initial step involve formation of β[beta]-strands which allow the protein to be stacked on one another utilizing hydrogen bonds of alternating C=O and N–H groups. This self-assembly process eventually forms a sheet of polypeptides. The sheets are then mated together via a dry interface through a process known as the steric zipper [8]. In this cross-β[beta] model, sheets are mated together to form protofilaments. Four to six protofilaments are then braided together to form a fibril. Congo red dye is capable of intercalating the space formed between the protofilaments in a fibril which allows it to be used as a diagnostic tool. Ultrastructurally, fibrils from different precursor proteins are indistinguishable from one another despite the fact that precursor proteins come in different sizes and tertiary structures.
The pathogenic process of amyloid fibril is still not completely understood. Obviously, deposition of the amyloid fibrils certainly plays a big role. In cardiac amyloidosis, the heart is concentrically thickened resulting initially in diastolic dysfunction and later as disease progresses, systolic dysfunction. In the kidney, degree of proteinuria is associated with location of amyloid deposition while, glomerular filtration rate is determined by amount of deposition in the glomerulus. However, recent evidence suggests fibril deposition may not be necessary for cellular toxicity. Exposure to the precursor protein alone is sufficient for cytotoxicity to occur in cardiac myocytes [9]. The same phenomenon has also been noted in the kidney. Patients with massive proteinuria have been known to have very little amyloid deposits in their kidney. In fact, some of these patients were initially diagnosed as minimal-change disease [10]. Correct diagnosis was made when their biopsy was reviewed for failure to respond to steroids. Conversely, large amyloid deposits have been found in patients who responded to therapy and have normal proteinuria [11]. Electron microscopy of these patients suggests repair of the glomerular basement membrane can occur despite the presence of amyloid fibril [12].
Diagnosis
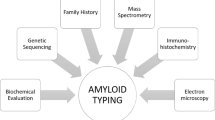
The diagnosis of amyloidosis requires the demonstration of the amyloid fibrils in the tissue (Fig. 21.1). The most commonly used method is Congo red staining [1, 3]. Congo red intercalates the fibrils and gives off an apple-green birefringence when viewed with polarized light. It is fairly sensitive but highly specific for amyloid fibrils. Thioflavin T is another stain that binds the β[beta]-sheet and gives off an enhanced fluorescence. However, it is considered to be less sensitive and specific than Congo red. Fibrils are also detected by electron microscopy. Characteristically, it is randomly arranged and has a diameter of 7–12 nm. These characteristics can be used to distinguish AL from other renal diseases with fibrillary deposits [13]. They include fibrillary glomerulonephritis, immunotactoid glomerulonephritis, cryoglobulinemia, hereditary nephropathies with fibronectin, and collagenofibrotic glomerulopathy. Fibrillary collagen can also be found in other glomerulopathies, most commonly in diabetes nephropathy, focal glomerulosclerosis, membranoproliferative glomerulonephritis, crescentic glomerulonephritis, and lupus. These diseases can be distinguished from amyloidosis by Congo red staining pattern and ultrastructural characteristics of the fibrils.

Diagnostic approach for renal biopsies suspected of amyloidosis
Various tissues have been used for the diagnostic evaluation of amyloidosis. In patients with renal manifestations, amyloid can be detected on the kidney biopsy in virtually all cases [14]. Historically, renal biopsy was felt to be risky because of the possibility of amyloid angiopathy. Patients with AL can also develop an acquired factor X deficiency further increasing their risk of bleeding. However, a recent study of 101 patients with amyloidosis found the rate of post-biopsy hemorrhage was no different than patients without amyloidosis if they were approved by the standard screening tests used for all patients [15]. Other high-yield tissue included the heart, although it is much more invasive. Fat aspirate is often used. It is positive in approximately 70 % of the patients [14]. Rectal biopsy had been popular in the past. This is usually performed via a flexible sigmoidoscopy and has a sensitivity of 70–80 %. However, it is probably best to biopsy the organ which is symptomatic in order to maximize the odds of obtaining amyloid for typing.
In the kidney, amyloid can be seen in all three compartments. Amyloid initially appears along the glomerular basement membranes [3]. In more advanced cases, extensive deposition can be seen in the mesangium, becoming nodular in appearance in some cases. Vascular deposition is common, but in a small percentage of patients, it is the only place amyloid deposition occurs. On H&E stain, the deposits appear pink and amorphous. Amyloid deposits do not stain with periodic acid Schiff (PAS) or silver stain, but spicules can be seen along the glomerular basement membranes on silver stain (Fig. 21.2). In AL, immunofluorescence study should show a preferential staining for one of the light chains in areas where amyloid deposits have been identified. Light chain staining should be negative or equal and mild in intensity for all other types of amyloid.

Silver stain of a glomerulus. Arrows denote spicules along the glomerular basement membranes
Once amyloid is found in the tissue, typing of the amyloid protein is necessary. Treatment and prognosis differ for each subtype. Therefore, accurate typing is essential. Typing must be performed directly on the amyloid fibril. The use of surrogate markers such as circulating monoclonal protein or plasma-cell dyscrasia has led to misdiagnosis and treatment with cytotoxic agents [16]. It is paramount that AL or AH is confirmed before cytotoxic therapy is employed. Historically, potassium permanganate was used to distinguish AA from other forms of amyloid [3]. Applying potassium permanganate to the tissue prevents Congo red from binding to AA fibrils but not AL. However, immunohistochemical agents are now available. Antibodies to immunoglobulin light chains (κ[kappa] and λ[lamda]) and heavy chains, serum amyloid A protein, prealbumin (transthyretin), β[beta] 2-microglobulin, and fibrinogen are commercially available. Unfortunately, immunohistochemical identification is limited by the availability of the antibodies. Genetic testing has been used to identify many of the hereditary forms of amyloidosis. While this is helpful, caution is needed when interpreting the result. Differences in penetrance exist for different amyloidosis. The diagnosis is even more difficult when a monoclonal protein coexists or when dealing with senile amyloid where the amyloidogenic protein is wild type (non-mutated). Recently, the use of liquid chromatography–tandem mass spectrometry has made tremendous progress in the field of amyloid typing [17]. Tissues embedded on glass slides are dissected with a laser to capture amyloid-rich material. The tissue then undergoes tryptic digestion and is analyzed by the liquid chromatography–tandem mass spectrometry. The raw data are queried by multiple algorithms and the peptides are assigned a probability score. This technique allowed the identification of a new amyloid proteins [18]. The technique of SAP scintigraphy should be mentioned [19]. Serum amyloid P component is a molecule commonly found in all types of amyloid. Location and amount of amyloid deposits can be determined by injecting radiolabeled serum amyloid P component into the patient (Fig. 21.3). SAP scintigraphy is most useful in prognostication and assessment of response. And although a positive scan is virtually diagnostic for systemic amyloidosis, tissue biopsy is still required and should not be substituted [20].

