
Abstract
In the present study, electrochemical sensing electrode based on uniform poly(arginine)-modified carbon nanotube paste electrode (PAMCNTPE) is introduced for concurrent determination of caffeic acid and paracetamol. Scanning electron microscopy (SEM) and electrochemical impedance spectroscopy (EIS) were used to characterize the surface of the sensor. In addition, cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were used to investigate the performance of PAMCNTPE. The redox optimum current of caffeic acid was increased dramatically at the PAMCNTPE surface alongside bare carbon nanotube paste electrode (BCNTPE) and their overpotentials decreased. SEM images shows the morphological differences between bare and modified electrode which shows the evidence of modification. The effects of pH, polymerization cycle, and potential were investigated on the electrochemical redox response of caffeic acid based on response surface methodology. In addition, scan rate technique showed the process is adsorption controlled. Under the optimized conditions, linear response was obtained over the range of 10–200 μM, and also, detection limits were found to be 0.13 μM by DPV method on the surface of PAMCNTPE. The PAMCNTPE showed good stability, reproducibility, and repeatability as well as high recovery. The practical application of the PAMCNTPE was demonstrated by measuring the concentration of caffeic acid in apple juice as real sample. The proposed technique is simple, rapid, and cost-effective, serving as an important analytical tool for quality control in the pharmaceutical industry.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
One polyphenol that belongs to the hydroxycinnamic acid category is caffeic acid (CA, Scheme 1). Plants such as tomatoes, mulberries, strawberries, tea, red wine, and coffee contain caffeine [1]. Reactive oxygen species are scavenged, and the antioxidant properties of caffeine acid are shown [2, 3]. Furthermore, because of its potential as an antioxidant, caffeic acid has been referred to as a free radical scavenger [4]. CA has the pharmacological action of protecting against inflammation and cancer cells [5]. By reducing arterial inflammation and atherosclerosis, caffeine has an antioxidant effect on the circulatory system [6]. The calculation of CA is of great relevance due to these business purposes. Phenolic substances are identified by chromatographic techniques [7, 8] and colorimetric approaches [9], often identified by voltammetric measures [10,11,12,13]. Because of the well-defined redox processes and characteristics of caffeine [14, 15], electrochemical techniques can be employed to detect caffeine effectively. In addition, voltammetric methods yield excellent findings and have certain benefits over spectrophotometric methods for determining polyphenols. These advantages include a quicker response and reduced experimental cost when compared to chromatographic methods and the Folin–Ciocalteu reagent method [16,17,18,19].

As quick, easy, sensitive, and reasonably priced analytical methods, electrochemical techniques such as cyclic voltammetry (CV) and differential pulse voltammetry (DPV) are regarded as excellent substitutes for traditional methods when identifying organic molecules that experience oxidation or reduction reactions, such as pharmaceuticals and related molecules in pharmaceutical preparations and biological fluids [20,21,22,23,24,25]. Since their surfaces serve as the sites of oxidation–reduction processes, modified electrodes now represent the most significant alternative in electrochemistry due to the breadth of their uses [26,27,28]. Because of their physicochemical characteristics, modified electrodes are also employed as electrochemical sensors [29,30,31]. A common starting material for the production of traditional electrodes such as glassy carbon (GC), carbon nanotubes (CNT), or multiwall carbon nanotubes (MWCNT) is carbon [32,33,34,35,36]. It has a solid reputation for being a good, chemically stable electrical and thermal conductor [37]. Because of these factors, several researchers have modified the surfaces of carbon and its allotropic derivatives to further enhance their electrochemical detection capability, which allows them to be used as basic materials for sensors [38,39,40]. (Scheme 2).

In this work, we describe a novel strategy for the individual and simultaneous determination of CA and paracetamol (PA) using a novel electrochemical sensor based on a polymer-modified carbon nanotube paste electrode for the first time. This work is a continuation of our previous studies concerning the development of electrochemical sensors through electro-catalysis for the detection of some important biological compounds [41,42,43,44,45]. To obtain good sensitivity and stability, the suggested electrode’s composition and the operating environment are methodically tuned. Interestingly, this investigation looked at the possibility of reciprocal interference between PA and CA, indicating that their oxidation occurs separately at the PAMCNTPE sensor. Ultimately, the modified electrode was effectively used to determine CA and PA simultaneously in a variety of genuine samples.
