
Abstract
Low phosphorus (P) tolerance in rice is a biologically and agronomically important character. Low P tolerant Indica-type rice genotypes, Sahbhagi Dhan (SD) and Chakhao Poreiton (CP), are adapted to acidic soils and show variable response to low P levels. Using RNAseq approach, transcriptome data was generated from roots of SD and CP after 15 days of low P treatment to understand differences and similarities at molecular level. In response to low P, number of genes up-regulated (1318) was more when compared with down-regulated genes (761). Eight hundred twenty-one genes found to be significantly regulated between SD and CP in response to low P. De novo assembly using plant database led to further identification of 1535 novel transcripts. Functional annotation of significantly expressed genes suggests two distinct methods of low P tolerance. While root system architecture in SD works through serine-threonine kinase PSTOL1, suberin-mediated cell wall modification seems to be key in CP. The transcription data indicated that CP relies more on releasing its internally bound Pi and coping with low P levels by transcriptional and translational modifications and using dehydration response-based signals. Role of P transporters seems to be vital in response to low P in CP while sugar- and auxin-mediated pathway seems to be preferred in SD. At least six small RNA clusters overlap with transcripts highly expressed under low P, suggesting role of RNA super clusters in nutrient response in plants. These results help us to understand and thereby devise better strategy to enhance low P tolerance in Indica-type rice.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phosphorus (P) deficiency occurs widely in soils with a high native P-fixing capacity such as acid soils, acidic sulphate soils, and alkaline soils. In rice, P deficiency adversely affects general plant metabolism (Wasaki et al. 2003) that leads to low crop productivity. Previous studies on genetics of phosphorus deficiency tolerance in rice were primarily based on the analysis of genes involved in response to low levels of P (Richardson 2009), including a few studies on mapping populations derived from parents of contrasting level of P deficiency tolerance (Wissuwa et al. 1998; Koide et al. 2013). However, the association of most of the previously reported genes with tolerant and susceptible germplasm is lacking. The biparental studies have generated only limited information on the genes involved in tolerance to phosphorus deficiency. P deficiency tolerance is a complex quantitative trait and understanding of genetically diverse tolerance mechanism is limiting. Therefore, there is a need to generate genomic resources from different tolerant germplasm and understand the functional role of genes identified.
One important aspect for understanding low P tolerance is the response of root growth and development to low-P conditions (Lynch 2007). Roots are important for maintaining crop yields, vital when plants are grown in soils containing insufficient supplies of water or nutrients (Bengough et al. 2011). They are the primary sites for stress signal perception in response to low P that initiates a cascade of gene expression (Gamuyao et al. 2012). Furthermore, root architecture is a key trait for dissecting the genotypic differences in rice responses to P deficiency (Richardson 2009). A number of studies were carried out on the gene expression patterns of roots in rice under low P stress (Richardson 2009; Li et al. 2010). However, these studies focused on comparing gene expression profiles of genotypes at seedling stage in response to short duration stress condition using cDNA microarrays containing cDNA clones from Japonica rice cultivar Nipponbare. Major drawback using such array include: (1) study of gene expression will be limited to set of genes spotted on the array plate and (2) the probes on the array are designed from the sensitive variety, Nipponbare. This may result in the failure to identify genes essential for the phosphorous tolerance. A previous study by Heuer et al. (2009) has clearly demonstrated limitations of using probes designed from a low P susceptible genotype like Nipponbare for identification of low P tolerant/responsive genes.
Despite being poorly characterized at molecular level, Sahbhagi Dhan (SD) is a released Indian variety, showing good performance in poor soils of eastern India. Under acidic field conditions of North Eastern India, the performance of this genotype is as good as the local check (unpublished data). On the other hand, Chakhaou Poreiton (CP) is an old landrace from North eastern state of Manipur, with purple aleurone layer, unique taste and aroma.
In this study, seedling stage root-specific transcriptome profiling using RNAseq was carried out for both SD and CP Indica-type rice varieties grown under normal and long-term low P levels. It has been reported that plants respond to phosphorus deficiency with a typical sigmoid growth curve and showed phosphorus deficiency symptoms after 12–13 days (Postma and Lynch 2010). Therefore, to obtain maximum representation of genes involved in signalling and adaptation response under low P stress, a time point of 15 days stress was selected in our study. The 30-day old root tissue collected after 15 days of low P treatment has allowed us to target the crucial stage in low P response in roots, making this transcriptome dataset first of its kind to be generated in Indica-type rice. The analysis of transcriptome has led to identification low P responsive genes and pathways associated with long-term P deficiency tolerance, which will help in devising strategies to enhance low P tolerance in rice.
Materials and methods
Hydroponics experiment and measurement of P content
Seeds of both the genotypes were pre-germinated on wet filter paper at 30 °C for 3 days and were transferred to Yoshida solution for growth. After 15 days, the seedlings were divided into two groups: control seedlings, grown in the full strength Yoshida solution (0.31 mM Pi) (Yoshida et al. 1976), and treated seedlings, grown in Yoshida solution with 0.015 mM Pi (Yi et al. 2005). After 15 days of growth, plants were harvested. For half of the harvested samples (both control and treatment), fresh weight was taken and immediately frozen with liquid nitrogen and stored at −80 °C. The other half was dried; dry weight taken and P content in the leaves and roots of seedlings are determined by the phosphovanadate method (Hanson 1950) after digestion in a mixture of HNO3 and HClO4 (3:1). Three independent replicates were used per genotype. After harvesting, the roots were frozen in liquid nitrogen and stored at −80 °C.
Illumina GAIIx sequencing and mapping of Illumina reads
All the four samples were sent to the commercial service provider, Xcelris Genomics Ltd., Ahmedabad, India for sequencing. The extraction of total RNA was followed by RNA quality and quantity analysis using 1 % denaturing agarose gel and Bioanalyzer (Agilent Technologies, Singapore). Libraries were generated using standard library preparation (Bagnaresi et al. 2012). Mapping of Illumina reads was done using standard Illumina pipeline (Garg et al. 2013). Only reads passing the quality criteria were used for downstream processing (Bowtie). The spliced read mapper TopHat version 1.4.1 was used to map reads to Nipponbare (MSU release 6.1) genome. Mapped paired end reads were de-duplicated with Picard 1.63, and read counts were collected with HTSeq version 0.5.3 (http://www-huber.embl.de/users/anders/HTSeq) in the paired end and ‘union’ mode using Oryza sativa MSU 6.1 GTF file. De novo transcriptome assembly was performed using the CLC Genomics Workbench using default parameters, and assembled consensus sequences were exported into a fasta file (Fig. S1).
