
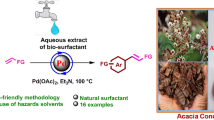
Abstract
In the present work, we have performed eco-benign strategy of in situ formation of PdNPs using aqueous extract of Acacia auriculiformis pods for Mizoroki–Heck coupling. A series of (E)-1-(3-argioallyl)indoline-2,3-diones have been synthesized from coupling of aryl halides with allyl isatins. The PdNPs were characterized by transmission electron microscopy (TEM) which revealed PdNPs size of around 6 nm. The key features of the present method are synthesis of novel derivatives of (E)-1-(3-argioallyl)indoline-2,3-diones, PdNPs in aqueous biosurfactant extract as catalytic system which can be recycled for four times without significant loss in the catalytic activity, no need of external ligand. The influence of various parameters such as the nature and amount of base, source of Pd, screening of available surfactants as well as the effect of temperature has been investigated.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cross-coupling reactions are extremely valuable tool for coupled moieties. The Heck cross-coupling has opened avenues for a variety of elegant and highly convergent routes to structurally complex molecules for instance pharmaceuticals, natural products, agrochemicals, organic polymers, dendrimers and material science [1,2,3,4,5]. In view of this, an organic chemist faces intimidating challenges to develop milder and operationally simpler procedures to carry out Mizoroki–Heck coupling.
The choice of solvent for reaction is a key factor in deciding the parameters such as toxicity, hazardous, cost, and waste generation of a particular process [6,7,8,9,10]. Water a ‘nature’s choice of solvent’ having several unique characteristics [11, 12] for organic synthesis is very attractive from both economic and environmental perspectives. Therefore, researchers focused on creating the most ecological experimental conditions such as use of water and in the presence of ligand‐free catalysts in tiny proportions, i.e. nanoparticles (NPs) [13]. One of the main limitations of the ligand-free approach is Pd leaching. This limitation can be partially avoided by the use of various surfactants and some additives [14, 15]. Some naturally occurring biodegradable surfactants have also been used as efficient additives for stabilizing the substrates and are innocuous to the ecosystem [16].
Although synthetic surfactant have wide application in synthetic and biological processes, their impact on environment such as destruction of aquatic microbial populations, reduction in photochemical energy conversion efficiency of plants and adverse effect on waste-water treatment processes are also considerable. Biosurfactants thus became attractive microbial products due to their environmental susceptible nature [17]. In this regards, Chahdoura et al. explored the significance of glycerol in synthesis of PdNPs using N-substituted PTA-based ligands as an original stabilizer [18]. Sarubbo and co-workers highlighted the advantages of biosurfactant over synthetic surfactant due to their amphipathic structure with hydrophilic and hydrophobic moieties [19]. Markande et al. enlighten the biochemistry and biosynthesis of biosurfactant from different microbial sources [20]. Kumbhar et al. performed Heck coupling employing Pd colloids generated in biosurfactant derived from Acacia concinna [21]. Molecules possessing isatin moiety found in many natural products and exhibits remarkable biological activities, viz. anticancer [22], antitubercular [23], anti-inflammatory [24], antifungal [25], antiviral [26] and anticonvulsant [27]. They are also present in structure of biologically active molecules such as donaxaridine [28], trikentramides A–D [29], and ammosamides [30]. In light of this and in continuation of our special efforts towards the development of an eco-friendly methodology using biodegradable materials in catalysis, herein we have investigated the novel and green method of Mizoroki–Heck coupling for synthesis of (E)-1-(3-argioallyl)indoline-2,3-diones.