Results and discussion
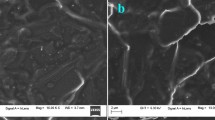
Morphologies of the BCNTPE and PAMCNTPE using SEM
The surface of the BCNTPE and PAMCNTPE is shown in the scanning electron microscopy (SEM) picture shown in Fig. 1a, which shows layers of multiwall carbon nanotubes with more shape. Following electropolymerization, discrete polymer films spread out over the electrode surface, exhibiting a much more sharp edged texture (Fig. 1b). This visible proof indicates that the electropolymerization procedure successfully changed the BCNTPE’s surface.

SEM images displaying (a) BCNTPE and (b) PAMCNTPE
Fabrication of PAMCNTPE
The PAMCNTPE electrode was created using the CV technique for electrochemical polymerization, operating within a potential ranging from − 1.0–1.5 V, as illustrated in Fig. 2a. A solution of 0.1 mM arginine in 0.2 M PBS (pH 7.0) was employed for the polymerization. To optimize the number of electrochemical polymerization cycles, experiments were conducted with 5, 10, 15, and 20 cycles, as shown in Fig. 2b. The current varied across these cycles, achieving maximum electrochemical activity at 10 cycles. Consequently, it was concluded that 10 scan cycles provide the optimal conditions for fabricating the PAMCNTPE electrode for further parameters.

(a) Electropolymerization in 7.0 pH of 0.2 M PBS on BCNTPE. (b) Graph of number of cycles vs. current
The EIS analysis
The process of electrochemical impedance spectroscopy (EIS) was examined to characterize each electrode surface. Consequently, a notable variation in the electrode responses was seen in the Nyquist plots for 1.0 × 10−3 mol dm−3 [K4Fe(CN)6] in 1.0 mol dm−3 KCl (Fig. 3). The semi-circular elements for BCNTPE (curve b) with a high diameter correlate to the measured charge transfer resistances (Rct), as this figure illustrates. However, the diameter of the semicircle was reduced when PAMCNTPE (curve a) electrodes were used. Obtained data from the Rct value simplified that, while employing the polymer-modified electrode, the charge transfer resistance of the electrode surface dropped and the charge transfer rate increased. Furthermore, comparing the plots for BCNTPE (curve b) and PAMCNTPE (curve a) revealed that the electrode Rct had an order of BCNTPE > PAMCNTPE. These findings suggested that adding polymer film to the carbon nanotube paste’s composition might enhance the electrode’s reactivity. Leading to an electrode with a high specific surface area when modified by the polymer film.

Nyquist plots showing (a) BGPE and (b) P(LT-Y)/MGPE
pH optimization
All pH ranges are susceptible to the electrochemical detection of CA; however, the pH range between 6.0 and 8.0 is particularly sensitive. Using 0.2 M phosphate buffer solution (PBS) at different pH values ranging from 6.0 to 8.0, CV was used to measure the voltammetry responses of CA at PAMCNTPE, as shown in Fig. 4a. The anodic peak potential went towards the negative potential as the pH value increased from 6.0 to 8.0, indicating that the proton had taken part in the redox process of CA. The greatest peak current of CA is around 7.61 µA at pH 7.0. The proton’s participation in the redox reaction of CA is shown by the reduction in the optimum current with an increase in the basicity of the solution (a rise in pH, Fig. 4b). We discovered that the ideal pH for determining CA previously thought to be 7.0. In addition, Fig. 4c demonstrates a strong direct correlation between pH and the anodic peak potential (Epa), with the regression equation for Epa/V = 0.744–0.065 pH (R2 = 0.99). The redox reaction at the surface of the electrode includes an equivalent amount of protons and electrons, as shown by the equation’s slope value of − 0.065, which is quite near the theoretical value [46].

(a) CV response of CA at PAMCNTPE across various pH levels ranging from 6.0 to 8.0 (0.2 M PBS) at a sweep rate of 0.1 V/s. (b) Graph of redox peak current (Ipa) vs. solution pH. (c) Plot of anodic peak potential (Epa) vs. pH
Electrochemical behavior of CA at BCNTPE and PAMCNTPE using K4[Fe(CN)6]
K4[Fe(CN)6] is used as the redox probe in CV to examine the electrochemical behavior of BCNTPE and PAMCNTPE (Fig. 5). PAMCNTPE (curve a) and BCNTPE (curve b) exhibit two distinct voltammetric peaks, as seen in Fig. 6. When compared, the electrochemically active sites are more in PAMCNTPE than BCNTPE. Thus, we can assume that the modification of the electrode is the reason behind the increased electron transfer.

