
Abstract
A series of N 1-(phosphonoalkyl)uracils was prepared in a two-step reaction sequence from ω-aminoalkylphosphonates and (E)-3-ethoxyacryloyl isocyanate followed by the uracil ring closure. Under standard conditions (NCS; NBS; I2/CAN) all N 1-(phosphonoalkyl)uracils were transformed into the respective 5-halogeno derivatives to be later benzoylated at N3. All compounds were evaluated in vitro for activity against a broad variety of DNA and RNA viruses. One compound was slightly active against human cytomegalovirus in HEL cell cultures (EC50 = 45 μM) while another showed weak activity against varicella-zoster virus (TK+ VZV strain OKA and TK− VZV strain 07-1) with EC50 = 43 and 53 µM, respectively. In addition, several compounds exhibited noticeable inhibitory effects on the proliferation of human cervical carcinoma cells (HeLa) at a concentration lower than 200 μM.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acyclic nucleoside and nucleotide analogues have been used in clinical practice for almost 50 years and have become cornerstones in the treatment of patients with viral infections [1–3]. Nucleoside analogues such as acyclovir, ganciclovir, and penciclovir as well as acyclic nucleoside phosphonates (ANPs), namely adefovir, cidofovir, and tenofovir, belong to the most widely used antiviral drugs (Fig. 1) [4–6]. The ribose moiety in natural nucleosides/nucleotides was replaced by linear or branched aliphatic units to obtain analogues with specific activity following activation by viral and/or cellular kinases. The antiviral activity of these compounds relies on enzyme inhibition or on incorporation as active metabolites into the viral nucleic acids resulting in inhibition of viral genome replication.

Acyclic nucleoside and nucleotide analogues
In the last decade, several new drugs, including nucleoside and nucleotide analogues, have been approved for the treatment of viral infections and in cancer chemotherapy. Many nucleoside/nucleotide analogues and their derivatives are currently under preclinical and clinical trials [7] which demonstrates how rapidly this area of research grows. However, some of these analogues have poor oral bioavailability and are associated with toxicity, limiting their application for treatment of cancer and viral infections. Moreover, a long-term treatment with antiviral drugs may result in lower sensitivity of the viruses to chemotherapeutics (drug resistance) [8, 9]. For this reason, a search for new compounds with higher antiviral and antitumor activities, good bioavailability, better solubility, and proper balance between efficacy and long-term toxicity still continues.
The known synthetic strategies to ANPs commonly apply alkylation of heterocyclic bases [10] while the construction of nucleobase skeletons from the appropriate terminal primary amines has been less frequently used [11–15]. Herein, we report on the efficient synthesis and biological evaluation of a new series of acyclic nucleotide analogues by application of the latter approach (Scheme 1).

Results and discussion
ω-Aminoalkylphosphonates can be prepared by several methods. Arbuzov reaction [16] of N-(ω-bromoalkyl)phthalimides with trialkyl phosphites seems to be most frequently applied. Alternatively, ω-aminoalkylphosphonates can be synthesized from nitriles [17] or from ω-azidophosphonates employing hydrogenolysis [18].
Diethyl aminoalkylphosphonates 2a–2d (aminomethyl-, 2-aminoethyl-, 3-aminopropyl-, and 4-aminobutylphosphonates), used in this study, are known and have already been described in the literature [19–27]. Thus, diethyl aminomethylphosphonate (2a) was prepared in total 90 % yield from N-(bromomethyl)phthalimide followed by the treatment with hydrazine [19–21] whereas ω-aminoalkylphosphonates 2b–2d were synthesized from the corresponding ω-azidoalkylphosphonates 1b–1d [28–32] by catalytic hydrogenation [23, 26]. However, several literature reports [33, 34] noticed the formation of significant amounts of symmetrical secondary amines as by-products during hydrogenolysis of azides depending on the azide concentration and the azide to catalyst ratio. When ω-azidophosphonates 1b–1d were subjected to hydrogenation in the presence of 10 % Pd–C, symmetrical secondary amines 3b–3d contaminated (11–33 %) the major phosphonates 2b–2d as judged from the 31P NMR spectra (Scheme 2). The mixtures of ω-aminoalkylphosphonates 2b–2d containing various amounts of symmetrical secondary amines 3b–3d were separated on silica gel columns.