SAP scintigraphy showing amyloid deposits in the liver and spleen. Images provided by Dr. Julian Gillmore
AL
Previously referred to as primary amyloidosis, AL is the most common form of amyloidosis in industrialized countries [14]. The age adjusted incidence ranges from 5.1 to 12.8/million/year which appears to be stable. The median age at presentation is 64 years with a range of 32–90 years. It is almost always the result of a plasma-cell dyscrasia, but rarely it can occur with a lymphoma or lymphoplasmacytic lymphoma (Waldenström’s macroglobulinemia). By immunofixation, a monoclonal protein is detectable in the serum and urine in 72 % and 73 % of patients, respectively. In the urine, the majority of the protein is made up of albumin with monoclonal protein representing only a small component. Eleven percent of the patients will have a negative monoclonal protein study by protein electrophoresis or immunofixation. These patients usually have a monoclonal light chain that exists in low levels. The sensitivity can be increased to 99 % by measuring serum-free light chain levels along with serum immunofixation [21]. Recent studies also showed serum-free light chain had a better correlation with outcomes of AL patients after treatment than the entire immunoglobulin [22]. Thus, measurement of serum-free light chain levels is essential in anyone suspected of having AL. A predilection toward lambda light chain exists in AL [14]. More than two-thirds of the patients with AL have a monoclonal lambda whereas the ratio is reversed in multiple myeloma. Approximately 18 % of the patients have >20 % plasma cells in the bone marrow, but coexistence with true multiple myeloma as defined by hypercalcemia, anemia, and lytic bone lesions is rare. AL is the most aggressive of the amyloidoses. Median survival is 13 months if left untreated with patients with advanced cardiac involvement or multiple myeloma faring the worst [23].
Kidney and heart are the most commonly affected visceral organs, but all visceral organs may be involved (liver, gut, spleen, and lung) as well as peripheral nerves, central nervous system, and soft tissues [14]. In a study of 474 patients, 73 % of patients presented with proteinuria and nearly half with renal insufficiency. The proteinuria was in the nephrotic range in approximately one-third of the patients. In a study of 145 patients, patients with lambda light chain were more likely to develop renal manifestations than ones with kappa [24]. The kappa to lambda ratio was 1:12 in patients with renal amyloidosis vs. 1:4 in those without (p = 0.02). Patients with lambda light chain also appeared to have more severe proteinuria. Median proteinuria of lambda patients was 3.6 g/day vs. 0.7 g/day in kappa patients (p = 001). When only patients with renal amyloidosis were analyzed, the disparity in proteinuria was maintained (7.2 g/day in lambda and 2.9 g/day in kappa). Elevated creatinine (p = 0.01) and higher proteinuria (p = 0.03) were risk factors for progression to end-stage renal disease (ESRD). ESRD eventually developed in 42 % of patients who presented with renal manifestations vs. 5 % of those without. This was similar to the rate (39 %) of ESRD reported in a recent Italian study of 198 biopsy-proven renal AL patients [25]. In the largest study to date with 923 patients from the UK, the rate of ESRD was slightly less at 23.9 % [26]. However, factors influencing progression to ESRD were similar (higher CKD stage and lower serum albumin). In this study, patients who had a hematologic response were less likely to require dialysis. Patients who progressed to ESRD had a significantly shorter survival [24]. Data for the USA and Europe showed a median survival of 11 months for patients with ESRD. These data may represent bias since approximately 20 % of the patients died within the first month many of whom withdrew from dialysis. A more recent study with more effective treatment found a median survival of 39 months with many patients surviving long enough to receive kidney transplantation [26].
Histologically, AL deposits can be found in all three compartments of the kidney [3]. Within the glomerulus, deposits may range from minimal to massive. Deposits can also be found in the interstitium as well as in the wall of blood vessels. Patients who have vascular limited deposits on renal biopsy have less proteinuria (<1 g) and typically present with unexplained renal insufficiency [4, 5]. The deposits in AL should stain preferentially for just one of the immunoglobulin light chains [3]. Immunofluorescence staining with antibodies to immunoglobulin light chains is therefore essential for the diagnostic evaluation. In AL variants, the deposits can be composed of immunoglobulin heavy chain (AH) or both immunoglobulin light and heavy chain amyloidosis (ALH) [27]. The immunoglobulin subclass should be identified to insure the heavy chain is monotypic. Antibodies are available for IgG, IgA, and IgM subclasses. In ALH, both the immunoglobulin heavy chain and light chain should be monotypic. Deposits in the kidney must match the isotype of circulating monoclonal protein detected in the blood or urine. In cases where the light chains are not well stained with immunofluorescence, immunoperoxidase may be helpful. Liquid chromatography–mass spectrometry (LC–MS) has been found to be very accurate in typing amyloidosis and is extremely helpful in cases where immunohistochemistry stains are unrevealing or equivocal. It is now considered the gold standard for amyloid typing (Fig. 21.4) [17].

(a) Photograph of a Congo red positive glomerulus traced for laser dissection and capture. (b) The glomerulus is dissected and captured for trypsin digestion and subsequent proteomic analysis by liquid chromatography and mass spectrometry. Photographs provided by Dr. Sanjeev Sethi
The treatment of AL had advanced considerably over the past two decades. The first effective treatment was melphalan and prednisone (MP). In two separate randomized controlled trials, MP extended median survival from 13 to 18 months [28–30]. Responders experienced significant reduction in proteinuria and alkaline phosphatase, but improvement in cardiac function was less common. Overall response rate was low in the 20–30 % range. Most of these patients achieved a hematologic partial response defined as >50 % reduction in the serum monoclonal protein, while hematologic complete response as defined by disappearance of the monoclonal protein was rare. In an attempt to reduce toxicity, high-dose dexamethasone was introduced. It was found to have some activity against AL. In a phase II trial, the median overall survival was 13.8 months [31]. The response was minor but was felt to be potentially beneficial if combined with other therapies. A median survival of 31 months was reported in patients treated with high-dose dexamethasone and undergone maintenance therapy with dexamethasone and alpha interferon [32].
The first therapy that significantly improved patient survival was high-dose melphalan followed by autologous stem cell transplantation (HDM–SCT) [33]. HDM–SCT was capable of achieving hematologic complete response at a high rate. In a study of 312 patients, a complete response was achieved in 40 % of the patients. In another large series of 270 patients, the partial response rate was 71 % with a complete response rate of 33 %. [34]. These high hematologic response rates helped extend the median overall survival to more than 4.6 years, and responders enjoy an even longer survival [33]. The factors associated with survival were the depth of the hematologic response and the severity of cardiac involvement at baseline. Improvement in proteinuria was noted in patients particularly those who had hematologic complete response. Of the patients with renal involvement, 63 % had a renal response after achieving a hematologic complete response. In the patients without complete response, the renal response rate of 11 %. Overall, 31.6 % of the patients with renal involvement had a renal response.