CVs of 1.0 mM K4[Fe(CN)6] in 0.1 M KCl at (a) PAMCNTPE and (b) BCNTPE

Curve (a) illustrates the CA response at BCNTPE, curve (b) depicts the CV response of CA at PAMCNTPE in 0.2 M PBS at pH 7.0, and curve (c) shows the CV response of 0.2 M PBS (pH 7.0) without CA at PAMCNTPE
From the Randles–Sevcik equation Ip = 2.69 × 105 A D1/2 n3/2 ν1/2 Co, the surface area of the BCNTPE and PAMCNTPE can be determined by substituting the values of D, A, n, and C. A denotes the active surface area of the electrode (cm2), n represents the number of total electrons involved in the redox reaction, Co signifies the concentration in mM, D denotes the coefficient of diffusion, and ν is the scan rate (V/s); thus, the surface areas of the PAMCNTPE and BCNTPE are determined to be 0.647 and 0.362 cm2, respectively. When compared to electrode reactions utilizing BCNTPE, all of these data clearly demonstrated that PAMCNTPE may significantly improve the current responses of K4[Fe(CN)6] and make their electrode reactions more reversible. These results are recorded and displayed in Fig. 5. Therefore, the PAMCNTPE’s electrochemical reaction to CA was significantly enhanced, and this might be utilized going forward for sensitive CA detection.
Electro-catalytic characteristic of CA at BCNTPE and PAMCNTPE
The electrochemical properties of CA were compared between PAMCNTPE and BCNTPE in a 0.1 M PBS solution (pH 7.0) containing 1 × 10−4 M CA at a scan rate of 0.1 V/s (Fig. 6). Electrochemical reactions occurring at the electrode surface typically involve proton exchange with the solution. Notably, PAMCNTPE (curve b) exhibited peak current compared to BCNTPE (curve a). The modified electrode demonstrated the best performance, with a peak current of approximately 7.61 µA and a potential of around 0.165 V. This observation suggests that the biosensor displays a selective response towards CA, indicating the beneficial impact of the polymer film on enhancing biosensor sensitivity. Figure 6 shows the CVs of both the blank solution and the solution with the electroactive species on the modified electrode (curve b) at a scan rate of 0.1 V/s in 0.1 M PBS (pH 7.0). Notably, the blank solution (curve c) exhibited no oxidation peak. However, upon addition of the CA solution, an anodic peak emerged at a potential of 0.165 V with a current response of 7.61 µA. This result underscores the remarkable catalytic activity of the polymer film, which plays an essential role in the electrooxidation of CA at the surface of the fabricated electrode.
Influence of scan rate
The study aimed to investigate the impact of varying scan rates on the redox peak current response in a solution containing 0.1 mM CA at pH 7.0 (0.2 M PBS) using the PAMCNTPE electrode with the CV technique. The aim was to understand how changes in scan rate affect the behavior of CA in this specific solution. Scan rates ranged from 0.025 to 0.425 V/s, as depicted in Fig. 7a, with a potential window from 0.3 to 0.5 V.

(a) CV for CA at PAMCNTPE in pH 7.0 under multiple sweep rates ranging from 0.025 to 0.425. (b) Graph of Epa vs. log υ. (c) Graph of log Ipa versus log υ. (d) Graph of Ipa versus υ
Figure 7c illustrates the relationship among the logarithm of the anodic peak current (log Ipa) and the logarithm of the scan rate (log υ), showing a clear linear correlation as described by the equation log Ipa/μA = 0.812 log υ–3.41 (R2 = 0.99). The analysis illustrated in Fig. 7c reveals a slope of 0.8 in the log Ipa versus log υ plot, suggesting that adsorption significantly impacts the process. The coefficient of determination (R2) is very near to the ideal value of one. Figure 7d presents a linear correlation between the anodic peak current (Ipa) and the sweep rate (ʋ). In addition, the graph in Fig. 7b shows a clear linear relationship between log υ and Epa, with the corresponding linear regression formula Epa/μA = 0.0809 + 0.233 log υ/V/s (R2 = 0.99). The total number of electrons involved in the redox process of CA was determined using Laviron’s equation and Bard–Faulkner’s equation mentioned as follows:
The total number of electrons is denoted by n, and the electrochemical charge transfer is represented by α. Ep signifies the optimum potential, while Ep/2 indicates the potential at half-peak current. The calculated value is approximately 1.827, which rounds to 2, indicating that two electrons participated in the redox reaction of CA.