Alkylation of nucleobases with substituted alkylphosphonates is generally recognized as a capricious process since dealkylation of phosphonate esters accompany the formation of a carbon-nucleobase bond and this was the primary reason to replace O,O-dimethyl and O,O-diethyl esters with O,O-diisopropyl phosphonates [35, 36]. Based on this experience and that of other groups, we concluded that the alternative approach to independently construct the pyrimidine ring through functionalization of ω-aminoalkylphosphonates is more feasible than the alkylation.
The N 1-(phosphonoalkyl)uracils 5a–5d were synthesized from ω-aminoalkylphosphonates 2b–2d in a two-step procedure which involved reaction with (E)-3-ethoxyacryloyl isocyanate [37] in situ generated from (E)-3-ethoxyacryloyl chloride [38–40] and silver cyanate followed by the uracil ring closure in the presence of 2 M H2SO4 (Scheme 3) [18, 39, 40].

The conversion of uracil phosphonates 5a–5d into 5-chlorouracils 6a–6d and 5-bromouracils 8a–8d was achieved by treatment with N-chlorosuccinimide (NCS) [41] and N-bromosuccinimide (NBS) [41] respectively, whereas 5-iodouracils 9a–9d were synthesized using iodine and cerium(IV) ammonium nitrate (CAN) [42] (Scheme 4). Although iodination and bromination of uracils 5a–5d proceeded regioselectively at C5, chlorination of uracil phosphonates 5c, 5d under the applied conditions provided mixtures of C5 and C6 regioisomers 7c, 7d in an approximately 8:2 ratio.

Moreover, based on the literature report [43] and on our previous observations regarding the effect of the benzoyl substituent at N-3 of a nucleobase on the antiviral activity [29, 44], the N 3-benzoyluracil analogues 10a–10d were synthesized from phosphonates 5a–5d as shown on Scheme 4.
Structures and purity of all synthesized compounds were established by 1H, 31P and 13C NMR and IR techniques as well as by elemental analysis.
Antiviral activity and cytotoxic evaluation
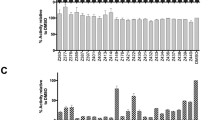
All the synthesized compounds 5a–5d, 6a–6d, and 8a–10d were evaluated for their antiviral activities against a wide variety of DNA and RNA viruses, using the following cell-based assays: (a) human embryonic lung (HEL) cells: herpes simplex virus-1 (KOS), herpes simplex virus-2 (G), herpes simplex virus-1 (TK− ACVr KOS), vaccinia virus and vesicular stomatitis virus, human cytomegalovirus (AD-169 strain and Davis strain), varicella-zoster virus (TK+ VZV strain OKA and TK− VZV strain 07-1); (b) CEM cell cultures: human immunodeficiency virus (HIV-1 and HIV-2); (c) Vero cell cultures: para-influenza-3 virus, reovirus-1, Sindbis virus, Coxsackie virus B4, Punta Toro virus; (d) HeLa cell cultures: vesicular stomatitis virus, Coxsackie virus B4 and respiratory syncytial virus (RSV); (e) Crandell-Rees Feline Kidney (CRFK) cell cultures: feline corona virus (FIPV) and feline herpes virus (FHV); (f) Madin Darby Canine Kidney (MDCK) cell cultures: influenza A virus H1N1 subtype (A/PR/8), influenza A virus H3N2 subtype (A/HK/7/87) and influenza B virus (B/HK/5/72). Ganciclovir, cidofovir, acyclovir, brivudin, (S)-9-(2,3-dihydroxypropyl)adenine [(S)-DHPA], Hippeastrum hybrid agglutinin (HHA), Urtica dioica agglutinin (UDA), dextran sulfate (molecular weight 5000, DS-5000), ribavirin, oseltamivir carboxylate, amantadine, and rimantadine were used as the reference compounds. The