Despite its advantages HDM–SCT does have one major drawback, treatment-related mortality (TRM). This is defined as mortality within 100 days of initiating treatment. TRM can be as high as 40 % in some centers, but it is ~10 % at major amyloidosis centers [35, 36]. Patients with severe cardiac involvement, multiorgan involvement, or advanced age are at the highest risk. Unfortunately, organ involvement may be hard to define, and cardiac assessment based on echocardiography tends to be operator dependent. A risk assessment scoring system using cardiac biomarkers troponin T (cTnT) and N-terminal pro-brain natriuretic peptide (NT-pro-BNP) developed by the Mayo Clinic has been found to accurately predict TRM [37]. This significantly simplified the risk assessment since these biomarkers are easy to obtain and measure. They also would not be influenced by the operator. The use of this scoring system has been credited with lowering the TRM down to 8–9 % in some centers [38].
In early 2000, melphalan and dexamethasone (MDex) was found to be an effective treatment for AL. In a small study of 46 patients not eligible for HDM–SCT, 66 % achieved a hematologic response with 33 % complete response [39]. Long-term follow-up of these patients revealed a median overall survival of 5.1 years [40]. Renal response was noted in 48.3 % of the patients with renal involvement. Median time to hematologic response was 4.5 months. This is a concern since disease progression can occur before the therapeutic effects take place. To evaluate its efficacy against HDM–SCT, a randomized trial was conducted on 100 patients. This study found the median survival of MDex-treated patients (56.9 months) was longer than that of HDM–SCT-treated patients (22.2 months, p = 0.04) [41]. However, critics of the study pointed out that the HDM–SCT had extraordinary high early mortality. Ten patients died prior to HDM–SCT, and another 24 % of the 37 patients who underwent the procedure died within the first 100 days. Together 19 of the 50 patients randomized to HDM–SCT died within the first 130 days. Compare with the MDex group where seven patients died within the first 130 days with only two dying prior to therapy. The difference in mortality was most profound in patients considered high risk by the Mayo Clinic criteria (p < 0.0001). Overall survival was similar in patients considered low risk regardless of treatment (p = 0.12). The impact of the early mortality was demonstrated in the landmark analysis which found no differences in survival between the two groups after 6 months, p = 0.38. These results highlight the importance of patient selection when considering aggressive treatment in these patients. In one study, the TRM was reduced with 50 % with patient selection alone [42]. Thus HDM–SCT may still be useful in the treatment of AL, but its use should be limited to low-risk individuals [38].
Recently, two new classes of drugs have demonstrated efficacy against multiple myeloma and have made their way to AL therapy. The first are the immunomodulatory drugs (IMiDs) represented by thalidomide and lenalidomide. The other is bortezomib, a proteosome inhibitor. These novel agents have shown remarkable activity against multiple myeloma especially when used in combination with other agents [43]. Lenalidomide and dexamethasone produced an overall hematologic response rate of 67 % with a complete response rate of 29 % in a small study of 34 patients [44]. Forty-one percent of the patients with renal involvement had a renal response. Because lenalidomide is renally cleared, dosage adjustment was necessary. Significant treatment-related toxicities were noted with the usual dose of 25 mg a day. Most of the toxicities improved when the dose was reduced to 15 mg a day. Worsening of renal function was also noted in 59 % of the patients. Majority of these patients had renal involvement from AL. Serum creatinine increased to >2.0 mg/dL in 38 % of the patients. Thalidomide had been studied in combination with cyclophosphamide and dexamethasone. In this study, patients with New York Heart Association class II or higher or patients with significant fluid retention were started on a lower dose of thalidomide (50 mg a day vs. 100 mg a day) and dexamethasone (20 mg a day for 4 days vs. 40 mg a day for 4 days) [45]. Hematologic response rates were similar between the dosing regimens, and a hematologic response by free light chain criteria was achieved in 72 % of the patients with 32 % complete response. Overall survival after starting treatment was 41 months but had not been reached when calculated from diagnosis since majority of the patients had had prior treatments. Toxicity was significant. In the study, 40 % experience fatigue or somnolence and 21 % had worsening fluid retention or congestive heart failure. The combination of bortezomib and dexamethasone had been reported in small study with 18 patients from a single center [46]. Of the 16 evaluable patients, 94 % achieved a hematologic response with 44 % complete response. Renal response was noted in 14 % of patients with renal involvement. The major toxicities were thrombocytopenia, fatigue, neurotoxicity, and orthostatic hypotension. Two multicenter retrospective studies had been published. In one study of 26 patients, overall hematologic response rate was 54 % with complete response rate of 31 %. Organ improvement was noted in 12 %, stabilization in 76 %, and progression in 12 %. Thrombocytopenia and hyponatremia were reported as grades 3 and 4 toxicity, and grade 2 neurotoxicity was reported in 42 %. In the other with 94 patients, the hematologic response rate was 71 % with 25 % in complete response. Organ response was noted in 29 % of patients with heart involvement and 19 % of patients with kidney involvement. Hematologic response and reduction in NT-pro-GNP were independently associated with better survival [47]. In all of the reports involving therapies with novel agents, the follow-ups were relatively short. Longer follow-ups are needed in order to fully evaluate the true effectiveness of these therapies.
While it is important to achieve hematologic response in AL, achievement of organ response is the ultimate goal. Much of this has to do with the differences between multiple myeloma and AL. First, the majority of patients with AL have only a low-grade plasma-cell dyscrasia. Few present or will ever progress to multiple myeloma [48]. Therefore, the tumor burden is not the problem but rather their byproduct, the monoclonal light chains. Second, patients with AL do not die from bone marrow failure as in multiple myeloma; they died of progressive organ failure which is the result of the amyloidogenic light chains. Obviously, eradicating the monoclonal light chains would be ideal but is not always possible. However, it is important to recognize that organ response can occur in the absence of complete hematologic response, while in some cases even a hematologic complete response does not guarantee organ response [49]. Therefore, it is important to measure the organ response along with hematologic response when making decision on further treatment. One of the easiest organ responses to measure is renal response defined as >50 % reduction (minimal of 0.5 g/day) in proteinuria with <25 % decline in renal function [20]. It has been reported in up to 60 % of the patients after HDM–SCT [49]. Patients who achieved renal response have a significant survival advantage over those who do not. In a study of 122 AL patients with renal involvement who had a minimum follow-up of 12 months after HDM–SCT, hematologic response was noted in 72.1 % and renal response in 43.4 % [50]. Hematologic response was noted in 96.2 % of the patients with renal response. The median survival had not been reached in patients with a both a hematologic and renal response. Patients who had either a renal or hematologic response had a median survival of 66 months vs. nonresponders who had a median survival of 4.7 months (Fig. 21.5). Renal response was noted after a median of 10–12 months, and complete resolution of proteinuria was seen at a median of 24 months. Independent features that predicted a renal response after HDM–SCT were lower proteinuria and lower cardiac troponin T.

