
Abstract
A new type of six-armed π-conjugated structure was built up from benzene as the central core and electron-rich triazine rings as the peripheral group. The hexasubstituted product was liquid and did not exhibit any liquid crystal properties while the equimolar mixtures in 1:1 ratio of the hexasubstituted compound and 4-(dodecyloxy)benzoic acid resulted in an organic salt exhibiting a columnar mesophase characterized with a dendritic growing texture. Examination of the relationship between structure and properties and the presence of the number of carbon atoms in the alkyl chain suggested that, the higher increase led to the phase transition at low temperature to form a columnar mesophase as an ionic liquid crystal composed of cations and anions. These are often stabilized by resonance with strongly delocalized charges. The liquid crystalline properties of the organic salt were investigated by differential scanning calorimetry (DSC) and polarizing optical microscopy (POM).
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Heterocyclic compounds containing nitrogen have been found in a wide spectrum of scientific research and industrial applications. Triazine [1] with alternating nitrogen and carbon atoms on a six-membered ring is a basic building block of many chemical materials [2,3,4], and commonly present in natural and synthetic compounds with significant bioactivity profiles [5,6,7] as antitumor agents [8], CRF receptor antagonists [9], antimicrobial and anti-inflammatory agents [10], antitumor [11] and anti-AIDS agents [12].
Triazine-based materials are rich in nitrogen and show higher thermal stability as compared to carbon-based materials [13, 14]. Some derivatives of triazines such as melamine, cyanuric chloride, and cyanuric acid are produced on industrial scale. In addition, inexpensive production cost makes triazine-based donor–π–acceptor structures suitable candidates for developing organic dyes that do not contain metals. Such structures are excellent photo stimulants and have a high molar absorption coefficient. Such materials are easy to synthesize [15] and present good electrical, optical properties [16,17,18,19,20]. In search of developing novel molecular architectures with unique structural, optical and electronic properties significant attention has been devoted to 1,3,5-triazine-based materials, due to the favorable LUMO levels of triazine moiety [21].
Cyanuric chloride and cyanuric fluoride can be subjected to sequences of nucleophilic substitution reactions in a controlled manner to prepare derivatives of triazine in aliphatic, aromatic, alkynic nature. It is possible to connect three different C-nucleophiles, N-nucleophiles, S-nucleophiles and/or O-nucleophiles in a desired order to the triazine unit. Many examples of this have been reported in the literature [22,23,24,25,26].
The basic principle in the design and production of synthetic materials is to bring together the building blocks possessing certain principles such as hydrogen bonding, van der Waals, π–π stacking, etc. to introduce desired functionalities to the final structures. In this context, triazine-based structures have important places in the literature and play key roles in imparting various functionalities to the molecular architectures [27,28,29].
Star-shaped macromolecules possessing well-defined and monodisperse architecture have found potential application as materials of OLED and electro-optic devices [30, 31]. Such macromolecules with rigid arms directing radially symmetrical away from the center have void regions where appropriate guest molecules can be hosted via electrostatic or hydrogen bonding motif resulting materials with unique properties [19, 20].
In an ongoing investigation to develop π-conjugated functional materials containing triazine units, which are strategically placed in the stare-shaped macromolecular, we have developed tri-armed mesogens of 2,4,6-tris[[4,6-bis(dodecyloxy)-1,3,5-triazine-2-yl]ethynyl]-1,3,5-triazine [19] and 2,4,6-tris[[4,6-bis((S)-citronellyloxy)-1,3,5-triazine-2-yl]ethynyl]-1,3,5-triazine [20] in which alkoxy-substituted triazines were connected to the central core with rigid acetylenic bridges, which demonstrated Smectic C (SmC) mesophase at low temperatures.
In this work, we present a six-armed macromolecule 1 with C3 symmetrical arrangements of the rigid side group on the central aromatic core. Chiral dialkoxy substituted three triazines were connected to the central benzene at 1,3,5-positions via rigid bridges. The remaining alternating position of the central benzene possessed three acetylenic thiophene units. We also prepared organic salt of 2 by mixing macromolecule 1 with 4-(dodecyloxy)benzoic acid (4-DBA) (Scheme 1) [20], which shows enantiotropic mesophases, ‘inequimolar’ ratio under ultrasonic conditions.