antiviral activity was expressed as the EC50: the compound concentration required to reduce virus-plaque formation (VZV) or virus-induced cytopathogenicity by 50 % (other viruses). It was found that the acyclic phosphonate analogues 10a and 10d containing the N 3-benzoyluracil group as a modified nucleobase exhibited noticeable activity. Thus, compound 10a showed activity against cytomegalovirus (AD-169 strain) in human embryonic lung (HEL) cells (EC50 = 45 µM), whereas compound 10d proved to be active against VZV (TK+ strain OKA and TK− strain 07-1) with EC50 = 43 and 53 µM, respectively. The antiviral activity of compound 10d against VZV TK− strain 07-1 appeared to be slightly better than the activity of acyclovir and brivudin used as reference compounds (EC50 = 160 and 103 µM, respectively). To rationalize the observed activity of the N 3-benzoyl derivatives 10a and 10d one can refer to their easier transport through cell membranes in comparison to the precursors 5a and 5d which appeared inactive. After entering into the cell debenzoylation would provide nucleoside analogues capable of Watson-Crick bas pairing. Our previous results [44] support this conclusion.
The cytotoxicity of the test compounds toward uninfected host cells was defined as the minimum cytotoxic concentration (MCC) that causes a microscopically detectable alteration of normal cell morphology. The 50 % cytotoxic concentration (CC50), causing a 50 % decrease in cell viability was determined using a colorimetric 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay system. None of the tested compounds affected cell morphology of HEL, HeLa, Vero, MDCK, and CRFK cells (MCC or CC50) at compound concentrations up to 100 μM.
Evaluation of cytostatic activity
The cytostatic activity of the tested compounds was defined as the 50 % inhibitory concentration (IC50), or compound concentration causing a 50 % decrease in cell proliferation. IC50 values were determined against murine leukemia L1210 cells, human lymphocyte CEM cells and human cervical carcinoma HeLa cells. Several synthesized compounds showed marginal inhibitory effect on the proliferation of HeLa cells at concentrations lower than 200 μM (Table 1).
Conclusion
A series of N 1-(phosphonoalkyl)uracils 5a–5d has been efficiently obtained from ω-aminoalkylphosphonates 2a–2d in a two-step process which involved a reaction with (E)-3-ethoxyacryloyl isocyanate followed by the uracil ring closure. The uracil phosphonates 5a–5d were successfully transformed into 5-chloro-, 5-bromo-, and 5-iodouracils as well as N 3-benzoyluracils. All synthesized compounds were tested in vitro for their antiviral activities against a broad variety of DNA and RNA viruses and compound 10a was slightly active against human cytomegalovirus in HEL cell cultures (EC50 = 45 μM) while 10d showed activity against both TK+ and TK-VZV strains with EC50 values of 43 µM (Oka strain) and of 53 µM (07-1 strain), which was two to threefold better than the potency of acyclovir and brivudin against 07-1 (EC50 = 160 and 103 µM, respectively).
Among them, several compounds exhibited a very poor inhibitory effect on the proliferation of human cervical carcinoma cells (HeLa) at a concentration lower than 200 μM. In contrast, none of the compounds affected the proliferation of CEM cells and L1210 cells up to a concentration of 250 µM.