Cumulative overall survival of patients with AL amyloidosis after autologous stem cell transplantation. Survival was calculated from day of stem cell transplantation. HR hematologic response defined by ≥50 % reduction in the M protein. RR renal response defined by ≥50 % reduction in proteinuria with <25 % reduction in renal function
Kidney transplantation had been successfully performed in patients with AL. Unfortunately, the early experience was discouraging due to high mortality rates [51]. Patients were often dying from cardiac complications, progression of disease, and infection. One study of patients from 1987 to 2008 found 21 patients who had undergone kidney transplantation. The status of their AL was not reported. Median follow-up was 50 months and the median estimated survival was 89 months. Nine patients had died, with two from progression of their extrarenal amyloidosis, four from infection, one from gastrointestinal bleed, and two from unknown causes [26]. However, as treatment of AL improved, so did the outcomes after kidney transplantation. In one study, 15 patients with ESRD underwent HDM–SCT. The median survival for these patients was 25 months [52]. Three patients eventually received kidney transplantation with survival of 5.3–6.0 years. An alternative approach was to perform kidney transplantation first followed by HDM–SCT. The Mayo Clinic reported eight patients who underwent kidney transplantation prior to HDM–SCT. Two of the patient died prior to HDM–SCT, but the other six successfully underwent HDM–SCT. Median survival had not been reached after a median follow-up of 41 months. Recurrence occurred in one patient whose HDM–SCT was delayed for more than 3 years after kidney transplantation. This patient was able to achieve a hematologic complete response and the allograft remains in good function. Despite the immense immunosuppression of HDM–SCT, acute rejection can occur. One patient developed steroid-resistant cellular rejection immediate after leukocyte engraftment that required antithymocyte globulin. Patients can also receive kidney transplantation after achieving hematologic response without HDM–SCT. Neither patient nor allograft survival was different from patients who received HDM–SCT [53]. One of five patients developed recurrence in the allograft and was successful treated with additional AL therapy. With 30–40 % of the patients developing ESRD and the poor survival of patients on dialysis, kidney transplantation is an attractive alternative. It should be considered in patients who have minimal cardiac involvement who had achieved a hematologic complete response.
AA Amyloid
The fibrils in AA are derived from serum amyloid A (SAA) protein, an acute-phase reactant. It is synthesized by hepatocytes under the control of proinflammatory cytokines [54]. Elevated SAA levels can be seen in infectious, inflammatory, and malignant conditions; however, in order for amyloid formation to occur, the overproduction of SAA needs to be sustained over time. Common conditions that cause AA include chronic inflammatory arthritis such as rheumatoid arthritis, juvenile idiopathic arthritis, and ankylosing spondylitis. It can also be the result of chronic infections. Examples include bronchiectasis, osteomyelitis, infected pressure sores, or urinary tract infection in paraplegics. Malignant conditions such as lymphoma, mesothelioma, and Castleman’s disease have also been associated with AA. Finally, a hereditary form is seen in patients with periodic fever syndromes [55].
Four types of periodic fever syndromes have been identified to cause AA [55]. The most common is the Familial Mediterranean Fever (FMF) which is an autosomal recessive disease resulting from the mutation in the MEFV gene on chromosome 16p. As its name implies, it affects people around the Mediterranean Sea, most commonly Armenians, Sephardic Jews, Greeks, Turks, and Arabs. It is characterized by periodic attacks of abdominal pain, serositis, arthritis, and fever. TNF-receptor-associated periodic fever syndrome (TRAPS) is the second most common and is the result of mutations in the TNFRSF1A gene which regulates the type 1 tumor necrosis factor (TNF) receptor. Over 40 mutations have been identified and the disease has an autosomal dominant pattern. It was first described in a boy with Scottish–Irish ancestry and was initially named Familial Hibernian Fever. Later studies reveal it is well distributed in many ethnicity and countries of origin. Its symptoms include but are not limited to recurrent abdominal pain, migratory myalgia, rash, periorbital edema, and testicular pain. Muckle–Wells syndrome is one of the cryopyrinopathies that includes familial cold autoinflammatory syndrome and multisystem inflammatory disease. They are characterized by urticaria like rash, arthralgia which is the result of mutations in the CIAS1 gene. Muckle–Wells syndrome is characterized by hearing loss and is the only cryopyrinopathy that is associated with amyloidosis. Most have an autosomal-dominant pattern of inheritance. Hyper IgD syndrome is an autosomal recessive disease involving the mutation of the mevalonate kinase (MVK) gene. Its distinguishing features are high levels of IgD and attacks triggered by vaccination. It is prominent in the Netherlands.
The most common presentation in AA is renal dysfunction [54]. In a study of 374 patients, 97 % presented with either proteinuria of >500 mg/day, serum creatinine >1.5 mg/dL, or both. Median proteinuria was 3.9 g/day and 12 % excretes >10 g/day. Median creatinine was 1.2 mg/day and 75 % had a serum creatinine of <3 mg/dL at diagnosis. ESRD was present in 11 % of the patients with another 33 % developing it during the course of their disease. Median time to ESRD was 256 months. Risk factors for ESRD include presence of Crohn’s disease, chronic infection, hepatic amyloid deposits, and poor renal function at baseline. Other clinical features include hepatomegaly which was noted in 9 % of the patients. However, if hepatic involvement was assessed by SAP scintigraphy, then the number increased to 23 %. Cardiac involvement was only noted in 2 % of the patients by echocardiogram and only 1 % developed clinically significant cardiac failure. This may explain the long median survival of 133 months. Factors associated with poor survival include elevated SAA concentration, older age, and ESRD. In this study, no patient developed autonomic neuropathy. Adrenal amyloid deposits were found on SAP scintigraphy on 41 % of the patients, but only five required adrenocorticoid replacement.
Histologically, the amyloid deposition pattern in AA is indistinguishable from other types of renal amyloidosis [3]. One feature that is particular but not unique to AA is the formation of crescents [56]. This occurs most commonly in patients whose AA is secondary to rheumatoid arthritis. It is also more common in female than male. The coexistence of crescents on the kidney biopsy is often associated with rapidly progressive glomerulonephritis and rapid loss in renal function. Corticosteroids have been found to be effective at stabilizing and reversing the rapid loss in renal function [57].
Treatment of AA involves eliminating or controlling the underlying disease [54]. Complete remission of the renal amyloidosis has been reported after resection of the Castleman’s disease, treatment of the periodic fever syndrome, or eradication of the infection [54, 58]. Steroids and TNF inhibitors are effective treatment for TRAPS [55]. Anakinra, an interleukin-1 receptor antagonist, is effective in Muckle–Wells and Hyper IgD syndromes [59]. Statins (HMG Co-A inhibitor) inhibit the production of mevalonate and have been found to be beneficial in Hyper IgD syndrome [60]. Colchicine is effective at preventing or reducing the symptoms of FMF in approximately 90 % of patients [59]. However, its benefits are limited once amyloidosis has developed. Eprodisate is a negatively charged, low molecular weight sulfonated molecule which can inhibit the interactions between amyloidogenic proteins in animal studies. A randomized placebo control trial was performed in patients with AA [61]. The primary endpoint of the study was a composite assessment of renal function or death. Secondary endpoints included slope of creatinine clearance, change in proteinuria, resolution of diarrhea, and change in amyloid content of abdominal fat. In this study, patients treated with eprodisate were less likely to have worsening renal function (27 %) than the placebo group (40 %, p = 0.06). There was no significant difference in death rate between the two groups. Treatment with eprodisate also significantly decreased the slope of creatinine clearance (−10.9 ± 5.1 mg/min/1.73 m2) vs. placebo (−15.6 ± 4.0 mL/min/1.73 m2, p = 0.02). There were no significant differences in the change in proteinuria, diarrhea, and amyloid content in abdominal fat. A second randomized placebo-controlled trial is current underway to further clarify the results.