Differential gene expression
Raw read counts obtained were normalized to reads per kilobase per million (RPKM), and genes above the 0.1 RPKM threshold were considered expressed. The RPKM values (RPKM_control and RPKM_treated) were further used to calculate the log fold change as log2 (RPKM_treated/RPKM_control). The categorization of differentially expressed genes (DEG) was done as mentioned previously by Bagnaresi et al. (2012). Based on the statistical significance (p value greater than the false detection rate (FDR) after Benjamin-Hochberg correction for multiple-testing), a set of expressed genes were identified. Genes having a log2-based ratio ≥2 were considered differentially expressed genes (DEG). Heat maps showing expression profiles (log2 fold change) were generated using the MultiExperiment Viewer (MeV, v4.8).
Analysis of metabolic pathway and gene ontology
To characterize the metabolic pathway involved in P deficiency in rice adapted to acidic soils, the statistically significant genes were classified into different categories based on the RiceCyc database and the Plant Metabolic Network (Garg et al. 2013). Using the locus search tool of Rice Genome Annotation Project, GO Classification was determined.
GO enrichment analyses
For identifying terms that were present in significant numbers, goseq analyses were performed (Bagnaresi et al. 2012). Briefly, gene lengths from Nipponbare cDNA (O. sativa MSU6.12) were retrieved with BiomaRt queries using FDR cutoff of 0.05. Gene ontology terms for biological processes and molecular functions were retrieved.
QRT-PCR analysis for confirmation of expression of selected differentially expressed genes
To validate the results from the transcriptome experiment, 10 randomly selected differentially expressed genes (DEGs) were analyzed using qRT-PCR. Total RNA was extracted from roots using Trizol reagent (SIGMA) according to the manufacturer’s protocol. The first strand cDNA synthesis was carried out as previously described by Dkhar et al. (2014). The rice ubiquitin gene (Jain et al. 2006) was used as reference gene for normalization. Amplification was confirmed by melt curve analysis and threshold values (Ct) for each of the genes calculated. Three technical replicates were performed. The correlation between expression profiles of selected genes obtained from qPCR and RNA-seq data analyses was determined using MS Excel.
1Kb upstream analysis
Upstream sequences (1 kb) of highly up-regulated genes were extracted and analyzed for cis-acting elements using PLACE database.
Results
Differential response of rice genotypes SD and CP to low P and transcriptome sequencing
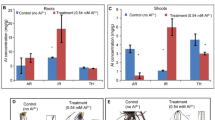
The growth of rice genotypes, SD and CP after 30 days under control (+P; 0.031 mM) and low P (−P; 0.015 mM) conditions, resulted in variation in root length and biomass (Fig. 1a; b). In SD, low P conditions led to more root biomass while the root biomass decreased in case of CP (Fig. 1c). In both CP and SD, the P content in the seedlings grown under low P conditions was comparable to the seedlings grown under +P conditions (Fig. 1d). Both the rice genotypes show features of low P tolerance, but the physiological approach and, thereby, underlying molecular mechanism responsible for low P tolerance seem to be significantly different. Therefore, we investigated the effects of low P treatment in roots of these two Indica-type rice genotypes. The approach used was generation of cDNA library from root tissues of both control and treated samples of SD and CP genotypes followed by next-generation sequencing.

Growth and P analysis of 30-day-old seedlings of two rice genotypes (SD and CP) grown under low P conditions for 15 days. a Biomass of roots and shoots in SD. b Biomass of roots and shoots in CP. c Length of rice roots in SD and CP. d Total P concentration in roots and shoots of rice genotypes. Significance was defined as P < 0.05. Error bars indicate standard deviation. SD, Sahbhagi Dhan; CP, Chakhao Poreiton
The size range of RNA-seq libraries was 250 to 400 bp (Fig. S2). A total of more than 131 million reads from above four samples were generated. After quality filtering, about 121 million high-quality reads (ranging from about 8 to 22 million reads for each sample) were obtained (Table 1).
Transcriptome assembly and differential gene expression analysis
The annotated cDNA sequences of Nipponbare (Japonica rice) are available (MSUv6.1; http://rice.plantbiology.msu.edu/) and were used as reference. Nipponbare is by far one of the most accurately assembled and annotated genomes. The same approach of using Japonica rice genome for assembly of RNA seq data generated on “indica” has also been reported previously (Garg et al., 2013). The genome assembly led to mapping of 53–59 % of the high-quality reads (Fig. S1) obtained from the four different samples (Table 1) with about 56 % of rice proteins represented in the assembly. More percentage of SD reads (58.61 %) mapped to reference genome compared with CP (54.64 %). After mapping of reads, 38,653 expressed genes were identified for SD and 41,126 for CP respectively (Fig. S3). A total of 5571 (14.4 %) and 4536 (11 %) transcripts for SD and CP, respectively, were identified as low P responsive based on p value. A set of 1620 and 1051 genes were differentially expressed (showing ≥2-fold changes) under low P conditions in SD and CP, respectively (Fig. 2a) (Table S1).

Functional annotation of differentially expressed genes under low P condition. a The number of up- and down-regulated genes under low P conditions in the two rice genotypes Shahbhagi Dhan (SD) and Chakhao Poreiton (CP). The total number of genes differentially expressed under each condition is given on the top of each bar. b The correlation of gene expression results obtained from quantitative-PCR analysis and RNA-seq for 10 selected genes. c Characterization of SD and CP transcripts expressed in low P conditions using GO Slim term assignment based on biological process. d Characterization of SD and CP transcripts expressed in low P conditions using GO Slim term assignment based on molecular function. Transcripts having p < 0.05 were considered
The results obtained from differential gene expression analysis were validated by qRT–PCR analysis for 10 randomly selected genes. The analysis revealed similar expression pattern for all the selected genes in qPCR analysis as observed from RNA-seq data. The statistical analysis also showed significant association (r 2 = 0.673) between the results of qPCR and RNA-seq data analyses (Fig. 2b).