Experimental
1H NMR was taken in CDCl3 on the following spectrometers: Varian Mercury-300 and Bruker Avance III (600 MHz) with TMS as an internal standard; chemical shifts δ in ppm with respect to TMS; coupling constants J in Hz. 13C NMR spectra were recorded for CDCl3 solution on the Varian Mercury-300 and Bruker Avance III (600 MHz) spectrometer at 75.5 and 151 MHz, respectively. 31P NMR spectra were taken in CDCl3 on the Varian Mercury-300 and Bruker Avance III (600 MHz) spectrometer at 121.5 and 243 MHz, respectively. IR spectra were measured on an Infinity MI-60 FT-IR spectrometer as KBr pellets or as films, and absorptions are given in cm−1. Melting points were determined on a Boetius apparatus. Elemental analyses were performed by Microanalytical Laboratory of this Faculty on Perkin Elmer PE 2400 CHNS analyzer and their results were found to be in good agreement (±0.3 %) with the calculated values. The following adsorbents were used: column chromatography, Merck silica gel 60 (70–230 mesh); analytical TLC, Merck TLC plastic sheets silica gel 60 F254.
Applied reagents such as silver cyanate (Alfa Aesar, catalogue number: 45411), CAN (Tokyo Chemical Industry (TCI) catalogue number: C1806) and inorganic reagents are commercially available and were used as received. Solvents were dried according to the literature methods. (E)-3-Ethoxyacryloyl chloride was obtained according to the known protocol [18]. Diethyl aminomethylphosphonate (2a) was prepared according to literature (colorless oil, yield 90 %) [19–24].
General procedure for the preparation of aminoalkylphosphonates 2b–2d
The ω-azidoalkylphosphonates 1b–1d (1.0 mmol) were dissolved in 10 cm3 ethanol and 10 % Pd–C (16 mg) was added. The suspension was stirred under hydrogen atmosphere (balloon) at room temperature for 20 h. The catalyst was filtered off through a layer of Celite and the solvent was evaporated to give a mixture of ω-aminoalkylphosphonates 2b–2d and 3b–3d (ca. 8:2). The crude products were chromatographed on silica gel columns with chloroform–methanol mixtures (50:1, 20:1, 10:1, v/v) to give pure ω-aminoalkylphosphonates 2b–2d and pure compounds 3b–3d.
Diethyl 2-aminoethylphosphonate (2b) [22–24]
Colorless oil; yield 80 %.
Bis[2-(O,O-diethylphosphoryl)ethyl]amine (3b) [45]
Pale yellow oil; yield 11 %.
Diethyl 3-aminopropylphosphonate (2c) [19, 24–26]
Yellowish oil; yield 80 %.
Bis[3-(O,O-diethylphosphoryl)propyl]amine (3c, C14H33NO6P2)
Pale yellow oil; yield 13 %; IR (film): \( \bar{\nu } \) = 3436, 2984, 2936, 2910, 1648, 1227, 1053, 1027, 964 cm−1; 1H NMR (600 MHz, CDCl3): δ = 4.14–4.04 (m, 8H, 4 × POCH 2CH3), 2.68 (t, J = 6.4 Hz, 4H, 2 × CH 2CH2CH2P), 2.03 (br s, 1H, NH), 1.82–1.73 (m, 8H, 2 × CH 2CH 2P), 1.32 (t, J = 7.1 Hz, 12H, 4 × POCH2CH 3) ppm; 13C NMR (151 MHz, CDCl3): δ = 61.5 (d, J = 6.4 Hz, POC), 49.7 (d, J = 16.7 Hz, CCCP), 23.4 (d, J = 142.1 Hz, CP), 22.8 (d, J = 4.7 Hz, CCP), 16.4 (d, J = 5.7 Hz, POCC) ppm; 31P NMR (243 MHz, CDCl3): δ = 32.20 ppm.
Diethyl 4-aminobutylphosphonate (2d) [19, 27]
Yellowish oil; yield 70 %.