End-stage renal disease is a significant risk factor for mortality in AA [62]. Survival rates on dialysis are 82 %, 46 %, and 37 % at 1, 2, and 3 years. At autopsy, 10 of 13 patients were noted to have amyloid infiltration of the heart, much higher than suggested in the non ESRD AA patients [63]. Whether this represents advance-stage disease in the ESRD population or acceleration of amyloid deposition in the heart from ESRD remains undetermined. Kidney transplantation is an option for these patients. However, concerns had been raised about high mortality after kidney transplantation. Some studies suggested an increase in early mortality with 1-year patient survival of 69–79 % comparing with 97–100 % in the non-amyloidosis patients [64, 65]. Five-year survival was 52–69 % vs. 87–100 %. The causes of death were sepsis and cardiac related. Others reported no differences in the 1-year (93 % vs. 94 %) or 5-year (89 % vs. 90 %) survival rate between AA patients and non-amyloidosis patients, respectively [66]. This study is more recent and may reflect better management of immunosuppression. Recurrence of AA has been reported in the renal allograft. The true prevalence is unknown and may depend on the underlying disease. In FMF, colchicine has been found to be effective at preventing recurrence [67]. Patients who received 1.5 mg/day of colchicine had less proteinuria than patients receiving 0.5 mg/day suggesting a dose effect. Finally, recently anakinra was found to be effective in a patient who was resistant to high-dose colchicine [68].
Hereditary Renal Amyloidoses
Fibrinogen Alpha A[Alpha]-Chain Amyloidosis (AFib)
First sequenced in 1993, AFib is one the most common types of hereditary systemic amyloidosis involving the kidney in Northern Europe [69]. Families with AFib have also been identified in Peru, Mexico, and Africa. The disease is inherited via an autosomal-dominant pattern. Six mutations have been identified in the fibrinogen Aα-chain gene that are associated with amyloid formation [70]. Initially, AFib was thought to be one of the hereditary nonneuropathic systemic amyloidosis, but later descriptions discovered peripheral neuropathy was possible with certain mutations. Renal manifestations had been universal in this form of amyloidosis. In a study of 71 patients from multiple countries of origin, proteinuria or hypertension was documented in 72 % of the patients at presentation and renal insufficiency is present in 54 % [71]. Amyloid deposits had been found by radiolabeled SAP scintigraphy in the adrenal gland and spleen. Whether cardiac involvement is a feature in AFib is controversial. In one report, none of the 63 patients who underwent radiolabeled SAP scan demonstrated cardiac deposition or echocardiographic features of amyloid heart disease [71]. However, a smaller series of 22 patients found 52 % of the patients had echocardiographic evidence for amyloid infiltration [70]. In this study, three of the four patients who underwent endomyocardial biopsy were found to have a substantial amount of amyloid deposition. The deposits were also found on atheromatous plagues, but the significance of this was unclear. Median age of presentation was 58 years with a range of 33–83 years [71]. ESRD developed in 62 % and the median time to progression to ESRD of 4.6 years. Median survival from diagnosis was 10.9 years.
Recurrence is common in patients with AFib who received a kidney transplant alone. In a series of ten patients with 12 allografts, three grafts failed immediately due to technical problems [71]. In the nine remaining grafts, four had documented recurrence of which three subsequently failed at a median of 6.7 years. Only 1 graft remained functioning 12.2 years after transplantation despite having recurrence documented 7 years after transplant. Faster graft loss had been reported [72]. While fibrinogen synthesis has been identified in other cell types, in human, it appears to be exclusively produced in the liver [73]. As a result, orthotopic liver transplantation may completely eliminate the production of the mutant fibrinogen Aα-chain and prevent recurrence and disease progression. Although the data is limited, combined liver–kidney transplantation appeared to be effective at preventing recurrence in the kidney allograft. Results from the two largest series revealed none of the 12 patients who survived the perioperative period had a documented recurrence [70, 71]. On the other hand, regression of visceral amyloid deposits was noted on serial radiolabeled SAP imaging. In addition, patients had improvement of their symptoms from GI immobility improved and native kidney function. However, perioperative mortality was higher than non-amyloidosis patients especially those with ESRD and were on long-term dialysis prior to transplantation. Because of the higher morbidity and mortality, recommendation of preemptive liver transplantation in these patients remains controversial.
Lysozyme Amyloidosis (ALys)
First described in 1993, ALys is one of the nonneuropathic (Ostertag) forms of systemic amyloidosis [74]. Lysozyme is a ubiquitous bacteriolytic enzyme which is produced by the macrophages, gastrointestinal cells, and hepatocytes. Currently, four mutations have been identified to be involved in amyloid formation. All of the mutations are located on exon 2. They are I56T, F57I, W64R, and D67H. In addition, a case of compound heterozygosity has been reported involving exon 2 (T70N) and exon 4 (W112R) [75]. Ethnic origins of families with ALys mutations include English, Italian, French, and Scandinavian descent. Diagnosis can be made by immunohistochemistry, but genetic confirmation is recommended.
The main feature of patients with ALys is GI manifestations. These include abdominal pain/discomfort to malabsorption syndrome [76]. Bleeding of the GI tract is also a common feature. One of the more serious complications is spontaneous rupture of the liver which has been reported in one family involving the mother and daughter [77]. Renal manifestations are the next most common. The presentation ranges from hypertension, proteinuria, to nephritic syndrome and ESRD. Time to ESRD is also quite variable ranging from 3 months to 18 years. Kidney transplantation has been successfully performed, but recurrence has also been reported. Unlike in AFib where liver is the exclusive source of the mutant protein, the mutant lysozyme is produced by cells in addition to hepatocytes. Thus, liver transplantation will not impede the production of the mutant protein and is not indicated in these patients.
AApoAI and AApoAII Amyloidoses
Apolipoproteins (Apo) are cofactor for lecithin cholesterol acyltransferase and are components of high-density lipoprotein (HDL). They are produced by cells of the liver and intestine and catabolized in the liver and kidney. AApoAI and AApoAII are part of the hereditary nonneuropathic systemic amyloidosis described by Ostertag in 1932.