Detection limit and calibration curve
We chose the CV mode because it produced sharper, more distinct peaks even at lower CA concentrations, which made it ideal for developing a voltammetric method for determining the CA. Based on the acquired outcomes, this method might be utilized for the quantitative assessment of CA. As the method produced a maximal peak current at a pH of 7.0 (0.2M PBS) was used as the supporting electrolyte for the quantification of CA. As depicted in Fig. 8a, peak current increased linearly with increasing concentrations of CVs obtained with increasing concentrations of CA. Linear calibration curves for CA were produced using the optimal conditions previously stated (Fig. 8b). It was Ipa = 2.89 × 10−5 + 0.579 [CA] (R2 = 0.99), a linear equation. From the various determinations, relevant statistical data for the calibration curves were gathered. 1.3 × 10−7 M was the limit of detection (LOD) and 4.1 × 10−7 M was the limit of quantification (LOQ). The following formulas were used to get the LOD and LOQ: LOD = 3 s/m; LOQ = 10 s/m: here, m is the calibration curve’s slope and s represents the standard deviation of the blank’s peak currents (five runs). Table 1 tabulates the detection limits for CA reported at various paste electrodes. When compared to other electrochemical approaches that have been disclosed, this one performed better [48, 49].

(a) Analyzing a range of CA concentrations (10 μM to 200 μM) on PAMCNTPE in pH 7.0 (0.2 M PBS) using DPV. (b) Represents the correlation between CA concentration and current response
Electrochemical behaviors of a mixture of CA and PA at the PAMCNTPE
Our current work’s primary goal is to determine CA and PA in PBS simultaneously. The DPV for CA and PA coexisting at pH 7.0 (0.2 M PBS) at BCNTPE and PAMCNTPE is displayed in Fig. 9. The DPV produced at the BCNTPE for the solution containing CA and PA combinations is depicted by the dotted line curve (b). For both analytes, the cyclic voltammogram showed a single wide peak. The observation unequivocally shows that each voltammetric signal from CA and PA was not successfully separated by the BCNTPE. The single peak for the combination of CA and PA is caused by the oxidized products of CA and PA fouling the electrode surface.

The DPV responses of CA and PA at PAMCNTPE (curve (a)) and at BCNTPE (curve (b))
The DPV of 0.1 mM CA and 1 mM PA in PBS at the PAMCNTPE is displayed by the solid line curve (a). Two distinct oxidation peaks of CA and PA at PAMCNTPE were seen at 0.0674 V and 0.272 V, respectively, and they corresponded to CA and PA. The catalytic oxidation of PA by the oxidized form of CA was completely abolished because the oxidation peak potential of PA was altered to a less positive potential than that of CA. As a result, the PAMCNTPE was able to determine both PA and CA in PBS simultaneously. The oxidation peak potentials of PA and CA differed by 0.204 V. The updated sensor shows excellent separation.
Investigation of interfering substances
The impact of a few interfering substances on the detection of CA was assessed utilizing the general approach under optimal circumstances in order to look into the selectivity of the suggested method, a crucial parameter for the recently created electrochemical sensor. In the presence of many fold excesses of organic chemicals such as ASC (ascorbic acid), RUT (rutin), RF (riboflavin), VLN (vanillin), and DOP (dopamine), and inorganic ions such as Cd2+, K+, Mg2+, and Fe2+, the selectivity of the PAMCNTPE for the measurement of CA was studied. The anodic peak current fluctuation due to interferences was not more than ± 5%, as indicated in Fig. 10, and it had no discernible effect on the oxidation current of CA. As a result, the suggested sensor has good selectivity and may be applied to the voltammetric analysis of actual samples to determine the content of CA.