Bis[4-(O,O-diethylphosphoryl)butyl]amine (3d, C16H36NO6P2)
Pale yellow oil; yield 23 %; IR (film): \( \bar{\nu } \) = 3441, 2982, 2936, 1642, 1230, 1052, 1026, 962 cm−1; 1H NMR (600 MHz, CDCl3): δ = 4.13–4.03 (m, 8H, 4 × POCH 2CH3), 2.59 (t, J = 7.1 Hz, 4H, 2 × CH 2CH2CH2CH2P), 2.00 (br s, 1H, NH), 1.76–1.70 (m, 4H, 2 × CH2P), 1.66–1.54 (m, 8H, 2 × CH 2CH 2CH2P), 1.31 (t, J = 7.1 Hz, 12H, 4 × POCH2CH 3) ppm; 13C NMR (151 MHz, CDCl3): δ = 61.4 (d, J = 6.5 Hz, POC), 49.3 (s, CCCCP), 30.9 (d, J = 16.0 Hz, CCCP), 25.6 (d, J = 140.9 Hz, CP), 20.3 (d, J = 5.2 Hz, CCP), 16.4 (d, J = 6.2 Hz, POCC) ppm; 31P NMR (243 MHz, CDCl3): δ = 32.07 ppm.
General procedure for the preparation of acryloylureas 4a–4d
A solution of 1.009 g 3-ethoxyacryloyl chloride (7.50 mmol) in 20 cm3 dry toluene was refluxed under an argon atmosphere with 2.173 g silver cyanate (14.50 mmol) previously dried in vacuo for 2 h with protection from light. After 45 min, the mixture was allowed to cool and then was transferred via cannula to a solution of the respective ω-aminoalkylphosphonate 2 (2.50 mmol) in 10 cm3 dry DMF at 0 °C. Solid AgCl was washed with dry toluene (2 × 8 cm3) to remove the residual 3-ethoxyacryloyl isocyanate which was added to the reaction flask. The reaction mixture was stirred at room temperature overnight. The solid residue was filtered through a layer of Celite and the solvent was evaporated. The crude product was chromatographed on a silica gel column with chloroform–methanol mixtures (100:1, 20:1 v/v) to afford phosphonates 4a–4d.
Diethyl (E)-[3-(3-ethoxyacryloyl)ureido]methylphosphonate (4a, C11H21N2O6P)
Creamy solid; yield 79 %; m.p.: 56–58 °C; IR (KBr): \( \bar{\nu } \) = 3428, 3239, 3133, 2984, 2936, 1684, 1617, 1563, 1304, 1229, 1168, 1120, 1054, 1023, 979, 807 cm−1; 1H NMR (300 MHz, CDCl3): δ = 10.00 (br s, 1H, NH), 9.01 (dt, J = 5.9, 1.9 Hz, 1H, HNCH2), 7.66 (d, J = 12.3 Hz, 1H, EtOCH=CH), 5.39 (d, J = 12.3 Hz, 1H, EtOCH=CH), 4.22–4.12 (m, 4H, 2 × POCH 2CH3), 3.97 (q, J = 6.9 Hz, 2H, OCH 2CH3), 3.77 (dd, J = 11.9, 5.9 Hz, 2H, CH2P), 1.37 (t, J = 6.9 Hz, 3H, OCH2CH 3), 1.34 (t, J = 7.2 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 168.3 (s, C=O), 162.9 (s, EtOCH=CH), 155.5 (d, J = 6.6 Hz, C=O), 98.0 (s, EtOCH=CH), 67.7 (s, OCH2CH3), 62.7 (d, J = 6.3 Hz, POC), 35.3 (d, J = 157.2 Hz, CP), 16.6 (d, J = 5.7 Hz, POCC), 14.7 (s, OCH2 CH3) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 23.25 ppm.