To date, 13 mutations have been identified to cause amyloid formation in AApoAI and 4 in AApoAII [78, 79]. Renal dysfunction is the most common feature of both AApoAI and AApoAII, but other visceral organs can be involved including liver, heart, spleen, gonads, and skin [78–80]. Involvement of peripheral nerves is not a feature. Visual impairment has been reported in patients with severe hepatic involvement. The typical age of presentation for AApoAI is between 18 and 55 years of age, but this can vary depending on the mutation. Patients with Leu75Pro tend to present late in their seventh decade and can live into their 90s. Levels of HDL, ApoAI, and ApoAII are not elevated in these patients.
No treatment is currently available for either type of amyloidosis. Organ transplantation has been successfully performed. In one study, eight of ten patients with AApoAI who received a kidney transplantation remain alive 4–28 years after transplant [80]. Two patients died at 2 months (CMV) and 13 years. Recurrence was noted in five patients, but only one graft loss was reported 6.5 years after the recurrence was documented and 25 years after kidney transplantation. The other grafts remained functional at 5.3–8 years after recurrence was documented. Two patients underwent liver transplantation. Both patients experienced regression of their visceral amyloid deposits by SAP scintigraphy. Their overall well-being also improved and to date no sign of recurrence. This is interesting since liver is not the only source of ApoAI.
Latest Amyloidosis
LECT2 Amyloidosis (ALECT2)
ALECT2 is the newest member of amyloidosis family. The native protein is leukocyte chemotactic factor 2 (LECT2) [6]. Commercial immunohistochemistry testing is currently not available so diagnosis must be made by tandem mass spectrometry. Only a dozen cases have been described in the literature so far. In one kidney biopsy series where 285 of 21,598 biopsies were congophilic, 31 could not be typed by immunohistochemistry [81]. Of these, seven were later identified to be ALECT2. It was the third more common amyloid (2.5 %) in this series behind AL and AA. In the largest series to date, the mean age of presentation was 67 years [18]. Seven of ten patients were Mexican or Mexican-American. In the kidney, ALECT2 is highly congophilic and is deposited extensively in all compartments. Renal dysfunction appears to be the main clinical feature although ALECT2 has been identified in liver, spleen, colon, and adrenal gland. The significance of the extrarenal deposition is unknown. LECT2 level measured in limited number of patients has been either normal or undetectable suggesting systemic overproduction is not part of the pathophysiology. Systemic infection or inflammation is also not identified in these patients. Genetic analysis has not revealed a mutation, but so far, all patients tested are homozygous for the G allele of the LECT2 gene. Further genetic analysis however is needed before any conclusion can be made. No treatment is currently available for ALECT2.
Summary
Despite the similarity in the physical characteristics of the fibrils, amyloidosis is a tremendously heterogeneous group of diseases. Important differences are noted ranging from amyloidogenesis to organ tropism. These patterns reflect the origin of the amyloidogenic protein and provide insights into the pathogenesis. Tremendous advances have been made in the field of diagnostic and typing of amyloid fibrils. Advances have also being made in the treatment of amyloidosis which has resulted in significant increases in the survival of these patients.
References
Merlini G, Bellotti V. Molecular mechanisms of amyloidosis [see comment]. N Engl J Med. 2003;349(6):583–96.
Xing Y, Higuchi K. Amyloid fibril proteins. Mech Ageing Dev. 2002;123(12):1625–36.
Dember LM. Amyloidosis-associated kidney disease. J Am Soc Nephrol. 2006;17(12):3458–71.
Falck HM, Tornroth T, Wegelius O. Predominantly vascular amyloid deposition in the kidney in patients with minimal or no proteinuria. Clin Nephrol. 1983;19(3):137–42.
Uda H, Yokota A, Kobayashi K, Miyake T, Fushimi H, Maeda A, et al. Two distinct clinical courses of renal involvement in rheumatoid patients with AA amyloidosis. J Rheumatol. 2006;33(8):1482–7 [Research Support, Non-U.S. Gov’t].
Benson MD. LECT2 amyloidosis. Kidney Int. 2010;77(9):757–9.
Jadoul M, Noel H, van Ypersele de Strihou C. Beta 2-microglobulin amyloidosis in a patient treated exclusively by continuous ambulatory peritoneal dialysis. Am J Kidney Dis. 1990;15(1):86–8 [Case Reports].
Liu C, Sawaya MR, Eisenberg D. Beta-microglobulin forms three-dimensional domain-swapped amyloid fibrils with disulfide linkages. Nat Struct Mol Biol. 2011;18(1):49–55 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.].
Liao R, Jain M, Teller P, Connors LH, Ngoy S, Skinner M, et al. Infusion of light chains from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts. Circulation. 2001;104(14):1594–7.
Hetzel GR, Uhlig K, Mondry A, Helmchen U, Grabensee B. AL-amyloidosis of the kidney initially presenting as minimal change glomerulonephritis. Am J Kidney Dis. 2000;36(3):630–5.
Kyle RA, Wagoner RD, Holley KE. Primary systemic amyloidosis: resolution of the nephrotic syndrome with melphalan and prednisone. Arch Intern Med. 1982;142(8):1445–7.
Mandreoli M, Casanova S, Vianelli N, Pasquali S, Zucchelli P. Remission of nephrotic syndrome due to AA amyloidosis and initiation of glomerular repair after surgical resection of localized Castleman’s disease. Nephron. 2002;90(3):336–40 [Case Reports].
Iskandar SS, Herrera GA. Glomerulopathies with organized deposits. Semin Diagn Pathol. 2002;19(3):116–32.
Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45–59.
Soares SM, Fervenza FC, Lager DJ, Gertz MA, Cosio FG, Leung N. Bleeding complications after transcutaneous kidney biopsy in patients with systemic amyloidosis: single-center experience in 101 patients. Am J Kidney Dis. 2008;52(6):1079–83.
Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346(23):1786–91.
Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen 3rd HR, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114(24):4957–9.
Murphy CL, Wang S, Kestler D, Larsen C, Benson D, Weiss DT, et al. Leukocyte chemotactic factor 2 (LECT2)-associated renal amyloidosis: a case series. Am J Kidney Dis. 2010;56(6):1100–7.
Hawkins PN, Myers MJ, Lavender JP, Pepys MB. Diagnostic radionuclide imaging of amyloid: biological targeting by circulating human serum amyloid P component. Lancet. 1988;1(8600):1413–8.
Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 Apr 2004. Am J Hematol. 2005;79(4):319–28 [Consensus Development Conference Review].
Katzmann JA, Abraham RS, Dispenzieri A, Lust JA, Kyle RA. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem. 2005;51(5):878–81.
Kumar S, Dispenzieri A, Katzmann JA, Larson DR, Colby CL, Lacy MQ, et al. Serum immunoglobulin free light-chain measurement in primary amyloidosis: prognostic value and correlations with clinical features. Blood. 2010;116(24):5126–9.
Kyle RA, Greipp PR, O’Fallon WM. Primary systemic amyloidosis: multivariate analysis for prognostic factors in 168 cases. Blood. 1986;68(1):220–4.