Result and discussion
The synthesis of the targeted macromolecule 1 was accomplished by a cross coupling reaction between the key intermediates 5 and 9 in the presence of Pd(PPh3)4 (Scheme 2). Intermediate 5 was prepared in two steps starting from 2,4,6-trichloro-1,3,5-triazine (3), which was reacted with two equivalents of (S)-(-)-π-citronellol followed by treating the resulting compound 4 with trimethylsilyl acetylene in the presence of palladium catalyst. The ethynylthiophene 8 was prepared as outlined in Scheme 2. Intermediate 9 was prepared by a cross coupling reaction between 1,3,5-trichloro-2,4,6-triiodobenzene, which was prepared with a reported procedure [32] from 1,3,5-trichlorobenzene, and 2-ethynylthiophene (8) in the presence of tetrakis(triphenylphosphine)palladium catalyst.

Six-armed shape persistent material 1 was found to be colourless oily substance at room temperature and converted to an organic salt by mixing with 4-(dodecyloxy)benzoic acid in a 1:1 ration in dry THF and sonicating the mixture at room temperature for 15 min. Macromolecule 1 and organic salt 2 were characterized by 1H NMR, 13C NMR, FT-IR, and QTOF analyses (Schemes 3, 4).


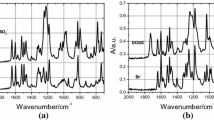
The formation of organic salt between six-armed π-conjugated system and the mesogenic carboxyl group took place by ionic interaction and mainly studied by FT-IR (Fig. 1). Asymmetric stretching of the sharp carbonyl peak of the carboxylic acid 4-DBA at 1680 cm−1 shifted to 1570 cm−1.

FT-IR spectra of compound 10, the organic salt 2 and benzoic acid 4-DBA
The formation of organic salt 2 was also followed by NMR spectroscopy. The signals belong to the aromatic protons of the alkoxy benzoate unit shifted from 8.05, 6.95 ppm to 7.85, 6.70 ppm, respectively, due to changes in electron density of the aromatic ring. Similarly, the signals of oxymethylene protons of 4-DBA shift to higher field 3.8 ppm as compared with the signals of pure 4-DBA at 4.05 ppm (Fig. 2). Additionally, the signals of thiophene protons in pure macromolecule 1 at 7.32, 6.95 ppm were shifted after the formation of the salt with alkoxybenzoic acid organic salt 2 to 7.15, 6.82 ppm. The changes in chemical shift values are considered as a result of the complex formation between macromolecule 1 and 4-(dodecyloxy)benzoic acid. However, the signals of oxymethylene protons which belong to compound 4-DBA experienced a lesser shift from 4.25 to 4.13 ppm due to the fact that their electronic environment did not change much. Furthermore, the 13C NMR spectra show that the carbonyl carbon shifted to a higher field 166.8 ppm as compared with the signals of pure 4-DBA at 171.6 ppm. On the other hand, slight shifting was observed of the triazine ring carbon from 171.5 to 170.8 ppm (Fig. 3).

The comparison of 1H NMR spectra (in CDCl3) of macromolecule 1, organic salt 2 and benzoic acid 4-DBA

13C NMR spectra in CDCl3 of macromolecule 1, organic salt 2 and benzoic acid 4-DBA
The mesomorphic properties of the six-armed organic salt 2 were investigated by differential scanning calorimeter (DSC) and using polarized optical microscope (POM). Compound 4-DBA with a n-dodecyloxy terminal chain which were previously reported [19, 20], shows enantiotropic tilted smectic phase (SmC) as well as nematic (N) phase. The phase transitions of the corresponding molecules are given in Table 1.
As shown in Fig. 4a, the differential thermograms of 4-DBA show three endotherms for a phase transition sequence of crystal (Cr)—smectic C (SmC)—nematic (N)—isotropic phase (iso). On cooling from isotropic phase, the same behaviour of reverse transitions was observed. Additionally, a calorimetric peak corresponding to Cr2–Cr1 transition at 65.86 °C was detected in cooling DSC thermogram.

DSC thermogram of compounds 4-DBA (a) and OS 2 (b) on 1st heating and cooling (10 °C min−1)
On heating DSC, the organic salt 2 shows transition from crystal to isotropic state at 86.13 °C. On cooling from isotropic, a dendritic growing texture started to appear at 72 °C and the texture was preserved until 51 °C under POM. This observation was in agreement with the two exotherms at 65.24 °C and 54.84 °C, respectively, corresponding to a phase transition sequence of isotropic phase (iso)-liquid crystalline (LC) mesophase-crystal (Cr) on DSC cooling curve (see Fig. 4b). The liquid crystalline mesophase was assigned as Col mesophase which the columns are stacked in either rectangular or hexagonal 2D lattices. Typical textures of organic salt 2 observed on cooling are given in Fig. 5.