The biosurfactant used in this study is a saponin from aqueous extract of Acacia auriculiformis pods commonly named as ‘Australian Babul’ [31, 32]. In present study, we used pods as a green and environmentally benign reaction medium for Pd catalyzed Mizoroki–Heck coupling of allyl isatins and aryl halides without using external ligand in aqueous medium at 100 °C (Scheme 1). To the best of our knowledge, this is the first report on synthesis of N-substituted indoline-2,3-diones employing coupling route. Previously, Shmidt et al. reported microwave assisted synthesis of 1-cinnamylindoline-2,3-diones by N-alkylation of isatin with 3-bromopropenyl benzene [33]. Shrestha and co-workers synthesized said compound by a copper-mediated reaction of 3-diazoquinoline-2,4-diones via ring contraction through domino Wolff rearrangement, decarboxylation, bromination, substitution, and dehydration [34]. Sele obtained 1-cinnamylindoline-2,3-diones together with indolic N′-allylindirubin and N,N′-diallylindirubin derivatives by cascade allylation of indirubin using cinnamyl bromide in the presence of base [35]. Jha et al. used N-cinnamyl derivatives of isatin as a precursors for synthesis of N-allyloxindoles employing Wolff–Kishner reduction. The starting compounds were synthesized by N-allylation of isatin derivatives with cinnamyl bromide in the presence of K2CO3 in DMF [36]. Vaidya and co-workers synthesized 1-cinnamylindoline-2,3-diones one of the derivatives via allylic cross-amination reaction by sequential and selective activation of acyl/allyl C–O bonds under additive-free nickel catalysis following the borrowing carbonate principle [37].

Result and discussion
Preparation of aqueous extract of Acacia auriculiformis dry pods
Pods of Acacia auriculiformis are commonly known as ‘Australian Babul’ and used as detergent in India. The fruits of A. auriculiformis give copious froth on shaking with water in powder form, indicating the presence of saponin. It contains tannins and terpenoids along with the polyphenols. Initially, 100 g of dry fruit pericarp of A. auriculiformis was converted into powder form and dissolved in 1000 cm3 of distilled water. The mixture was stirred for 3 h using a magnetic stirrer. The supernatant solution was then centrifuged for 45 min. The liquid solution was filtered, collected, and referred as aqueous extract (Fig. 1).

Preparation of aqueous extract of Acacia auriculiformis pods
Optimization of reaction conditions for Mizoroki–Heck coupling in aqueous extract of biosurfactant
At the onset feasibility studies were performed for PdCl2 catalyzed Mizoroki–Heck coupling of aryl halide and allyl isatin employing biosurfactant as solvent. The reaction conditions, viz type of catalyst, surfactant, reaction time, and temperature study were screened.
To select the suitable Pd source initially, the model reaction of allyl isatin with iodobenzene was carried out in the presence of Pd(OAc)2 in water. The reaction was not completed and formation of a sticky material is observed (Table 1, entry 1). The reaction with Pd/C did not resulted desired product even after prolonged reaction time (after TLC screening; Table 1, entry 2). The model reaction was also performed with other palladium sources such as Pd(PPh3)4 and PdCl2 (2 mol %) at 100 °C in water, but reaction failed to furnish product in good yield (Table 1, entries 3, 4). Next we employed Pd(OAc)2 and PdCl2 in aqueous extract of biosurfactant and surprisingly it gave exciting results (Table 1, entries 5, 6). Furthermore, the effect of amount of PdCl2 was investigated (Table 1, entries 6–11). The best result was obtained for 3 mol % of PdCl2 with 87% of desired product.
We also checked the effect of other ionic surfactants such as sodium dodecyl sulfate (SDS), cetyltrimethylammonium bromide (CTAB), and sodium dioctyl sulfosuccinate (SDOSS) for the model reaction. Even though these form good CMC in aqueous medium, reactions give very sluggish results (Table 1, entries 12–14). This might be because of no reduction of Pd(II) to Pd(0), which is essential for the progress of reaction. When we used Triton X-100 with PdCl2 (3 mol %), the yield of the reaction was improved gradually up to 80% (Table 1, entry 15); however, the results are not considered due to undesired nature of Triton X-100.