Gertz MA, Leung N, Lacy MQ, Dispenzieri A, Zeldenrust SR, Hayman SR, et al. Clinical outcome of immunoglobulin light chain amyloidosis affecting the kidney. Nephrol Dial Transplant. 2009;24(10):3132–7.
Bergesio F, Ciciani AM, Manganaro M, Palladini G, Santostefano M, Brugnano R, et al. Renal involvement in systemic amyloidosis: an Italian collaborative study on survival and renal outcome. Nephrol Dial Transplant. 2008;23(3):941–51.
Pinney JH, Lachmann HJ, Bansi L, Wechalekar AD, Gilbertson JA, Rowczenio D, et al. Outcome in renal Al amyloidosis after chemotherapy. J Clin Oncol. 2011;29(6):674–81.
Sethi S, Theis JD, Leung N, Dispenzieri A, Nasr SH, Fidler ME, et al. Mass spectrometry-based proteomic diagnosis of renal immunoglobulin heavy chain amyloidosis. Clin J Am Soc Nephrol. 2010;5(12):2180–7.
Kyle RA, Greipp PR. Primary systemic amyloidosis: comparison of melphalan and prednisone versus placebo. Blood. 1978;52(4):818–27.
Kyle RA, Greipp PR, Garton JP, Gertz MA. Primary systemic amyloidosis. Comparison of melphalan/prednisone versus colchicine. Am J Med. 1985;79(6):708–16.
Skinner M, Anderson J, Simms R, Falk R, Wang M, Libbey C, et al. Treatment of 100 patients with primary amyloidosis: a randomized trial of melphalan, prednisone, and colchicine versus colchicine only. Am J Med. 1996;100(3):290–8.
Gertz MA, Lacy MQ, Lust JA, Greipp PR, Witzig TE, Kyle RA. Phase II trial of high-dose dexamethasone for untreated patients with primary systemic amyloidosis. Med Oncol. 1999;16(2):104–9.
Dhodapkar MV, Hussein MA, Rasmussen E, Solomon A, Larson RA, Crowley JJ, et al. Clinical efficacy of high-dose dexamethasone with maintenance dexamethasone/alpha interferon in patients with primary systemic amyloidosis: results of United States Intergroup Trial Southwest Oncology Group (SWOG) S9628. Blood. 2004;104(12):3520–6.
Skinner M, Sanchorawala V, Seldin DC, Dember LM, Falk RH, Berk JL, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140(2):85–93.
Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar SK, Leung N, et al. Effect of hematologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Haematologica. 2007;92(10):1415–8.
Moreau P, Leblond V, Bourquelot P, Facon T, Huynh A, Caillot D, et al. Prognostic factors for survival and response after high-dose therapy and autologous stem cell transplantation in systemic AL amyloidosis: a report on 21 patients. Br J Haematol. 1998;101(4):766–9.
Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar SK, Dingli D, et al. Autologous stem cell transplant for immunoglobulin light chain amyloidosis: a status report. Leuk Lymphoma. 2010;51(12):2181–7.
Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, et al. Prognostication of survival using cardiac troponins and N-terminal pro-brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood. 2004;104(6):1881–7.
Gertz MA, Lacy MQ, Dispenzieri A, Kumar SK, Buadi FK, Dingli D, et al. Trends in day 100 and 2-year survival after auto-SCT for AL amyloidosis: outcomes before and after 2006. Bone Marrow Transplant. 2011;46(7):970–5.
Palladini G, Perfetti V, Obici L, Caccialanza R, Semino A, Adami F, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103(8):2936–8.
Palladini G, Russo P, Nuvolone M, Lavatelli F, Perfetti V, Obici L, et al. Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood. 2007;110(2):787–8.
Jaccard A, Moreau P, Leblond V, Leleu X, Benboubker L, Hermine O, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357(11):1083–93.
Mollee PN, Wechalekar AD, Pereira DL, Franke N, Reece D, Chen C, et al. Autologous stem cell transplantation in primary systemic amyloidosis: the impact of selection criteria on outcome. Bone Marrow Transplant. 2004;33(3):271–7.
Richardson PG, Laubach J, Mitsiades CS, Schlossman R, Hideshima T, Redman K, et al. Managing multiple myeloma: the emerging role of novel therapies and adapting combination treatment for higher risk settings. Br J Haematol. 2011. doi:10.1111/j.1365-2141.2011.08791.x.
Sanchorawala V, Wright DG, Rosenzweig M, Finn KT, Fennessey S, Zeldis JB, et al. Lenalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 2 trial. Blood. 2007;109(2):492–6 [Clinical Trial, Phase II Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t].
Wechalekar AD, Goodman HJ, Lachmann HJ, Offer M, Hawkins PN, Gillmore JD. Safety and efficacy of risk-adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood. 2007;109(2):457–64 [Clinical Trial Research Support, Non-U.S. Gov’t].
Kastritis E, Anagnostopoulos A, Roussou M, Toumanidis S, Pamboukas C, Migkou M, et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. 2007;92(10):1351–8 [Clinical Trial].
Kastritis E, Wechalekar AD, Dimopoulos MA, Merlini G, Hawkins PN, Perfetti V, et al. Bortezomib with or without dexamethasone in primary systemic (light chain) amyloidosis. J Clin Oncol. 2010;28(6):1031–7.
Rajkumar SV, Gertz MA, Kyle RA. Primary systemic amyloidosis with delayed progression to multiple myeloma. Cancer. 1998;82(8):1501–5.
Leung N, Dispenzieri A, Fervenza FC, Lacy MQ, Villicana R, Cavalcante JL, et al. Renal response after high-dose melphalan and stem cell transplantation is a favorable marker in patients with primary systemic amyloidosis. Am J Kidney Dis. 2005;46(2):270–7.
Leung N, Dispenzieri A, Lacy MQ, Kumar SK, Hayman SR, Fervenza FC, et al. Severity of baseline proteinuria predicts renal response in immunoglobulin light chain-associated amyloidosis after autologous stem cell transplantation. Clin J Am Soc Nephrol. 2007;2(3):440–4.
Pasternack A, Ahonen J, Kuhlback B. Renal transplantation in 45 patients with amyloidosis. Transplantation. 1986;42(6):598–601.
Casserly LF, Fadia A, Sanchorawala V, Seldin DC, Wright DG, Skinner M, et al. High-dose intravenous melphalan with autologous stem cell transplantation in AL amyloidosis-associated end-stage renal disease. Kidney Int. 2003;63(3):1051–7.
Herrmann SM, Gertz MA, Stegall MD, Dispenzieri A, Cosio FC, Kumar S, et al. Long-term outcomes of patients with light chain amyloidosis (AL) after renal transplantation with or without stem cell transplantation. Nephrol Dial Transplant. 2011;26(6):2032–6.
Lachmann HJ, Goodman HJ, Gilbertson JA, Gallimore JR, Sabin CA, Gillmore JD, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356(23):2361–71 [Research Support, Non-U.S. Gov’t].