To assign functional categories to the stress-responsive genes, gene ontology (GO) enrichment analysis was performed. In the case of SD, the largest number of transcripts belonged to biosynthetic response (19.4 %), response to stress (17.5 %), metabolic activity (16.5 %) and response to abiotic stress (12 %) among the biological function GOSlim terms (Fig. 2c). In CP, in response to low P levels, majority of enriched biological function GOSlim terms belonged to biosynthetic response (26.4 %), metabolic activity (18.4 %) and response to endogenous stimulus (14 %). Among the molecular function terms, the transcripts related to enzyme regulator activity (16.1 %), nucleotide binding (11.3 %), ion binding (11 %), DNA binding (10 %) and transporter activity (10 %) were the largest in number (Fig. 2d) in SD. In CP, transcripts related to nucleotide binding (17 %), DNA binding (15 %), enzyme regulator activity (14.2 %) and ion binding (14 %) were the largest in number. Terms associated with various metabolic processes (GO:0008152; GO:0044237) and biosynthetic processes (GO:0006633; GO:0046949) were most enriched in the genes up-regulated under stress conditions (Table S2). In addition, the GO terms associated with chromatin changes (GO:0006333) were also found enriched in low P conditions. CP showed enrichment for GO terms associated with transport (GO:0015031; GO:0006857) and signal transduction (GO:0007165) in low P conditions (Fig. 2c) while GO terms associated with response to stress (GO:0009651; GO:0009737) were enriched in SD. Enzyme regulatory activity (GO:0050662) was enhanced in both SD and CP (Fig. 2d). Terms enriched with ion-binding activities (GO:0008270; GO:0046872) were found more in CP. Although transport activity was enhanced in CP under low P conditions, the activity of transporters was more in SD (Fig. 2c, d). Interestingly, under low P conditions in SD, we detected enrichment of GO terms implicated in freezing stress response (GO:0042309). GO terms associated with cell wall modification (GO:0042545), cutin biosynthesis (GO:0010143) and cellulose biosynthesis (GO:0030244) were also found up-regulated in low P conditions.
In response to low P, number of genes up-regulated (1318) was more when compared with downregulated genes (761). To further characterize the DEGs, a t test (assuming equal variance and α (overall threshold p value) = 0.01) was performed. This led to identification of 821 genes which were significantly different between the two genotypes, SD and CP (Fig. S4). Furthermore, K-means clustering analysis was performed and 5 clusters with different gene expression patterns across two genotypes were identified (Fig. S5). Among the clusters generated, 3 groups of genes showed distinct pattern of expression (Fig. 3). Cluster I genes were SD specific. Cluster II genes were CP specific. Cluster III genes were upregulated in low P conditions.

K-means clustering of differentially expressed genes under low P conditions. The clustering was performed on log2 fold change for each gene under low P condition (T) when compared with control condition (C). Genes exhibiting similar pattern have been grouped in distinct groups. The scale at the bottom shows log2 fold change
Identification of unannotated transcripts in response to low P
Bacterial artificial chromosome (BAC) clones for an indica-type rice genotype, Kasalath, which is known to carry several favourable alleles for performance under poor soils (Gamuyao et al. 2012), are available. To find similarities between the three indica cultivars for response to low P, raw reads from treated samples of SD and CP were mapped on Kasalath BAC clones (Fig. S1), resulting in identification of an additional set of genes (16 %) which had previously remained undetected in Nipponbare reference genome (Table S3).
Fifty-three to 59 % rice proteins showed a significant hit with the reference genome-based assembly generated from high-quality reads, suggesting that the assembly was optimal. As SD and CP are indica-type rice and Nipponbare is japonica-type rice, the possibility of increasing the number of mapped reads exists. Therefore, de novo assembly of unused reads from treated tissues of both genotypes in the first step of reference-based assembly was done. Using the CLC Genomics Workbench, a total of 7069 and 26,115 contigs for SD (T) and CP (T), respectively, was identified using non-redundant database (Tables S4–S5). The minimum and maximum contig length varied from 200 to 7938 bp. Based on the accession hit with functionally annotated transcript contigs, a total of 1533 annotated transcript contigs were identified (Fig. S3) (Table S6).
Characterization of de novo assembled reads expressed under low P conditions
One of the genes playing role in low P tolerance under field conditions is PSTOL1 (Gamuyao et al. 2012). The expression of PSTOL1 was up-regulated under low P stress in SD, consistent with previous findings (Tyagi et al. 2012).
The de novo assembly led to identification of: (a) expressed novel transcripts and (b) transcripts showing alignment to other species of Oryza, like Oryza minuta, Oryza rufipogon, Oryza officianalis, etc. We found that many transcripts in SD and CP showed similarity with other plant species like Vitis, sorghum, Arabidopsis, etc. More number of contigs (1116) were obtained from SD and included transcripts associated with sugar transport, transcription factor, etc. These contigs from SD and CP which were not identified in the reference genome, Nipponbare, may be specifically transcribed and function only in tolerant genotypes under low P conditions or these transcripts could be Indica-type specific.
Functional annotation of highly up-regulated transcripts under low P conditions for SD and CP roots
In response to low P, highly up-regulated (more than 20-fold increase) genes in SD and CP roots were identified and functionally annotated (Fig. 4, Table S7). In SD and CP, 18 and 7 transcripts respectively were highly up-regulated (Fig. 4b, c). A common set of eight transcripts enriched in GO terms associated with phosphate starvation (phosphate transporter activity) and alteration in lipid processes (glycolipid synthesis) were found to be highly up-regulated in roots of both genotypes (Fig. 4a).