Optical textures of organic salt 2 as observed between crossed polarizers in ordinary glass plates on cooling; a dendritic texture of Col phase at 62 °C and b 57 °C; c crystalline phase at 42 °C
Herein, we would like to mention the mesomorphic properties of the organic salt 12 (see Fig. 6) prepared from the tri-armed macromolecule with chiral citronellyloxy side groups and 4-DBA by mixing 1:1 ratio [20]. The comparison of organic salts 2 and 12 with two chiral citronellyloxy side groups attached to three 1,3,5-triazine units at the periphery shows that the structural change on macromolecule of organic salt from the tri-armed to six-armed macromolecule based on central benzene core with three acetylenic thiophene units lead to the formation of columnar mesophase which was traditionally observed for disc-shaped molecules. As a result of the decreased noncovalent intermolecular interaction due to steric hindrance at the periphery, the thermal range of columnar mesophase of organic salt 2 is about 10 °C whereas SmC phase range of organic salt 12 is significantly much higher as well as the mesophase is more stable. One point we noted here for organic salt 2 that the introducing of three acetylenic thiophene units which leads to six-armed structure gives rise to a drastic change on melting and clearing temperatures as well as inducing columnar phase at lower temperatures.

The macromolecule structure of organic salt 2,4,6-tris[(4,6-bis(S)-citronellyloxy)ethynyl]-1,3,5-triazine (12)
Conclusion
In summary, we have successfully synthesized and characterized a six-armed macromolecule, which is composed of a benzene unit as the central core, three thiophene units and three 1,3,5-triazine rings carrying two (S)-citronellyloxy branched chains which are connected to the benzene core by acetylene bridges. The presence of three triazines substituted with chiral citronellyloxy and three thiophene rings on the central molecular was unable to induce mesomorphism of triazines derivatives. Therefore, the final product was mixed with 4-DBA through hydrogen-bonded interaction that may increase the ability to form liquid crystal material. The organic salt of the six-armed macromolecule with 4-(dodecyloxy)benzoic acid prepared by mixing 1:1 ratio in THF exhibited a monotropic columnar mesophase at lower temperatures in a narrow mesomorphic range.
Experimental
The following reagents and solvents were acquired without additional purification from Merck: cyanuric chloride, ethynylbenzene, 1,3,5-trichlorobenzene, 2-iodo-thiophene, 2-methylbut-3-yn-2-ol, copper iodide, potassium carbonate, tetrakis(triphenylphosphine)palladium, tetrahydrofuran, and dioxane. Thin-layer chromatography was carried out on aluminum plates (20 × 20 cm) covered with silica gel 60 F254 (Merck), whereas column chromatography was carried out on Merck's silica gel 60 (0.063–0.200 mm).
High-resolution mass spectrometry (HRMS), FT-IR (Shimadzu Prestige-21, KBr discs), 1H NMR (500 MHz) and 13C NMR (125 MHz) (CDCl3, standard internal TMS) spectrometers were used to determine the structure of compounds. Differential scanning calorimetry (DSC) and polarized optical microscope (POM) was used to evaluate the organic salts’ liquid crystalline state.
The preparation of compound 4, 5, 7, 8, and 9 were reported in the references [20, 26, 31], respectively.