Simon A, van der Meer JW. Pathogenesis of familial periodic fever syndromes or hereditary autoinflammatory syndromes. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R86–98 [Research Support, Non-U.S. Gov’t Review].
Masutani K, Nagata M, Ikeda H, Takeda K, Katafuchi R, Hirakata H, et al. Glomerular crescent formation in renal amyloidosis. A clinicopathological study and demonstration of upregulated cell-mediated immunity. Clin Nephrol. 2008;70(6):464–74 [Comparative Study Multicenter Study].
Nagata M, Shimokama T, Harada A, Koyama A, Watanabe T. Glomerular crescents in renal amyloidosis: an epiphenomenon or distinct pathology? Pathol Int. 2001;51(3):179–86 [Research Support, Non-U.S. Gov’t].
Keven K, Nergizoglu G, Ates K, Erekul S, Orhan D, Erturk S, et al. Remission of nephrotic syndrome after removal of localized Castleman’s disease. Am J Kidney Dis. 2000;35(6):1207–11 [Case Reports].
Church LD, Churchman SM, Hawkins PN, McDermott MF. Hereditary auto-inflammatory disorders and biologics. Springer Semin Immunopathol. 2006;27(4):494–508 [Review].
van der Hilst JC, Kluve-Beckerman B, Bodar EJ, van der Meer JW, Drenth JP, Simon A. Lovastatin inhibits formation of AA amyloid. J Leukoc Biol. 2008;83(5):1295–9.
Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, et al. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med. 2007;356(23):2349–60 [Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.].
Lachmann HJ, Gallimore R, Gillmore JD, Carr-Smith HD, Bradwell AR, Pepys MB, et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. Br J Haematol. 2003;122(1):78–84.
Ylinen K, Gronhagen-Riska C, Honkanen E, Ekstrand A, Metsarinne K, Kuhlback B. Outcome of patients with secondary amyloidosis in dialysis treatment. Nephrol Dial Transplant. 1992;7(9):908–12.
Haq A, Hussain S, Meskat B, Mohan P, Conlon P, Hickey DP. Complications of renal transplantation in patients with amyloidosis. Transplant Proc. 2007;39(1):120–4.
Sherif AM, Refaie AF, Sobh MA, Mohamed NA, Sheashaa HA, Ghoneim MA. Long-term outcome of live donor kidney transplantation for renal amyloidosis. Am J Kidney Dis. 2003;42(2):370–5.
Keven K, Sengul S, Kutlay S, Ekmekci Y, Anadol E, Nergizoglu G, et al. Long-term outcome of renal transplantation in patients with familial Mediterranean fever amyloidosis: a single-center experience. Transplant Proc. 2004;36(9):2632–4.
Ozdemir BH, Ozdemir FN, Sezer S, Sar A, Haberal M. Does colchicine have an antifibrotic effect on development of interstitial fibrosis in renal allografts of recipients with familial Mediterranean fever? Transplant Proc. 2006;38(2):473–6.
Moser C, Pohl G, Haslinger I, Knapp S, Rowczenio D, Russel T, et al. Successful treatment of familial Mediterranean fever with Anakinra and outcome after renal transplantation. Nephrol Dial Transplant. 2009;24(2):676–8 [Case Reports].
Benson MD, Liepnieks J, Uemichi T, Wheeler G, Correa R. Hereditary renal amyloidosis associated with a mutant fibrinogen alpha-chain. Nat Genet. 1993;3(3):252–5 [Case Reports Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.].
Stangou AJ, Banner NR, Hendry BM, Rela M, Portmann B, Wendon J, et al. Hereditary fibrinogen A alpha-chain amyloidosis: phenotypic characterization of a systemic disease and the role of liver transplantation. Blood. 2010;115(15):2998–3007.
Gillmore JD, Lachmann HJ, Rowczenio D, Gilbertson JA, Zeng CH, Liu ZH, et al. Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen A alpha-chain amyloidosis. J Am Soc Nephrol. 2009;20(2):444–51.
Mousson C, Heyd B, Justrabo E, Rebibou JM, Tanter Y, Miguet JP, et al. Successful hepatorenal transplantation in hereditary amyloidosis caused by a frame-shift mutation in fibrinogen Aalpha-chain gene. Am J Transplant. 2006;6(3):632–5 [Case Reports].
Tennent GA, Brennan SO, Stangou AJ, O’Grady J, Hawkins PN, Pepys MB. Human plasma fibrinogen is synthesized in the liver. Blood. 2007;109(5):1971–4 [Research Support, Non-U.S. Gov’t].
Pepys MB, Hawkins PN, Booth DR, Vigushin DM, Tennent GA, Soutar AK, et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 1993;362(6420):553–7 [Research Support, Non-U.S. Gov’t].
Rocken C, Becker K, Fandrich M, Schroeckh V, Stix B, Rath T, et al. ALys amyloidosis caused by compound heterozygosity in exon 2 (Thr70Asn) and exon 4 (Trp112Arg) of the lysozyme gene. Hum Mutat. 2006;27(1):119–20 [Research Support, Non-U.S. Gov’t].
Granel B, Valleix S, Serratrice J, Cherin P, Texeira A, Disdier P, et al. Lysozyme amyloidosis: report of 4 cases and a review of the literature. Medicine. 2006;85(1):66–73 [Case Reports Research Support, Non-U.S. Gov’t Review].
Loss M, Ng WS, Karim RZ, Strasser SI, Koorey DJ, Gallagher PJ, et al. Hereditary lysozyme amyloidosis: spontaneous hepatic rupture (15 years apart) in mother and daughter. role of emergency liver transplantation. Liver Transpl. 2006;12(7):1152–5 [Case Reports].
Eriksson M, Schonland S, Yumlu S, Hegenbart U, von Hutten H, Gioeva Z, et al. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: identification of three novel mutations in the APOA1 gene. J Mol Diagn. 2009;11(3):257–62 [Case Reports Research Support, Non-U.S. Gov’t].
Yazaki M, Liepnieks JJ, Barats MS, Cohen AH, Benson MD. Hereditary systemic amyloidosis associated with a new apolipoprotein AII stop codon mutation Stop78Arg. Kidney Int. 2003;64(1):11–6 [Case Reports Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S.].
Gillmore JD, Stangou AJ, Lachmann HJ, Goodman HJ, Wechalekar AD, Acheson J, et al. Organ transplantation in hereditary apolipoprotein AI amyloidosis. Am J Transplant. 2006;6(10):2342–7 [Multicenter Study. Research Support, Non-U.S. Gov’t].
Larsen CP, Walker PD, Weiss DT, Solomon A. Prevalence and morphology of leukocyte chemotactic factor 2-associated amyloid in renal biopsies. Kidney Int. 2010;77(9):816–9.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Leung, N. (2014). Amyloidosis. In: Fervenza, F., Lin, J., Sethi, S., Singh, A. (eds) Core Concepts in Parenchymal Kidney Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8166-9_21
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8166-9_21
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8165-2
Online ISBN: 978-1-4614-8166-9
eBook Packages: MedicineMedicine (R0)