Heat map representation of genes highly expressed under low P conditions across two different rice genotypes (CP and SD). The RPKM values obtained from RNA-seq data were used to calculate fold change and heat map generated using TIGR MeV v4.1.1 software. a Genes highly expressed in both SD and CP. b Genes highly expressed only in SD. c Genes highly expressed only in CP. The highly expressed genes were identified based on fold values. Bar at the top represents fold values; thereby values 0, 20 and 40 represent low, high and very high expression, respectively
The highly expressed genes include high-affinity phosphate transporter, OsPT6 (Os08g45000), members of SPX domain containing protein (Os02g10780, Os03g29250). A set of uncharacterized but expressed proteins (Os09g26670, Os03g51350, Os04g45510, Os01g67364, Os01g02450 and Os05g08830) were also identified. Different protein phosphatases were up-regulated under low P conditions. A member of phosphoethanolamine/phosphocholine phosphatases (Os10g52230) was up-regulated in both SD and CP, though the level of up-regulation was more in CP than in SD roots. Member of serine/threonine phosphatase family (Os10g02750) was highly up-regulated in CP. Jacalin-related lectin (JRL) domain containing protein (Os01g25160) was highly up-regulated only in CP roots under low P conditions. Members of dehydrin family (Os11g26570, Os11g26790 and Os11g26750), LEA proteins (Os3g07180), MYB (Os5g50350), WRKY19 (Os05g49620) and AP2 transcription family (Os09g35010) are highly up-regulated in SD. Topoisomerase 6 subunit A was up-regulated in low P conditions in SD.
The regulation of gene expression can be affected by non-coding RNAs including microRNAs (miRNAs). An in silico analysis (http://sundarlab.ucdavis.edu/cgi-bin/smrna_browse/rice2/) to identify role of small RNAs in regulating the highly expressed genes led to identification of seven genes (Os01g04920, Os01g67364, Os05g31670, Os06g18820, Os03g51350, Os08g20420 and Os09g15240) being part of small RNA supercluster (Fig. S7), while most of the other highly up-regulated genes (31) show small RNA clusters of 3–12 (Fig. S7).
Expression pattern of known genes/factors for low P response in SD and CP
A number of signalling molecules play a role in development of root system architecture in response to low P (reviewed by Niu et al. 2013). Transcript abundance for genes involved in signals like auxins, sugar and metals like Al, Fe and copper in response to low P in roots was investigated. Our analysis of 30 early auxin-responsive Aux/IAA genes of rice revealed up-regulation of 16 out of 25 genes in SD (Fig. 5). Five out of eight auxin response factors were up-regulated in SD. From a total of 15 trehalose biosynthetic genes, 7 were up-regulated in SD and 4 in CP. Suberin is a primary constituent of cell wall, and it has been suggested that changes in the degree of suberization may result in improved stress tolerance in plants (Garg et al. 2013). All the 5 genes (Os01g63540, Os01g70025, LOC_Os05g38350, Os10g34480 and Os11g31090) reported for suberin biosynthesis were highly up-regulated in CP, suggesting an important role in root response to low P (Fig. 5). It has been demonstrated that P deficiency leads to higher accumulation of metals like Fe, Al and copper in Arabidopsis and rice (Ohkama-Ohtsu and Wasaki 2010; Rouached et al. 2010). For Al homeostasis, 3 out of 4 genes (Os01g65080, Os05g02750 and Os06g50910) were up-regulated under low P conditions in CP and SD. When transcript level of genes involved in Fe homeostasis was checked, all the 4 transcripts (Os04g36720, Os06g47910, Os11g01530 and Os12g01530) showed at least 2-fold or higher expression in roots of CP under low P conditions (Fig. 5). Up-regulation of copper oxidases was also observed under stress conditions. Fifty percent of copper oxidase genes were up-regulated only in SD in response to low P and remaining genes were only up-regulated in CP (Fig. 5; Table S8).

The relative expression of reported genes under low P conditions in SD and CP genotypes. Selected genes (P < 0.05) related with metal homeostasis (aluminum and iron homeostasis and copper oxidases), lateral root development; root morphology; auxin response; trehalose, callose and suberin biosynthesis; genes induced by micro RNAs; and Pi transporters in SD and CP genotypes are shown. Bar at the top represents log2 transformed values; thereby values 0, 5.6 and 11.25 represent low, intermediate and high expression, respectively
In roots, one would not only expect genes involved in internal P utilization, but also genes involved in effective P uptake to be up-regulated by long-term P deficiency to account for uptake of P against concentration gradient. In fact, in plants, two P uptake systems with multiple P transporters of each system have been identified—a high-affinity system that is either increased or de-repressed under P deficiency and a low affinity system that is constitutively expressed (Rausch and Bucher 2002). Our data suggests that at least three P transporters (OsPT3, OsPT5 and OsPT10) show genotype-specific variation. Our study is first reported, in our knowledge, to implicate a correlation between activities of P transporters with genotype in P acquisition efficiency. The up-regulated transcripts include many Pi-related genes such as PHR, WRNKY75, LTN, etc. (Richardson 2009) (Fig. 5). Increase in lateral roots in response to P deficiency is a desirable trait as it leads to better foraging of limited nutrient in the top soil. We found significant enrichment of the genes involved in the lateral root development under stress conditions in CP and SD (Fig. 5).
Several miRNA, like osa-miR399 and osa-miR169, have been reported in low P stress. Analysis of expression levels of predicted target genes of miR399 and miR169 in treated roots revealed that while both the targets of miR399 (Os05g48390, Os04g55230) are down-regulated, four of the seven predicted targets of miR169 (including Os03g29760, Os03g44540, Os03g48970 and Os07g41720) were up-regulated at 15 days after low P stress in both SD and CP (Fig. 5). In addition, Os07g06470 is up-regulated only in SD and Os02g19970 and Os02g18064 only in CP after low P stress.
Cis-regulatory elements in the 5′UTR of highly expressed genes suggests cross-talk between signalling pathways
To investigate whether the similar or differential expression pattern observed for genes between the two genotypes was due to similarity or differences, respectively in the cis regulatory elements, individual 5′ untranslated regions (5′UTR) of highly expressed genes were analyzed using the database of plant promoters (PLACE) (http://www.dna.affrc.go.jp/PLACE), which searches for common motifs in putative promoters. Putative cis-acting regulatory elements were examined in the genes up-regulated or down-regulated under P stress condition using Nipponbare as genome reference. Due to large number of motif counts identified and a large number being significant (p < 0.01), a cutoff of 20 % was set for short-listing the putative motifs. From this set of motifs, the level of occurrence was calculated under P stress for both SD and CP.