Graph illustrating the fluctuation in CA potential on PAMCNTPE in the presence of different interferents
Repeatability, reproducibility, and stability assessment
The precise calculation of CA is influenced by a number of factors, with repeatability being one of the most important. In ideal circumstances, CVs assess the electrochemical sensor’s reproducibility. Five times in a row, CA solutions were identified using the same modified electrode. The very reproducible electrochemical responses of the modified electrode to CA were demonstrated by the relative standard deviations (RSDs) of 5.2%. In the meanwhile, it was imperative to consider the electrochemical sensor’s endurance. Over the course of a month, the modified electrode’s durability was assessed. Eight experiments were carried out in parallel at this time. After a month of room temperature storage for the modified electrodes, the Ipa values of CA remained elevated at 95.8%. As a result, the altered electrode had excellent stability as well. In addition, five separately built electrodes were able to measure the same CA concentration with RSDs of 4.6% and 5.1%, respectively, demonstrating the electrode’s high degree of repeatability.
Real sample analysis
The analysis of the real sample was made using apple juice to demonstrate the adaptability and flexibility of PAMCNTPE for the quantification of CA by employing the DPV technique. The sample was analyzed with varying concentrations of apple juice from 100 μM to 250 μM in a 0.2 M PBS solution at pH 7.0. Data from the DPV technique were evaluated, showing a recovery rate between 96.38% and 98.52%, as indicated in Table 2. These findings demonstrate that the prepared electrode is excellent for analyzing CA in apple juice.
Conclusion
This study described the creation and use of an electrochemical sensor for cyclic voltammetry-based CA measurement that is based on a carbon nanotube paste electrode fabricated with a polymer. SEM images shows the evidence of modification of the electrode. As compared to other electrodes, this sensor’s performance actually offers benefits in terms of analytical figures of merit, including increased sensitivity, a lower detection limit, limit of quantification and a larger dynamic range. The current electrochemical sensor has been adjusted with respect to pH, the electrode’s polymerization cycle, and other factors in order to maximize the sensor’s electrocatalytic reaction to CA oxidation. With a very low detection limit of 0.13 μM, the DPV peak current increases linearly with the concentration of CA in the range of 10–200 μM. The real sample study shows that the electrode is capable of detecting CA using apple juice. The suggested sensor has the potential to be a practical, sensitive, and straightforward procedure for determining CA in a clinical lab.
Experimental
Without further purification, analytical-grade chemicals and reagents were utilized exactly as supplied. In order to obtain the desired pH range of 2.0–11.0, 0.2 M phosphate buffer (PBS) was prepared by mixing various volumes of sodium phosphate monobasic monohydrate (NaH2PO4·H2O, purity 99%) and sodium phosphate dibasic dihydrate (Na2HPO4·2H2O, purity 99.5%) from HiMedia, India, with the appropriate amount of 0.2 M NaOH (purity 98.5%) from Molychem, India. Distilled water and analytical-grade chemicals were used to make each solution. We bought CA (purity 98%) and PA (purity 99%) from Sigma-Aldrich. We bought silicon oil, potassium chloride (KCl) (purity 99.5%), and carbon nanotubes from Nice Chemicals in India.
For the voltammetric studies, the CH-Instrument-6038E (bought from the USA) was used. Throughout the experiment, a standard three-electrode arrangement was used, with a surface-modified carbon nanotube paste electrode and an unmodified carbon nanotube paste electrode serving as the working electrodes. The reference and auxiliary electrodes were a Pt wire and an Ag/AgCl 3 mol dm−3 KCl electrode.
Preparation of the carbon nanotube sensors
Getting ready, a thorough hand mix of carbon nanotube and very viscous silicone oil (60:40) was used to create carbon nanotube paste. A copper wire was attached to the electrical link after both components were homogenized to create a mixture that was then packed into a polytetrafluoroethylene tube with an internal diameter of 3.0 mm. When necessary, the 2 mm diameter carbon paste surface was mechanically refreshed by pressing about 0.5 mm of carbon paste out of the electrode surface and smoothing it with moist filter paper.
By electropolymerizing 0.5 × 10−4 M arginine on the BCNTPE surface during the course of ten consecutive cycles of potential scan ranges between − 1.0 and 1.5 V/s at a scan rate of 50 mV s−1, with 0.2 M PBS as the supporting electrolyte, the poly(arginine) modified CNTPE was created. To get rid of any physical residue left behind after the electropolymerization, deionized water was used to rinse the modified electrode.