2,2′,2″-[(2,4,6-Trichlorobenzene-1,3,5-triyl)-tris(ethyne-2,1-diyl)]trithiophene (10, C24H9Cl3S3)
A mixture of 0.5 g 1,3,5-trichloro-2,4,6-triiodobenzene (9, 0.89 mmol), 0.48 g 2-ethynylthiophene (8, 2.69 mmol), 0.1 g Pd(PPh3)4 (0.089 mmol), 0.03 g CuI (0.178 mmol), and 0.39 g K2CO3 (2.84 mmol) were dissolved in 10 cm3 of dioxane then stirred at 75 °C for 6 h. under argon atmosphere. The solution was poured into a mixture of 20 cm3 ethyl acetate and 20 cm3 water. The organic layer was washed and dried over sodium sulfate. The solvent was removed under vacuum and the residue was purified by column chromatography with hexane/ethyl acetate (5% EtOAc) as an eluent to give pale yellow powder. Yield 0.37 g (84%); m.p.: 124–127 °C; 1H NMR (500 MHz, CDCl3): δ = 7.33 (m, 3H, Ar–H), 7.15 (m, 3H, Ar–H), 6.75 (dt, J = 6.8, 3.4 Hz, 3H, Ar–H) ppm; 13C NMR (126 MHz, CDCl3): δ = 145.7, 140.8, 136.9, 131.5, 128.9, 126.3, 100.4, 98.1 ppm; HRMS: m/z calcd for C24H9Cl3S3 (M+) 499.88, found 499.8921 (M+) and 500.8859 ([M + H]+).
6,6′,6″-[[2,4,6-Tris(thiophen-2-ylethynyl)benzene-1,3,5-triyl]-tris(ethyne-2,1-diyl)]tris[2,4-bis[(3,7-dimethyloct-6-en-1-yl)-oxy]-1,3,5-triazine] (1, C99H123N9O6S3)
Compound 10 (0.14 g, 0.28 mmol), 0.41 g 2,4-bis[(3,7-dimethyloct-6-en-1-yl)-oxy]-6-[(trimethylsilyl)ethynyl]-1,3,5-triazine (5, 0.84 mmol), 0.032 g Pd(PPh3)4 (0.028 mmol), 0.01 g CuI (0.056 mmol), and 0.136 g K2CO3 (0.98 mmol) were dissolved in 10 cm3 of dioxane then stirred at 80 °C for 16 h under argon atmosphere. The solution was poured into a mixture of 20 cm3 ethyl acetate and 20 cm3 water. The organic layer was washed and dried over sodium sulfate. The solvent was removed under vacuum and the residue was purified by column chromatography with hexane/ethyl acetate (5% EtOAc) as an eluent to give colourless oil. Yield 0.39 g (86%); 1H NMR (500 MHz, CDCl3): δ = 7.32 (m, 6H, Ar–H), 6.95 (m, 3H, Ar–H), 5.07 (s, 6H), 4.37 (t, 12H, OCH2), 1.97 (m, 12H, CH2), 1.75 (m, 18H, CH3), 1.63 (m, 8H, CH2), 1.25–0.95 (m, 30H, CH2), 0.75 (m, 9H, CH3) ppm; 13C NMR (126 MHz, CDCl3): δ = 171.5, 170.2, 134.4, 131.1, 128.9. 127.4, 127.1, 124.8, 124.6, 121.9, 101.2, 93.1, 77.8, 65.6, 41.6, 37.7, 37.5, 36.4, 36.3, 31.9, 30.2, 29.9, 29.4, 25.7, 25.5, 22.7, 19.5, 17.6, 14.1, 13.5, 13.1 ppm; FT-IR: \(\overline{\nu }\) = 2922, 2853, 1734, 1568, 1622, 1522, 1503, 1458, 1430, 1412, 1382 cm−1; HRMS: m/z calcd for C99H123N9O6S3 (M+) 1631.29, found 816.4493 ([M + 2H]2+).
Organic salt 2 (C118H153N9O9S3)
4-DBA mesogenic unit 10 with a carboxyl group was added into a solution of compound 1 in 10 cm3 of dry THF with one-to-one ratio. The resulting solution was sonicated for 15 min until observing a transparent solution. Then, the solvent was removed in vacuum. 1H NMR (500 MHz, CDCl3): δ = 7.85 (m, 1H, Ar–H), 7.15 (m, 6H, Ar–H), 6.82 (m, 3H, Ar–H), 6.75 (m, 1H, Ar–H), 4.92 (s, 6H), 4.17 (t, 12H, OCH2), 3.82 (t, 2H, OCH2), 1.85 (m, 18H, CH3), 1.65–1.32 (m, 24H, CH2), 1.24–0.85 (m, 60H, CH2), 0.75 (m, 18H, CH3) ppm; 13C NMR (126 MHz, CDCl3): δ = 171.7, 170.6, 165.6, 163.6, 134.4, 132.2, 131.1, 128.9, 127.2, 124.7, 121.9, 121.2, 114.1, 68.2, 65.6, 41.5, 37.2, 37.1, 35.6, 35.8, 35.5, 31.9, 29.7, 29.6, 29.5, 29.4, 29.3, 29.2, 29.1, 25.9, 25.7, 25.4, 22.6, 19.4, 17.6, 14.1 ppm; FT-IR: \(\overline{\nu }\) = 2922, 2850, 1738, 1570, 1521, 1431 cm−1; HRMS: m/z calcd for C118H153N9O9S3 (M+) 1937.73, found 387.4189 (M5+) and 426.3900 ([M + 5 K]5+).