A set of 154 common motifs were observed in the 5′UTR of genes expressed under P stress in both rice genotypes (Fig. 6; Table S9). After using a cutoff of 6-mer (most of the transcription binding sites are less than 8-mer in length), 53 motifs in genes over-expressed under treatment conditions were identified (Table S9). Twenty-seven motifs were differentially represented in low P conditions in SD and CP (Fig. 6a).

cis-element enrichment within 1000 bp from the transcription start sites for differentially expressed genes in response to low P levels in SD (T) and CP (T). Seven categories (A to G) of cis-elements were identified. Motifs that were significant (P < 0.01) and had at least 20 % occurrence are shown. Bar at the top represents percentage occurrence; thereby, values 0, 22430 and 9000 represent low, intermediate and high occurrence, respectively
The motifs could be divided into seven categories (Fig. 6b). These 7 categories (A to G) have different number of cis-acting elements with maximum (17 occurrences) found in category A. Noticeably, the abiotic stress and auxin-related motifs were highly represented in genes under treatment conditions, suggesting that genes regulated in this two categories are most common. This is followed by over-representation of motifs related to biotic stress, sugar-response, tissue-specific as well as response to light. This analysis suggests a complex cross-talk among different abiotic stresses and auxin and sugar response in P limiting conditions and also a more global role by different genes having similar transcriptional regulation. The cis-regulating element analysis revealed motifs related to dehydration (A3, A13 and B3) and abiotic response (A6 and B3) were more abundant in CP, whereas motifs related to Myb transcription factor (A10), sugar (C1, C3) and SAUR (Small Auxin Up RNA) (B2) were present in more number in SD (T). This data along with set of highly expressed transcripts suggest diverse mechanism of tolerance in the two rice genotypes.
Discussion
Importance of understanding low P tolerance in Indica-type genotypes adapted to poor soils
Considerable efforts have been made to understand the molecular mechanism of low P tolerance/adaptation in plants (Wasaki et al. 2003, 2006; Li et al. 2006; Cai et al. 2012). However, only a few studies have focused on difference in tolerance mechanism due to diverse genetic backgrounds. It is known that Indica-type genotypes possess greater diversity in terms of response to low P (Rose et al. 2011). So far, Kasalath (a traditional landrace growing in poor soils of Eastern India) is the only Indica-type genotype in which low P tolerance mechanism has been studied extensively. Genotypes adapted to poor soils (soils lacking phytoavailability of macro and/or minor nutrients) harbour tolerant genes/alleles for various abiotic stresses including low P tolerance (Wissuwa and Ae 2001; Lynch 2011). The acidic soils of eastern and north eastern India due to high levels of Pi fixation cause P deficiency symptoms in the plants. Therefore, we identified two Indica-type genotypes (SD and CP) adapted to acidic soils of eastern and north eastern India to study the mechanism of low P tolerance. SD is a released variety performing well in the acidic soils and carries the PSTOL1 gene. CP, a short-grained traditional variety, is known for its purple grain and aroma and lacks the entire PUP1 region (Tyagi et al. 2012). We now know that SD carries a different allele across PupK20 gene (Yumnam et al. 2015), suggesting that though sequence of SD is exactly same as Kasalath for PSTOL1 gene, it harbours differences for at least one other gene reported for low P tolerance. Genome-wide marker study using SSRs has also suggested that SD is genetically distinct and diverse from Kasalath (Tiwari et al. 2015) and CP (Challam et al. 2013). Therefore, SD and CP were selected to study response to low P stress. Root tissues after 15 days of low P treatment from both control and treated samples were taken for studying gene expression. Roots are primary sensors of low P levels and there are only a few studies performed on effect of long-term low P response in roots. The importance of long-term low P deficiency study is now recognized as an appropriate physiological system to study molecular biology of stress tolerance and to utilize the important genes for the improvement of rice via molecular breeding/biotechnological approaches (Lynch 2011).
RNA-seq transcriptome-based approach was used for data generation so as to capture transcripts unique to these two tolerant genotypes. The assembly of significant transcripts was made using: (1) the reference Japonica genome, Nipponbare; (2) alignment to available BAC clones in Kasalath; and (3) de novo assembly using green plants database. This three-tier approach enabled us to identify a large number of transcripts from the two Indica-type rice genotypes. Our study, therefore, provides a gene expression dataset which can be used to identify potential targets for functional studies of genes involved in low P tolerance in Indica-type rice.
Differences in response to low P in SD and CP genotypes
Physiological response of SD and CP to low P conditions is very different. While SD shows increase in root length and root biomass, CP maintains its root length and shows decrease in root biomass. The increase in root biomass requires the investment of nutrients, and the time taken by roots for nutrient acquisition to compensate for these investments increases with decreasing soil fertility (Postma and Lynch 2010). The plant is more nutrient deficient during this period, which may have detrimental effects on shoot growth. However, a study on 14 Brassica cultivars, grown at a range of P levels in hydroponics, showed that P-efficient cultivars produced most shoot and total biomass and had the lowest shoot P concentrations (Akhtar et al. 2008).
Upon analysis of significantly regulated genes (Fig. 7), it was found that there appears to be distinct difference between SD and CP with respect to response to low P. While root system architecture changes in SD may be due to a cellular serine-threonine kinase, PSTOL1, suberin-mediated root cell wall modification seems to be key in CP. The role of OsPSTOL1 in P deficiency tolerance is well known (Gamuyao et al. 2012). However, further investigations are required to find the exact mechanism of P deficiency tolerance in different rice genotypes to unravel novel pathway(s). Suberin is a primary constituent of cell wall, and it has been suggested that changes in the degree of suberization may result in improved stress tolerance in plants (Garg et al. 2013). The up-regulation of genes (Os01g63540, Os01g70025, Os05g38350, Os10g34480 and Os11g31090) associated with cell wall metabolism and suberin (Fig. 7), especially in CP, suggests that these genes may be key players in response to low P stress.