After stirring the reaction mixture at 100 °C, we observed colour change in the reaction mixture from brown to black indicate the formation of PdNPs. After separation of the product from the reaction mixture, the aqueous layer consisting palladium was analysed by transmission electron microscope (TEM) analysis. TEM micrograph analysis (Fig. 2a, b) and size distribution curve confirmed the presence of PdNPs of 5.9 nm average size with spherical morphology. The –OH functionality of saponin is responsible for the reduction of Pd(II) to Pd(0), thus biosurfactant was found to act as a reductant and stabilizer during in situ formation of Pd NPs.

TEM micrograph (a and b) and size distribution curve (c) of palladium nanoparticles
After the initial success with palladium source, we next targeted to analyse role of base and effect of temperature on model reaction. Optimization was initially performed in a extracted biosurfactant as a solvent with 3 mol% of PdCl2 catalyst under base-free conditions at 100 °C under aerobic conditions. Unfortunately, the reaction did not furnish desired product (Table 2, entry 1). Therefore, we screened a variety of bases for the said reaction. Different inorganic and organic bases were screened. Poor conversions were noted (TLC monitoring), when inorganic bases such as NaOH, K3PO4, and K2CO3 were used (Table 2, entries 2–4). Organic bases like DBU and DABCO furnished improved yield of 70 and 75% product, respectively (Table 2, entries 5, 6). The excellent result was obtained in the presence of 2 mmol of triethyl amine (TEA) and 3 mol% of PdCl2 (Table 2, entry 7). No improvement in yield was observed when excess of TEA was used (Table 2, entries 9, 10). Effect of change in reaction temperature was also examined at room temperature and 75 °C (Table 2, entries 11, 12). However, no significant yield was obtained. The reaction was also performed in water which resulted only 42% yield (Table 2, entry 13).
After completion of the reaction, the product was isolated from reaction mixture by extracting with ethyl acetate and purified by column chromatography (stationary phase: silica mesh size 60–120 and mobile phase: ethyl acetate + petroleum ether). After purifying the product, it was characterized by different analytical techniques such as IR, 1H NMR, 13C NMR, and GCMS.
It is worthy to mention that in 1H NMR spectrum the characteristic two vinylic hydrogens exhibited 3JH–H value of 16 Hz and depicted excellent selectivity towards the formation of E-isomer. The vinylic hydrogens Hb and Ha are observed at δ = 6.16–6.20 and 6.66–6.70 ppm, respectively (Fig. 3). The N–CH2 (Hc and Hd) protons appeared as a multiplet at 4.52–4.54 ppm because of vicinal coupling with Hb and geminal coupling with each other. Rest of the all aromatic protons appeared between 6.95 and 7.38 ppm. In the 13C NMR spectrum, we observed a characteristic carbonyl carbon at δ = 183 ppm while the amide carbonyl appeared at 157 ppm. The methylene carbon (–CH2) exhibited signal at 42 ppm. All aromatic carbons including vinylic carbons appeared between 110 and 150 ppm. These spectroscopic data confirmed the formation of desired product.

Structure of 1-cinnamylindoline-2,3-dione (3a)
To extend the generality of this method, the Mizoroki–Heck coupling of various aryl halides with allyl isatin was also studied. It is noteworthy that this catalytic system was effective for the coupling with both activated and deactivated aryl iodides, bromides, and chlorides and allyl isatins (Table 3).
Allyl isatin with no substituent furnished greater yield in short reaction time with iodobenzene whereas chlorobenzene furnished slightly lower yield (Table 3, entries 3a, 3o). We checked reactivity of allyl isatin with aryl halide possessing both electron-withdrawing and -donating substituent. It was observed that reaction proceeds smoothly (Table 3, entries 3b, 3c). 5-NO2-allyl isatin reacts effectively with activated aryl iodide and gave sluggish results with deactivated aryl bromide and chloride (Table 3, entries 3d, 3e). We next checked the reactivity of 5-OMe-allyl isatin with both activated aryl iodides and deactivated aryl bromide. It is worthy to note that, reaction progressed easily with good yields (Table 3, entries 3f–3i). Subsequently, we examined the reactivity of halogen containing allyl isatins (5-F, 5-Cl, and 5-Br) with both aryl halides possessing electron-donating and electron-withdrawing groups (Table 3, entries 3j–3n).