References
Giacomelli G, Porcheddu A, Luca LD (2004) Curr Org Chem 15:1497
Shahbaz M, Urano S, LeBreton PR, Rossman MA, Hosmane RS, Leonard NJ (1984) J Am Chem Soc 106:2805
Li J, Tao L, Wang Y, Yao Y, Guo Q (2021) Front Chem 18:482
Kroke E, Schwarz M, Horath-Bordon E, Kroll P, Noll B, Norman AD (2002) New J Chem 26:508
Phillips RM (2016) Cancer Chemother Pharmacol 77:441
Ide M, Kato T, Nakata M, Saito K, Yoshida T, Awaya T, Heike T (2015) Brain Dev 37:825
Alazawi SK, Al-Jumaili MHA (2022) J Chem Res 7:1
Yan W, Zhao Y, He J (2018) Mol Med Rep 18:75
Abu-Aisheh NM, Mustafa SM, Mubarak SM, El-Abadelah MM, Voelter W (2012) Lett Org Chem 9:65
Hynes J Jr, Dyckman AJ, Lin S, Wrobleski ST, Wu H, Gillooly KM, Kanner SB, Lonial H, Loo D, McIntyre KW, Pitt S (2008) J Med Chem 10:4
Cascioferro S, Parrino B, Spanò V, Carbone A, Montalbano A, Barraja P, Diana P, Cirrincione G (2017) Eur J Med Chem 15:523
Singla P, Luxami V, Paul K (2015) Eur J Med Chem 102:39
Lim J, Kong SY, Yun YJ (2018) J Nanomater 25:2018
Wu C, Zhang H, Hu M, Shan G, Gao J, Liu J, Zhou X, Yang J (2020) Adv Electron Mater 6:2000253
Zhang H, Zang XF, Hong YP, Chen ZE (2021) Synth Met 280:116882
Li B, Lei Q, Qin T, Zhang X, Zhao D, Wang F, Li W, Zhang Z, Fan L (2021) CrystEngComm 23:8260
Zhang M, Ren S, Guo Q, Shen B (2021) Microporous Mesoporous Mater 326:111395
An ZF, Chen RF, Yin J, Xie GH, Shi HF, Tsuboi T, Huang W (2011) Chem Eur J 17:10871
Akkurt N, Al-Jumaili MH, Eran BB, Ocak H, Torun L (2019) Turk J Chem 43:1436
Akkurt N, Al-Jumaili MH, Ocak H, Cakar F, Torun L (2020) Turk J Chem 44:726
Bakr EA, Al-Jumaili MH (2020) Mol Cryst Liq Cryst 710:40
Sun C, Hudson ZM, Helander MG, Lu ZH, Wang S (2011) Organometallics 30:5552
Banerjee R, Brown DR, Weerapana E (2013) Synlett 24:1599
Steffensen MB, Hollink E, Kuschel F, Bauer M, Simanek EE (2006) Polym Chem 44:3411
Janairo JI, Sakaguchi T, Mine K, Kamada R, Sakaguchi K (2018) Protein Pept Lett 25:4
Al-Jumaili MH, Akkurt N, Torun L (2021) Monatsh Chem 152:551
Kouvetakis J, Grotjahn D, Becker P, Moore S, Dupon R (1994) Chem Mater 6:636
Couderc G, Hulliger J (2010) Chem Soc Rev 39:1545
Whitesides GM, Simanek EE, Mathias JP, Seto CT, Chin D, Mammen M, Gordon DM (1995) Acc Chem Res 28:37
Azhar U, Bashir MS, Babar M, Arif M, Hassan A, Riaz A, Mujahid R, Sagir M, Suri SU, Show PL, Chang JS (2022) Chemosphere 302:134792
Al-Jumaili M, Hamed A, Akkurt N, Torun L (2020) Indones J Chem 20:705
Sonoda M, Inaba A, Itahashi K, Tobe Y (2001) Org Lett 26:2419
Acknowledgements
Authors thank to Professor Belkis Bilgin Eran for valuable discussions on POM studies. The authors acknowledge the funding from the following agents. The Scientific and Technological research Council of Turkey (TUBITAK) with the project no. 114Z722 and The Office of Scientific Research of Yildiz Technical University, project no. FCD-2021-3892.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Al-Jumaili, M.H.A., Ocak, H. & Torun, L. Hydrogen-bonded ionic liquid crystals based on multi-armed structure: synthesis and characterization. Monatsh Chem 153, 939–947 (2022). https://doi.org/10.1007/s00706-022-02969-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-022-02969-x