Schematic diagram showing proposed mechanism of low P response. Preferred response mechanism in SD and CP (significant up-regulation under stress condition) are highlighted with box and circles, respectively. Solid lines indicate connections between low P levels, regulatory or molecular components. Dashed lines represent still undefined mechanisms. Arrows denote positive effects, whereas flat-ended lines indicate negative effects. Open arrowheads indicate an increase in metal ions upon P deficiency. PHT stands for P uptake transporters. References for proposed pathway are Rouached et al. 2010; Chiou and Lin 2011
Metal homeostasis is affected during low P conditions, leading to an increase in accumulation of cations like Al, Fe and Cu. These metal ions in turn affect primary root growth (Niu et al. 2013). In both SD and CP, there is similar up-regulation for both Al and Cu levels but genes involved in Fe homeostasis are differentially regulated in CP (Fig. 5).
Increasing P uptake from environment and/or releasing bound Pi from internal cellular pools are keys to better adaptation to low P levels. The gene set identified in our study suggests that genetically diverse genotypes employ different mechanism for stress adaptation (Fig. 7). This study indicated that CP copes with low P levels predominantly by: (a) releasing its internally bound Pi as suggested by up-regulation of phosphatases and high-affinity transporters; (b) by transcriptional and translational modification as suggested by enrichment of molecular GO terms related to nucleotide, DNA and RNA binding and protein modification (Fig. 2c); and (c) by using dehydration response-based signals (enrichment of DRE/CRT motifs; Fig. 6).
In contrast, SD appears to rely more on: (a) signalling components like Myb factors (enrichment of MYBZBM motif, Fig. 6) and (b) sugars like trehalose (Fig. 5; enrichment of sugar responsive motifs like SURE and SREATMSD, Fig. 6) to trigger increased root growth (enrichment in vascular response motifs, Fig. 6 and Fig. 1c) in response to low P.
Protein phosphatases are important component of P signalling pathway and play a vital role in P homeostasis (Chiou and Lin 2011). Members of phosphatases family were preferentially highly up-regulated in CP under low P conditions. Most probably, these phosphatases are involved in release of inorganic phosphate from intracellular sources especially under phosphate-deprived conditions. The role of Pi transporters in P uptake appear to be vital for CP to cope for low P stress. These genes include high-affinity phosphate transporter, OsPT6. Members of SPX domain containing protein (Os02g10780 and Os03g29250) were also found to be highly up-regulated. SPX domain-containing proteins have been implicated in phosphate homeostasis (Rouached et al. 2010). Members of dehydrin family, LEA proteins, MYB, WRKY19 and AP2 transcription family are highly up-regulated under low P conditions in SD. Dehydrin, LEA, MYB and AP2 transcription family members are reported to play role in conferring tolerance to abiotic stresses like salt, dehydration and freezing stress in transgenic rice and Arabidopsis (Su et al. 2010; Hanin et al. 2011; Lata and Prasad 2011). Role of members of WRKY and MYB family like WRKY71 and MYB3S2 are well elucidated in low P stress (Rouached et al. 2010). However, role of dehydrin and LEA members is not reported in low P stress. These transcription family members appear to be genotypic specific in terms of their role in P stress as they are not differentially expressed in CP roots (Fig. 4). Topoisomerase 6 subunit A is up-regulated in SD and can be a putative target for designing strategy for low P tolerance. Members of this family from rice have been reported to confer stress tolerance in rice (Jain et al. 2006). A protein family (jacalin-related lectin (JRL) containing domain) unique to CP roots has been identified. It has been suggested that plant JRLs are critical for plant adaptation to stressful environments (Song et al. 2014) with wheat JRLs showing gene level induced expression mainly in response to biotic and abiotic stresses. Whether a similar role is played by JRL rice ortholog in CP needs to be investigated.
Our analysis revealed that early auxin-responsive genes are up-regulated under P stress (Fig. 5). It has been reported that auxin induces glucanases, and the degradation of polysaccharides in the cell wall leads to cell elongation (Kotake et al. 2000). Previously, changes in expression of various genes involved in cell wall metabolism by Pi deficiency have been observed (Misson et al. 2005). Other components of plant cell wall have also been shown to alter stress tolerance. Expansins form a large gene family in cereal genomes and are involved in expansion of cell walls as the tissue grows. A beta-expansin precursor (Os09g29710) is one of the highly up-regulated genes in both SD and CP.
Though an average of 56.6 % similarity with reference Nipponbare rice proteins was obtained, 821 transcripts appeared genotype-specific. Clustering analysis revealed 3 distinct clusters based on gene expression profiles, and these can be targeted to further understand low P tolerance response in rice. De novo assembly led to identification of genes showing similarity to other species of rice and other plant species. This might prove to be a rich source of putative novel low P tolerant genes. A significant number of genes of unknown function were also differentially expressed under low P conditions, which might also play a crucial role in stress adaptation. An up-regulation of freezing stress tolerance genes under P deficiency stress was observed. Although the exact function of these freezing stress genes in P stress tolerance is unclear, their probable contribution to osmolyte accumulation may help in water balance, ion translocation and stress tolerance. A set of uncharacterized but expressed proteins (Os09g26670, Os03g51350, Os04g45510, Os01g67364, Os01g02450 and Os05g08830) were identified among the highly up-regulated gene set. These proteins could be studied in greater detail to identify new protein domains/families involved in low P stress tolerance.
The regulation of gene expression can be affected by non-coding RNAs including microRNAs (miRNAs). miRNAs operate mainly at the level of translation but can influence transcription indirectly by regulating translation of protein transcription factors. An important example is regulation of Hox genes in animal development. Seven genes in current study were found to be part of small RNA super clusters. This suggests that there are genic regions in rice which are regulated by non-coding RNAs. MiRNAs like miR399 and miRNA169 have been implicated in low P response in plants (Richardson 2009; Cai et al. 2012). Analysis of expression levels of predicted target genes of osa-miR399 and osa-miR169 in roots of SD and CP revealed differential regulation under low P stress. In addition, genotype-specific response for miRNA targets was detected as well. In our understanding, this is the first report suggesting the role of super clusters in regulation of nutrient response genes in plants. This study, therefore, opens door for better understanding of signalling pathway and its various components involved in low P stress.
In conclusion, we have obtained a transcript dataset using RNA seq approach from indica rice adapted to low P conditions prevalent in acidic soils. It will be interesting to investigate the precise role of genes involved in the two distinct pathways enriched during P deficiency stress. This rice dataset will provide a strong basis for future research on the response to P deficiency. In addition, a new set of candidate targets for functional studies on genes involved in P deficiency tolerance has been generated. Further work focusing on characterizing these genes could lead to a better understanding of the genetic basis of the rice response to this important environmental stimulus and would be essential for improving performance of rice in P deficient conditions.