Finally, the reusability of catalyst was investigated by performing model reaction under optimized reaction conditions. After completion of the reaction, the product was extracted by adding ethyl acetate to the reaction mixture. The aqueous layer consisting of biosurfactant and the palladium catalyst was washed with ethyl acetate and reused for subsequent runs. The results revealed that catalyst can be reused at least for 4 times resulted 86, 85, 83, 79% yields, respectively (Fig. 4).

Reusability of catalytic system
The general mechanism for Mizoroki–Heck cross-coupling is depicted in Fig. 5. The mechanism involves the oxidative addition of aryl halide, migratory insertion of olefin and β-hydride elimination to form the product. The regeneration of palladium(0) catalyst takes place using a base in the reductive elimination step.

Plausible mechanism for Mizoroki–Heck cross-coupling
Conclusion
In conclusion, an aqueous extract of the Acacia auriculiformis pods was utilized for Mizoroki–Heck coupling. The in situ generated PdNPs was employed as a highly efficient catalyst for Mizoroki–Heck coupling of various aryl iodides/bromides/chlorides with various allylisatins with good to excellent yields at 100 °C in 1.5–8 h. The reported method is superior with the added benefits of green chemistry, no use of external ligand, use of aqueous medium as well as natural resources and a green and economical method for the synthesis of PdNPs.
Experimental
Various substituted isatins (Sigma–Aldrich) and aryl halides (Spectrochem) were used as received. Melting points recorded were determined in open capillaries. IR spectra were recorded on ATR-IR-4600 spectrometer. NMR spectra were recorded on Bruker AV-400 spectrometer (400 MHz 1H NMR and 100 MHz for 13C NMR) in DMSO-d6 employing TMS as internal standard and δ values are expressed in ppm. GC–MS spectra were recorded on Shimadzu QP 2010 GCMS mass spectrometer.
General procedure for Mizoroki–Heck coupling reaction
In a 25 cm3 round bottom flask allyl isatin 1 (1.1 mmol), aryl halide 2 (1 mmol), triethyl amine (2 mmol), PdCl2 (3 mol %), and 5 cm3 aqueous extract of A. Auriculiformis were added. The mixture was stirred vigorously at 100 °C for the time mentioned in Table 3. After cooling to room temperature, the desired product was isolated from surfactant by extraction with ethyl acetate. The combined organic phases were dried over NaSO4 and evaporated using a rotary evaporator to give a crude product, which was purified by column chromatography on silica gel using pet ether/ethyl acetate (90:10 v/v) as the eluent to afford the corresponding derivative of 3.
(E)-4-[3-(2,3-Dioxoindolin-1-yl)prop-1-en-1-yl]benzonitrile (3b, C18H12N2O2)
Orange solid; m.p.: 196–198 °C; IR: \(\overline{\nu }\) = 3190, 3110, 2979, 2905, 2803, 2207, 1642, 1602, 1510, 1463, 1402, 1340, 1259, 1175, 1082, 908, 848, 794, 747 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 4.38–4.40 (2H, m), 5.83–5.89 (1H, m), 6.31–6.35 (1H, d, J = 16 Hz), 6.84–6.82 (2H, d, J = 8 Hz), 7.13–7.17 (1H, m), 7.49–7.56 (1H, m), 7.66–7.76 (4H, m) ppm; 13C NMR (CDCl3, 100 MHz): δ = 42.64, 112.61, 116.72, 118.98, 127.06, 128.18, 128.50, 128.62, 130.00, 132.08, 132.56, 132.67, 133.42, 140.52, 149.53, 151.02, 163.97, 181.75 ppm; GC–MS: m/z calcd. for C18H12N2O2 (M+) 288.31, observed 288.