Diethyl (E)-2-[3-(3-ethoxyacryloyl)ureido]ethylphosphonate (4b, C12H23N2O6P)
Colorless solid; yield 65 %; m.p.: 61–63 °C; IR (KBr): \( \bar{\nu } \) = 3297, 3231, 3149, 3102, 2984, 2938, 1705, 1680, 1618, 1536, 1227, 1164, 1032, 965, 846 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.81 (br s, 1H, NH), 8.92 (t, J = 5.7 Hz, 1H, HNCH2), 7.64 (d, J = 12.3 Hz, 1H, EtOCH=CH), 5.38 (d, J = 12.3 Hz, 1H, EtOCH=CH), 4.21–4.00 (m, 4H, 2 × POCH 2CH3), 3.97 (q, J = 7.2 Hz, 2H, OCH 2CH3), 3.58 (dt, J = 14.7, 7.2 Hz, 2H, CH 2CH2P), 2.08 (dt, J = 18.0, 7.2 Hz, 2H, CH2P), 1.36 (t, J = 7.2 Hz, 3H, OCH2CH 3), 1.34 (t, J = 7.2 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 168.1 (s, C=O), 162.7 (s, EtOCH=CH), 155.4 (s, C=O), 98.2 (s, EtOCH=CH), 67.6 (s, OCH2CH3), 62.0 (d, J = 6.6 Hz, POC), 34.2 (d, J = 3.1 Hz, CCP), 26.5 (d, J = 139.1 Hz, CP), 16.7 (d, J = 6.0 Hz, POCC), 14.8 (s, OCH2 CH3) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 29.76 ppm.
Diethyl (E)-3-[3-(3-ethoxyacryloyl)ureido]propylphosphonate (4c, C13H25N2O6P)
Yellow oil; yield 73 %; IR (film): \( \bar{\nu } \) = 3285, 3140, 2981, 1681, 1613, 1548, 1242, 1167, 1105, 1027, 966 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.67 (br s, 1H, NH), 8.76 (t, J = 6.0 Hz, 1H, HNCH2), 7.64 (d, J = 12.3 Hz, 1H, EtOCH=CH), 5.38 (d, J = 12.3 Hz, 1H, EtOCH=CH), 4.16–4.04 (m, 4H, 2 × POCH 2CH3), 3.98 (q, J = 7.0 Hz, 2H, OCH 2CH3), 3.38 (dt, J = 12.6, 6.0 Hz, 2H, CH 2CH2CH2P), 1.94–1.72 (m, 4H, CH 2CH 2P), 1.36 (t, J = 7.0 Hz, 3H, OCH2CH 3), 1.32 (t, J = 7.0 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 168.4 (s, C=O), 162.5 (s, EtOCH=CH), 155.5 (s, C=O), 98.1 (s, EtOCH=CH), 67.4 (s, OCH2CH3), 61.7 (d, J = 6.3 Hz, POC), 39.9 (d, J = 18.0 Hz, CCCP), 23.2 (d, J = 142.6 Hz, CP), 23.0 (d, J = 4.9 Hz, CCP), 16.6 (d, J = 6.0 Hz, POCC), 14.6 (s, OCH2 CH3) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 32.56 ppm.
Diethyl (E)-4-[3-(3-ethoxyacryloyl)ureido]butylphosphonate (4d, C14H27N2O6P)
Yellowish oil; yield 72 %; IR (film): \( \bar{\nu } \) = 3421, 3283, 3144, 3103, 2982, 2941, 1704, 1679, 1617, 1549, 1234, 1168, 1105, 1057, 1027, 965, 819 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.52 (br s, 1H, NH), 8.68 (t, J = 5.8 Hz, 1H, HNCH2), 7.63 (d, J = 12.2 Hz, 1H, EtOCH=CH), 5.36 (d, J = 12.2 Hz, 1H, EtOCH=CH), 4.15–4.03 (m, 4H, 2 × POCH 2CH3), 3.97 (q, J = 7.0 Hz, 2H, OCH 2CH3), 3.31 (dt, J = 6.4, 5.8 Hz, 2H, CH 2CH2CH2CH2P), 1.81–1.62 (m, 6H, CH 2CH 2CH 2P), 1.36 and 1.32 (2t, J = 6.9 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (151 MHz, CDCl3): δ = 168.4 (s, C=O), 162.4 (s, EtOCH=CH), 155.5 (s, C=O), 98.2 (s, EtOCH=CH), 67.2 (s, OCH2CH3), 61.5 (d, J = 6.6 Hz, POC), 38.9 (s, CCCCP), 30.4 (d, J = 15.5 Hz, CCCP), 25.2 (d, J = 141.8 Hz, CP), 19.8 (d, J = 5.5 Hz, CCP), 16.3 (d, J = 6.3 Hz, POCC), 14.4 (s, OCH2 CH3) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 32.19 ppm.