References
Akhtar MS, Yoko O, Tadashi A (2008) Intraspecific variations of phosphorus absorption and remobilization, P forms, and their internal buffering in Brassica cultivars exposed to a P-stressed environment. J Integr Plant Biol 50:703–716
Bagnaresi P, Biselli C, Orru L, Urso S, Crispino L, Abbruscato P, Piffanelli P, Lupotto E, Cattivelli L, Vale G (2012) Comparative transcriptome profiling of the early response to Magnaporthe oryzae in durable resistant vs susceptible rice (Oryza sativa L.) genotypes. PLoS One 7, e51609
Bengough AG, McKenzie BM, Hallet PD, Valentine TA (2011) Root elongation, water stress, and mechanical impedance: a review of limiting stresses and beneficial root tip traits. J Exp Bot 62:59–68
Cai HM, Lu YG, Xie WB, Zhu T, Lian XM (2012) Transcriptome response to phosphorus starvation in rice. Acta Physiol Plant 34:327–341
Challam C, Kharshing GA, Yumnam JS, Rai M, Tyagi W (2013) Association of qLTG3-1 with germination stage cold tolerance in diverse rice germplasm from the Indian subcontinent. Plant Genetic Resources 11:206–211
Chiou TJ, Lin SI (2011) Signaling network in sensing phosphate availability in plants. Annu Rev Plant Biol 62:185–206
Dkhar F, Rai M, Tyagi W (2014) Looking beyond PsTOL1: marker development for two novel rice genes showing differential expression in P deficient conditions. J Genet 93:573–577
Gamuyao R, Chin JH, Pariasca-Tanaka J, Pesaresi P, Catausan S, Dalid C, Slamet-Loedin I, Tecson-Mendoza EM, Wissuwa M, Heuer S (2012) The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 488:535–537
Garg R, Verma M, Agrawal S, Shankar R, Majee M, Jain M (2013) Deep transcriptome sequencing of wild halophyte rice, Porteresia coarctata, provides novel insights into the salinity and submergence tolerance factors. DNA Res 42:1–16
Hanin M, Brini F, Ebel C, Toda Y, Takeda S, Masmoudi K (2011) Plant dehydrins and stress tolerance; versatile proteins for complex mechanisms. Plant Signal Behav 10:1503–1509
Hanson WC (1950) The photometric determination of phosphorus in fertilizers using the phosphovanado-molybdate complex. J Sci Food Agric 1:172–173
Heuer S, Lu X, Chin JH, Tanaka JP, Kanamori H, Matsumoto T, De Leon T, Ulat VJ, Ismail AM, Yano M, Wissuwa M (2009) Comparative sequence analysis of the major quantitative trait locus phosphorus uptake 1 (Pup1) reveal a complex genetic structure. Plant Biotechnol J 7:456–471
Jain M, Nijhawan A, Tyagi AK, Khurana JP (2006) Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative realtime PCR. Biochem Biophys Res Commun 345:646–651
Koide Y, Tanaka JP, Rose T, Fukuo A, Konisho K, Yanagihara S, Fukuta Y, Wissuwa M (2013) QTLs for phosphorus deficiency tolerance detected in upland NERICA varieties. Plant Breed 132:259–265
Kotake T, Nakagawa N, Takeda K, Sakurai N (2000) Auxin-induced elongation growth and expressions of cell wall-bound exo- and endo-β-glucanases in barley coleoptiles. Plant Cell Physiol 41:1272–1278
Lata C, Prasad M (2011) Role of DREBs in regulation of abiotic stress responses in plants. J Exp Bot 62:4731–4748
Li Y-F, Luo A-C, Hassan MJ, Wei X-H (2006) Effect of phosphorus deficiency on leaf photosynthesis and carbohydrates partitioning in two rice genotypes with contrasting low phosphorus susceptibility. Rice Sci 13:283–290
Li L, Liu C, Lian X (2010) Gene expression profiles in rice roots under low phosphorus stress. Plant Mol Biol 72:423–432
Lynch JP (2007) Roots of the second green revolution. Aust J Bot 55:493–512
Lynch JP (2011) Root phenes for enhanced soil exploration and phosphorus acquisition: tools for future crops. Plant Physiol 156:1041–1049
Misson J, Raghothama KG, Jain A, Jouhet J, Block MA, Bligny R, Ortet P, Creff A, Somerville S, Rolland N, Doumas P, Nacry P, Herrerra-Estrella L, Nussaume L, Thibaud MC (2005) A genome-wide transcriptional analysis using Arabidopsis thaliana affymetrix gene chips determined plant responses to phosphate deprivation. Proc Natl Acad Sci U S A 102:11934–11939
Niu YF, Chai RS, Jin GL, Wang H, Tang CX, Zhang YS (2013) Responses of root architecture development to low phosphorus availability: a review. Ann Bot 112:391–408
Ohkama-Ohtsu N, Wasaki J (2010) Recent progress in plant nutrition research: cross-talk between nutrients, plant physiology and soil microorganisms. Plant Cell Physiol 51:1255–1264
Postma JA, Lynch JP (2010) Theoretical evidence for the functional benefit of root cortical aerenchyma in soils with low phosphorus availability. Ann Bot 107:829–841
Rausch C, Bucher M (2002) Molecular mechanisms of phosphate transport in plants. Planta 216:23–37
Richardson AE (2009) Regulating the phosphorus nutrition of plants: molecular biology meeting agronomic needs. Plant Soil 322:17–24
Rose TJ, Rose MT, Pariasca-Tanaka J, Heuer S, Wissuwa M (2011) The frustration with utilization: why have improvements in internal phosphorus utilization efficiency in crops remained so elusive? Front Plant Sci 2:73
Rouached H, Arpat AB, Poirier Y (2010) Regulation of phosphate starvation responses in plants: signaling players and cross-talks. Mol Plant 3:288–299
Song M, Xu W, Xiang Y, Jia H, Zhang L, Ma Z (2014) Association of jacalin-related lectins with wheat responses to stresses revealed by transcriptional profiling. Plant Mol Biol 84:95–110
Su CF, Wang YC, Hsieh TH, Lu CA, Tseng TH, Yu SM (2010) A novel MYBS3-dependent pathway confers cold tolerance in rice. Plant Physiol 153:145–158