(E)-1-[3-(4-Methoxyphenyl)allyl]indoline-2,3-dione (3c, C18H15NO3)
Red solid; m.p.: 200–204 °C; IR: \(\overline{\nu }\) = 3088, 2934, 2837, 2762, 1714, 1609, 1475, 1431, 1332, 1275, 1233, 1186, 1122, 997, 922, 874, 818, 764, 677, 618 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 3.81 (3H, s), 4.36–4.38 (2H, m), 5.82–5.92 (1H, m), 6.38–6.42 (1H, d, J = 15.2 Hz), 6.80–6.82 (2H, d, J = 8 Hz), 7.38–7.40 (2H, d, J = 8 Hz), 7.45–7.46 (1H, m), 7.49–7.51 (1H, m), 7.67- 7.71 (2H, m) ppm; 13C NMR (CDCl3, 100 MHz): δ = 42.64, 54.40, 112.60, 116.71, 118.98, 124.51, 126.06, 127.43, 128.19, 128.50, 128.48, 128.61, 130.01, 131.99, 132.18, 140.51, 160.28, 163.44, 179.84 ppm; GC–MS: m/z calcd. for C18H15NO3 (M+) 293.32, observed 294 ([M + 1]+).
(E)-1-[3-(4-Methoxyphenyl)allyl]-5-nitroindoline-2,3-dione (3e, C18H14N2O5)
Yellow solid; m.p.: 258–260 °C; IR: \(\overline{\nu }\) = 3161, 3073, 1713, 1603, 1525, 1505, 1428, 1379, 1332, 1288, 1196, 1122, 1023, 940, 902, 862, 810, 766, 705, 615, 592 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 3.88 (3H, s), 4.17–4.22 (2H, m), 5.37–5.47 (1H, m), 6.65–6.69 (1H, d, J = 16 Hz), 7.50–7.52 (2H, d, J = 7.6 Hz), 7.56–7.58 (2H, d, J = 7.2 Hz), 7.67–7.72 (3H, m) ppm; 13C NMR (CDCl3, 100 MHz): δ = 48.36, 56.81, 114.42, 115.32, 118.12, 123.48, 124.31, 128.47, 130.46, 132.08, 137.03, 145.17, 154.96, 159.87, 162.52, 180.03 ppm; GC–MS: m/z calcd. for C18H14N2O5 (M+) 338.32, observed 338.
(E)-1-[3-(4-Methoxyphenyl)allyl]-5-methylindoline-2,3-dione (3 h, C19H17NO3)
Brown solid; m.p.: 236–238 °C; IR: \(\overline{\nu }\) = 3049, 1725, 1610, 1546, 1484, 1432, 1330, 1277, 1187, 1116, 1038, 989, 939, 827, 765, 718, 686, 622 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 2.36 (3H, s), 3.81 (3H, s), 4.36–4.38 (2H, m), 5.82–5.90 (1H, m), 6.80–6.84 (1H, d, J = 16 Hz), 7.38–7.40 (2H, d, J = 8 Hz), 7.49–7.52 (1H, m), 7.56–7.58 (2H, d, J = 8.8 Hz), 7.67–7.73 (2H, m) ppm; 13C NMR (CDCl3, 100 MHz): δ = 20.67, 42.51, 55.03, 110.69, 118.52, 124.39, 125.73, 128.24, 128.58, 130.52, 132.18, 133.64, 138.67, 145.49, 158.41, 162.27, 179.72 ppm; GC–MS: m/z calcd. for C19H17NO3 (M+) 307.12, observed 308 ([M + 1]+).