General procedure for the preparation of uracil derivatives 5a–5d
To a solution of the respective acryloylurea 4a–4d (1.0 mmol) in 8 cm3 dioxane, 8 cm3 H2SO4 (2 M) was added and the mixture was refluxed for 20 h. NaOH (2 M) was added to reach pH 7 and the reaction mixture was concentrated to dryness. To the solid residue, chloroform was added and the mixture was stirred for 15 min, then filtered through a layer of Celite and concentrated. The product was purified by column chromatography with chloroform–methanol mixtures (50:1, 20:1 v/v) to give pure uracil derivatives 5a–5d.
Diethyl [3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]methylphosphonate (5a, C9H15N2O5P)
White solid; yield 58 %; m.p.: 122–124 °C; IR (KBr): \( \bar{\nu } \) = 3428, 3172, 3057, 2991, 2870, 2822, 1689, 1632, 1452, 1382, 1343, 1239, 1049, 977, 732 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.29 (br s, 1H, NH), 7.35 (d, J = 7.9 Hz, 1H, HC-6), 5.76 (dd, J = 7.9, 1.4 Hz, 1H, HC-5), 4.19 (dq, J = 7.9, 6.9 Hz, 4H, 2 × POCH 2CH3), 4.17 (d, J = 11.5 Hz, 2H, CH2P), 1.34 (t, J = 6.9 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 163.8 (s, C=O), 150.6 (d, J = 2.0 Hz, C=O), 144.2 (s, C-6), 102.9 (s, C-5), 63.5 (d, J = 6.6 Hz, POC), 42.0 (d, J = 156.3 Hz, CP), 16.6 (d, J = 6.0 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 19.35 ppm.
Diethyl 2-[3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]ethylphosphonate (5b) [46, 47]
Colorless oil; yield: 83 %.
Diethyl 3-[3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]propylphosphonate (5c) [46, 47]
Colorless oil; yield: 84 %.
Diethyl 4-[3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]butylphosphonate (5d) [46, 47]
Colorless oil; yield: 85 %.
General procedure for the chlorination and bromination reactions
To the solution of the respective phosphonate 5a–5d (0.30 mmol) in 5 cm3 pyridine, N-halogenosuccinimide (NCS or NBS, 0.45 mmol) was added and the mixture was stirred for 30 min at 100 °C. Saturated aqueous NaHCO3 solution (10 cm3) was added and the mixture was extracted with chloroform (3 × 20 cm3). The combined organic phases were dried over MgSO4, filtered, and the solvent was removed. The crude product was purified by column chromatography or crystallization.
Diethyl [5-chloro-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]methylphosphonate (6a, C9H14ClN2O5P)
Colorless solid; yield 72 % [after column chromatography (ethyl acetate) and crystallization from ethyl acetate-petroleum ether mixture]; m.p.: 136–138 °C; IR (KBr): \( \bar{\nu } \) = 3406, 3172, 3043, 2984, 2945, 2831, 1712, 1690, 1633, 1338, 1224, 1061, 1012, 971 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.92 (br s, 1H, NH), 7.61 (s, 1H, HC-6), 4.21 (dq, J = 7.9, 6.9 Hz, 4H, 2 × POCH 2CH3), 4.19 (d, J = 10.7 Hz, 2H, CH2P), 1.35 (t, J = 6.9 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 159.4 (s, C=O), 149.9 (d, J = 2.0 Hz, C=O), 141.0 (s, C-6), 109.4 (s, C-5), 63.6 (d, J = 6.3 Hz, POC), 42.2 (d, J = 156.0 Hz, CP), 16.6 (d, J = 6.0 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 18.80 ppm.