Data availability
Data will be made available on reasonable request.
References
Manach C, Scalbert A, Morand C, Remesy C, Jimenez L (2004) J Clin Nutr 79:727
Piazzon A, Vrhovsek U, Masuero D, Mattivi F, Mandoj F, Nardini M (2012) J Agric Food Chem 60:12312
Chang W, Huang D, Lo YM, Tee Q, Kuo P, Wu JS, Huang W, Shen S (2019) J Agric Food Chem 67:7684
Khan FA, Maalik A, Murtaza G (2016) J Food Drug Anal 24:695
Koga M, Nakagawa S, Kato A, Kusumi I (2019) Tissue Cell 60:14
Genaro-Mattos TC, Mauricio AQ, Rettori D, Alonso A, Hermes-Lima M (2015) PLoS ONE 10:e0129963
Zhang L, Li Y, Liang Y, Liang K, Zhang F, Xu T, Wang M, Song H, Liu X, Lu B (2019) Food Chem 276:538
Meinhart AD, Caldeirao L, Damin FM, Teixeira Filho J, Godoy HT (2018) J Food Compos Anal 73:76
Haghi G, Hatami A, Arshi R (2011) Food Chem 124:1029
David IG, Bizgan AMC, Popa DE, Buleandra M, Moldovan Z, Badea IA, Tekiner TA, Basaga H, Ciucu AA (2015) Food Chem 173:1059
Sordoń W, Salachna A, Jakubowska M (2016) J Electroanal Chem 764:23
Zhang Y, Liu Y, He J, Pang P, Gao Y, Hu Q (2013) Electroanalysis 25:1230
Erady V, Mascarenhas RJ, Satpati AK, Bhakta AK, Mekhalif Z, Delhalle J, D’Souza A (2019) Microchem J 146:73
Photinon K, Chalermchart Y, Khanongnuch C, Wang S-H, Liu C-C (2010) Sensors 10:1670
Gamella M, Campuzano S, Reviejo A, Pingarrón J (2006) J Agric Food Chem 54:7960
Tyszczuk K, Skalska-Kamińska A, Woźniak A (2011) Food Chem 125:1498
Makhotkina O, Kilmartin PA (2010) Anal Chim Acta 668:155
Rajendrachari S, Mahale RS, Adimule V, Gulen M, Mahesh V, Basavegowda N (2024) Inorg Chem Commun 164:112428
Mahale RS, Rajashekar V, Vasanth S, Peramenahalli Chikkegowda S, Rajendrachari S, Mahesh V (2024) ACS Omega 9:10660
Švorc Ľ, Strežova I, Kianičkova K, Stanković DM, Otřisal P, Samphao A (2018) J Electroanal Chem 822:144
Stanković DM, Švorc Ľ, Mariano JFML, Ortner A, Kalcher K (2017) Electroanalysis 29:2276
Švorc Ľ, Borovska K, Cinkova K, Stanković DM, Plankova A (2017) Electrochim Acta 251:621
Pysarevska S, Dubenska L, Plotycya S, Švorc Ľ (2018) Sens Actuators B 270:9
Halfadji A, Naous M, Rajendrachari S, Ceylan Y, Ceylan KB, Shekar PR (2024) J Mol Struct 1301:137318
Rajendrachari S, Basavegowda N, Vinaykumar R, Narsimhachary D, Somu P, Lee MJ (2023) Inorg Chem Commun 155:111010
de Barros A, Ferreira M, Constantino CJL, Bortoleto JRR, Ferreira M (2015) ACS Appl Mater Interfaces 7:6828
Mazloum-Ardakani M, Beitollahi H, Amini MK, Mirkhalaf F, Mirjalili BF (2011) Biosens Bioelectron 26:2102
Mazloum-Ardakani M, Beitollahi H, Mohseni MA, Benvidi A, Naeimi H, Nejati-Barzoki M, Taghavinia N (2010) Colloids Surf B: Biointerfaces 76:82
Zhang N, Qu F, Luo HQ, Li NB (2013) Biosens Bioelectron 42:214
Tajik S, Beitollahi H, Shahsavari S, Nejad FG (2022) Chemosphere 291:132736
Venkatramanan S, Chung SY, Ramkumar T, Selvam S (2015) Environ Monit Assess 187:505