(E)-4-[3-(5-Methoxy-2,3-dioxoindolin-1-yl)prop-1-en-1-yl]benzonitrile (3i, C19H14N2O3)
Dark brown solid; m.p.: > 300 °C; IR: \(\overline{\nu }\) = 3073, 2924, 2210, 1730, 1602, 1475, 1432, 1331, 1171, 1112, 921, 814, 707, 601 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 3.78 (3H, s), 4.54–4.59 (2H, m), 5.82–5.89 (1H, m), 6.81–6.84 (1H, d, J = 15.2 Hz), 7.49–7.51 (2H, d, J = 7.6 Hz), 7.54–7.58 (1H, m), 7.69–7.76 (3H, m), 8.15–8.17 (1H, d, J = 8 Hz) ppm; 13C NMR (CDCl3, 100 MHz): δ = 42.64, 55.76, 112.61, 116.72, 118.98, 124.87, 127.06, 128.18, 128.62, 130.00, 132.08, 132.67, 133.42, 140.52, 160.05, 161.95, 180.56 ppm; GC–MS: m/z calcd. for C19H14N2O3 (M+) 318.33, observed 318.
(E)-5-Bromo-1-[3-(4-methoxyphenyl)allyl]indoline-2,3-dione (3 k, C18H14BrNO3)
Yellowish solid; m.p.: 294–298 °C; IR: \(\overline{\nu }\) = 3185, 3072, 2972, 2923, 1730, 1602, 1471, 1331, 1261, 1172, 1111, 992, 925, 811, 710, 606 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 3.82 (3H, s), 4.38–4.40 (2H, m), 5.81–5.89 (1H, m), 6.81–6.84 (1H, d, J = 15.2 Hz), 7.49–7.51 (2H, d, J = 8 Hz), 7.56–7.58 (2H, d, J = 7.6 Hz), 7.69–7.72 (1H, m), 7.74–7.76 (1H, d, J = 8 Hz), 8.06–8.06 (1H, d, J = 2.4 Hz) ppm; 13C NMR (CDCl3, 100 MHz): δ = 48.62, 55.18, 114.30, 117.37, 118.98, 124.51, 127.43, 128.19, 128.61, 130.01, 132.02, 132.18, 140.51, 145.46, 147.06, 158.27, 160.42, 180.78 ppm; GC–MS: m/z calcd. for C18H14 BrNO3 (M+) 372.22, observed 372.
(E)-4-[3-(5-Bromo-2,3-dioxoindolin-1-yl)prop-1-en-1-yl]benzonitrile (3 l, C18H11BrN2O2)
Brown solid; m.p.: > 300 °C; IR: \(\overline{\nu }\) = 3185, 3095, 3036, 2929, 2226, 1730, 1597, 1464, 1432, 1326, 1261, 1176, 1112, 990, 942, 824, 707, 659 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 4.38–4.40 (2H, m), 5.82–5.89 (1H, m), 6.81–6.84 (1H, d, J = 15.2 Hz), 7.49–7.51 (2H, d, J = 7.2 Hz), 7.56–7.56 (1H, d, J = 2.8 Hz), 7.69–7.72 (1H, m), 7.75–7.77 (2H, d, J = 8 Hz), 8.13–8.13 (1H, d, J = 2.4 Hz) ppm; 13C NMR (CDCl3, 100 MHz): δ = 48.29, 112.61, 116.72, 118.98, 119.49, 119.74, 124.36, 128.18, 128.50, 132.08, 132.35, 132.67, 133.42, 140.52, 145.04, 147.35, 163.97, 179.67 ppm; GC–MS: m/z calcd. for C18H11 BrN2O2 (M+) 367.20, observed 367.