Tiwari K, Singh A, Pattanaik S, Sandhu M, Kaur S, Jain S, Tiwari S, Mehrotra S, Anumalla M, Samal R et al (2015) Identification of a diverse mini-core panel of Indian rice germplasm based on genotyping using microsatellite markers. Plant Breed. doi:10.1111/pbr.12252
Tyagi W, Rai M, Dohling A (2012) Haplotype analysis for Pup1 locus in rice genotypes of north eastern and Eastern India to identify suitable donors tolerant to low phosphorus. SABRAO J Breed Genet 44:398–405
Wasaki J, Yonetani R, Kuroda S, Shinano T, Yazaki J, Fuji F, Shimbo K, Yamamoto K, Sakata K, Sasaki T, Kishimoto N, Kikuchi S, Yamagishi M, Osaki M (2003) Transcriptomic analysis of metabolic changes by phosphorus stress in rice plant roots. Plant Cell Environ 26:1515–1523
Wasaki J, Shinano T, Onishi K, Yonetani R, Yazaki J, Fujii F, Shimbo K, Ishikawa M, Shimatani Z, Nagata Y, Hashimoto A, Ohta T, Sato Y, Miyamoto C, Honda S, Kojima K, Sasaki T, Kishimoto N, Kikuchi S, Osaki M (2006) Transcriptomic analysis indicates putative metabolic changes caused by manipulation of phosphorus availability in rice leaves. J Exp Bot 57:2049–2059
Wissuwa M, Ae N (2001) Genotypic variation for tolerance to phosphorus deficiency in rice and the potential for its exploitation in rice improvement. Plant Breed 120:43–48
Wissuwa M, Yano M, Ae N (1998) Mapping for phosphorus-deficiency tolerance in rice (Oryza sativa L.). Theor Appl Genet 97:777–783
Yi K, Wu Z, Zhou J, Du L, Guo L, Wu Y, Wu P (2005) OsPTF1, a novel transcription factor involved in tolerance to phosphate starvation in rice. Plant Physiol 138:2087–2096
Yoshida S, Forno DA, Cook JH, Gomez KA (1976) Laboratory manual for physiological studies of rice. International Rice Research Institute, Manila, pp 61–67
Yumnam JS, Rai M, Tyagi W (2015) Allele mining across PSTOL1 and Dirigent-like genes reveals novel haplotypes in rice genotypes adapted to acidic soils. Plant Genetic Resources. doi:10.1017/S1479262115000544
Acknowledgments
This work was supported by grant from Indian Council of Agricultural Research, National Agricultural Innovative Project [C30033/415101-036 to W.T.]. We are grateful to Drs. T. Mohapatra and A.K. Singh, NAIP, for helpful suggestions during initial design of the experiment and for sharing of seeds of SD, respectively. We are also grateful to Dr. P. Dasaradhi for fruitful discussions. Julia S. Yumnam and Aibanshan K. Dohling are acknowledged for technical assistance.
Note: The RNAseq data reported are available in NCBI sequence read archive under the accession number SRP058964 (PRJNA285664).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Bhumi Nath Tripathi
Supplementary materials
Below is the link to the electronic supplementary material.
Figure S1
Experimental design and bioinformatics flow. (PPT 168 kb)
Figure S2
cDNA library bioanalyzer profile on high sensitivity chip of the four samples. (PPT 422 kb)
Figure S3
Venn diagram showing the number of genes expressed under different conditions in SD (a) and CP (b). Venn diagrams display the overlap between expressed genes in roots following treatment with low P (green circles, right) compared with control (peach circles, left). The total gene count for each category is shown within each circle. (PPT 186 kb)
Figure S4
Volcano plot showing significantly different genes between the two genotypes, SD and CP. (PPT 126 kb)
Figure S5
K-means clustering of differentially expressed genes under low P conditions into five distinct clusters. (PPT 142 kb)
Figure S6
Global overview and hierarchical clustering of Sahbhagi Dhan (SD) and Chakhao Poreiton (CP) transcriptional changes. Mean expression versus log fold change plots (MA-plots) were computed for significantly expressed transcripts in SD and CP; normalized expression mean values are plotted versus log2 fold changes and called DEGs (FDR<0.05) are plotted in red. Bar at the top represents log2 transformed values, thereby values 0, 40 and 200 represent low, intermediate and high expression, respectively (PPT 388 kb)
Figure S7
Selected genomic origins showing target for small RNAs. The potential target transcript is written at the right side of each snapshot with locus ID (LOC_). The number of individual clusters within a region is indicated on top of the line. (PPT 244 kb)
Table S1
List of transcripts differentially regulated in SD and CP under low P conditions. (XLS 640 kb)
Table S2
List of GOSlim term assignment to SD and CP transcripts in different categories of biological process under control (C; normal P levels) and treatment conditions (T; low P levels). (XLS 204 kb)
Table S3
The mapping statistics of SD and CP treated samples on Kasalath BAC clones. (DOC 26 kb)
Table S4
Denovo assembly of unused reads for genotype, SD. (XLS 2104 kb)
Table S5
Denovo assembly of unused reads for genotype, CP. (XLS 7542 kb)
Table S6
List of functionally annotated transcripts based on denovo assembly. (XLS 240 kb)
Table S7
List and functional annotation of highly upregulated transcripts under low P conditions in genotypes, SD and CP. (XLS 33 kb)
Table S8
Log2 fold expression of putative copper oxidase transcripts under low P conditions in genotypes, SD and CP. (XLS 185 kb)
Table S9
List of cis-acting elements. (TXT 1 kb)
Rights and permissions
About this article

Cite this article
Tyagi, W., Rai, M. Root transcriptomes of two acidic soil adapted Indica rice genotypes suggest diverse and complex mechanism of low phosphorus tolerance. Protoplasma 254, 725–736 (2017). https://doi.org/10.1007/s00709-016-0986-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00709-016-0986-7