Wang J, Anik Kirgöz Ü, Mo J-W, Lu J, Nasser Kawde A, Muck A (2001) Electrochem Commun 3:203
Jashari G, Švancara I, Sýs M (2022) Electrochim Acta 410:140047
Narayana PV, Madhusudana Reddy T, Gopal P, Mohan Reddy M, Ramakrishna Naidu G (2015) Mater Sci Eng C 56:57
Tajik S, Taher MA, Beitollahi H (2014) Ionics 20:1155
Tajik S, Dourandish Z, Nejad FG, Beitollahi H, Jahani PM, Di Bartolomeo A (2022) Biosens Bioelectron 216:114674
Newcomb BA (2016) Compos Appl Sci Manuf 91:262
Shaoying H, Ping H, Zhang X, Xiaojuan Z, Liu K, Jia L, Dong F (2018) Anal Chim Acta 1031:75
Li Y, Ye Z, Zhou J, Liu J, Song G, Zhang K, Ye B (2012) J Electroanal Chem 687:51
Bhakta AK, Mascarenhas RJ, D’Souza OJ, Satpati AK, Detriche S, Mekhalif Z, Delhalle J (2015) Sci Eng C Mater Biol Appl 57:328
Manjunatha JG, Deraman M, Basri NH, Talib IA (2018) Arabian J Chem 11:149
Amrutha BM, Manjunatha JG, Bhatt AS, Hareesha N, Al-Kahtani AA, Tighezza AM, Ataollahi N (2022). Topics Catal. https://doi.org/10.1007/s11244-022-01567-9
Hareesha N, Manjunatha JG, Alothman ZA, Sillanpää M (2022) J Electroanal Chem 917:116388
Tigari G, Manjunatha JG (2020) J Sci Adv Mater Dev 5:56
Engin C, Yilmaz SE, Saglikoglu G, Yagmur S, Sadikoglu M (2015) Int J Electrochem Sci 10:1916
Lawrence NS, Beckett EL, Davis J, Compton RG (2002) Anal Biochem 303:1
Bhimaraya K, Manjunatha JG, Moulya KP, Tighezza AM, Albaqami MD, Sillanpää M (2023) Chemosensors 11:191
Manjunatha JG, Subbaiah NP, Hareesha N, Raril C, Tighezza AM, Albaqami MD (2023) Monatsh Chem 154:1235
Ren W, Luo HQ, Li NB (2006) Biosens Bioelectron 21:1086
Bounegru AV, Apetrei C (2020) Chemosensors 8:41
Shi Y, Xu H, Wang J, Li S, Xiong Z, Yan B, Wang C, Du Y (2018) Sens Actuators B Chem 272:135
Karabozhikova V, Tsakova V (2019) Electrochim Acta 293:439
Santos DP, Bergamini MF, Fogg AG, Zanoni MV (2005) Microchim Acta 151:127
Leite FR, Santos W, Kubota LT (2014) Sens Actuators B Chem 193:238
Fernando dSL, Nelson RS, Paulino OH (2008) Electroanalysis 20:1252
Erk N (2004) Anal Bioanal Chem 378:1351
Diaconu M, Litescu S, Radu G (2010) Sens Actuators B Chem 145:800
Li J, Jiang J, Liu M, Xu Z, Deng P, Qian D, Tong C, Xie H, Yang C (2017) Anal Chim Acta 985:155
Zhang Y, Liu Y, He J, Pang P, Gao Y, Hu Q (2013) Electroanalysis 25(5):1230–1236
Acknowledgements
Sumanth D. gratefully acknowledges the financial support from the SC/ST Cell for the SC/ST Fellowship (No. MU/SCT CELL/RSF/2022-23/SCT-3), Mangalore University. Dr. J.G. Manjunatha gratefully acknowledges the financial support from the VGST, Bangalore, under the Research Project. No. VGST/K-FIST L-2/2022-23/GRD-1020
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sumanth, D., Manjunatha, J.G. Detection of caffeic acid using a nanotube paste-based electrochemical sensor. Monatsh Chem 155, 937–946 (2024). https://doi.org/10.1007/s00706-024-03248-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-024-03248-7