References
Whitcombe NJ, HiiK KM, Gibson SE (2001) Tetrahedron 57:7449
Farina V (2004) Adv Synth Catal 346:1553
Hassan J, Sevignon M, Gozzi C, Schulz E, Lemaire M (2002) Chem Rev 102:1359
Albaneze Walker J, Murry JA, Soheili A, Ceglia S, Springfield SA, Bazaral C, Dormer PG, Hughes DL (2005) Tetrahedron 61:6330
Kertesz M, Choi CH, Yang S (2005) Chem Rev 105:3448
Simon MO, Li CJ (2012) Chem Soc Rev 41:1415
Anastas P, Eghbali N (2010) Chem Soc Rev 39:301
Carril M, Martin RS, Dominguez E (2008) Chem Soc Rev 37:639
Shaughnessy KH (2006) Eur J Org Chem 2006:1817
DeSimone JM (2002) Science 297:799
Savage PE (1999) Chem Rev 99:603
Gawande MB, Bonifacio VD, Luque R, Branco PS, Varma RS (2013) Chem Soc Rev 42:5522
Nasrollahzadeh M, Sajadi SM, Maham M (2015) J Catal A 396:297
LaSorella G, Strukul G, Scarso A (2015) Green Chem 17:644
Khazaei A, Rahmati S, Hekmatian Z, Saeednia S (2013) J Mol Catal A Chem 372:160
Puthiaraja P, Pitchumani K (2014) Green Chem 16:4223
Phillip J, Anna T, Victor S, Valerie JP (2021) Adv Colloid Interface Sci 288:102340
Chahdoura F, Favier I, Pradel C, Mallet-Ladeira S, Gómez M (2015) Catal Commun 63:47
Leonie AS, Maria GCS, Italo JBD, Karen GOB, Beatriz GR, Ivison AS, Matthew ST, Ibrahim MB (2022) Biochem Eng J 181:108377
Markandea AR, Patelb D, Varjanic S (2021) Bioresour Technol 330:124963
Patil SP, Jadhav SN, Rode CV, Shejwal RV, Kumbhar AS (2020) Transit Met Chem 45:403
Han K, Zhou Y, Liu F, Guo Q, Wang P, Yang Y, Song B, Liu W, Yao Q, Teng Y, Yu P (2014) Bioorg Med Chem Lett 24:591
Tarek AF, Bin-Jubair FAS (2010) Int J Res Pharm Sci 1:113
Matheus ME, Violante FA, Garden SJ, Pinto AC, Fernandes PD (2007) Eur J Pharmacol 556:200
Chohan ZH, Pervez H, Rauf A, Khan KM, Supuran CT (2004) J Enzyme Inhib Med Chem 19:417
Kilpin KJ, Henderson W, Nicholson BK (2007) Polyhedron 26:204
Verma M, Pandeya SN, Singh KN, Stables JP (2004) Acta Pharm 54:49
Rasmussen HB, MacLeod JK (1997) J Nat Prod 60:1152
Khokhar S, Feng Y, Campitelli MR, Quinn RJ, Hooper JNA, Ekins MG, Davis RA (2013) J Nat Prod 76:2100
Hughes CC, Fenical W (2010) J Am Chem Soc 132:2528
Asati N, Yadava RN (2014) Int J Pharm Res Bio-Sci 3:341
Mandal P, Sinha Babu SP, Mandal NC (2005) Fitoterapia 76:462
Shmidt MS, Reverdito AM, Kremenchuzky L, Perillo IA, Blanco MM (2008) Molecules 13:831
Shrestha R, Lee GJ, Lee YR (2016) RSC Adv 6:63782
Sele AM, Bremner JB, Willis AC, Haritakun R, Griffith R, Keller PA (2015) Tetrahedron 43:8357
Jha M, Shelke GM, Kumar A (2014) Eur J Org Chem 16:3334
Vaidya GN, Nagpure M, Kumar D (2021) ACS Sustain Chem Eng 9:1846
Jha M, Shelke GM, Kumar A (2014) Eur J Org Chem 2014:3334
Trost BM, Kalnmals CA, Ramakrishnan D, Ryan MC, Smaha RW, Parkin S (2020) Org Lett 22:2584
Acknowledgements
One of the authors, PMM is grateful to the Council of Scientific and Industrial Research (CSIR), New Delhi, Government of India, for the award of the Junior Research Fellowship (File no. 09/ 816(0040)/2017-EMR-I).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Patil, M.V., Mhaldar, P.M. & Pore, D.M. Mizoroki–Heck coupling: a novel approach for synthesis of (E)-1-(3-argioallyl)indoline-2,3-dione. Monatsh Chem 153, 1243–1250 (2022). https://doi.org/10.1007/s00706-022-02978-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-022-02978-w