Diethyl 2-[5-chloro-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]ethylphosphonate (6b, C10H16ClN2O5P)
Creamy solid; yield 69 % [after column chromatography (chloroform/methanol 100:1 v/v)]; m.p.: 149–151 °C; IR (KBr): \( \bar{\nu } \) = 3171, 3029, 2994, 2926, 2839, 1701, 1665, 1631, 1368, 1250, 1069, 1018, 963, 793 cm−1; 1H NMR (600 MHz, CDCl3): δ = 9.18 (br s, 1H, NH), 7.61 (s, 1H, HC-6), 4.19–4.10 (m, 4H, 2 × POCH 2CH3), 4.05 (dt, J = 16.1, 6.8 Hz, 2H, CH 2CH2P), 2.26 (dt, J = 18.2, 6.8 Hz, 2H, CH2P), 1.35 (t, J = 7.1 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (151 MHz, CDCl3): δ = 159.6 (s, C=O), 149.9 (s, C=O), 142.2 (s, C-6), 108.4 (s, C-5), 62.3 (d, J = 6.4 Hz, POC), 44.6 (d, J = 3.0 Hz, CCP), 25.0 (d, J = 140.9 Hz, CP), 16.4 (d, J = 5.7 Hz, POCC) ppm; 31P NMR (243 MHz, CDCl3): δ = 26.78 ppm.
Diethyl 3-[5-chloro-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]propylphosphonate (6c, C11H18ClN2O5P)
Colorless solid; yield 53 % (after column chromatography (ethyl acetate) and crystallization from ethyl acetate-petroleum ether mixture); m.p.: 64–66 °C; IR (KBr): \( \bar{\nu } \) = 3545, 3044, 2988, 1701, 1654, 1624, 1458, 1272, 1242, 1227, 1015, 962 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.95 (br s, 1H, NH), 7.63 (s, 1H, HC-6), 4.24–4.07 (m, 4H, 2 × POCH 2CH3), 3.90 (t, J = 7.2 Hz, 2H, CH 2CH2CH2P), 2.06 (dqu, J = 14.6, 7.2 Hz, 2H, CH 2CH2P), 1.81 (dt, J = 18.2, 7.2 Hz, 2H, CH2P), 1.36 (t, J = 6.9 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 159.7 (s, C=O), 150.2 (s, C=O), 141.6 (s, C-6), 108.8 (s, C-5), 62.2 (d, J = 6.6 Hz, POC), 49.1 (d, J = 14.6 Hz, CCCP), 22.4 (d, J = 142.8 Hz, CP), 22.3 (d, J = 4.9 Hz, CCP), 16.7 (d, J = 5.7 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 31.35 ppm.
Diethyl 3-[6-chloro-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]propylphosphonate (7c, C11H18ClN2O5P)
Colorless oil; yield 6 % [after column chromatography (ethyl acetate)]; IR (film): \( \bar{\nu } \) = 3229, 3077, 2925, 2852, 1745, 1696, 1471, 1240, 1098, 1024, 970 cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.18 (br s, 1H, NH), 5.14 (s, 1H, HC-5), 4.19–4.02 (m, 4H, 2 × POCH 2CH3), 3.86 (dt, J = 13.9, 7.0 Hz, 1H, CH aHbCH2CH2CH2P), 3.47 (dt, J = 13.9, 6.8 Hz, 1H, CHa H bCH2CH2CH2P), 2.18–1.84 (m, 4H, CH 2CH 2P), 1.34 and 1.32 (2 × t, J = 7.0 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (151 MHz, CDCl3): δ = 161.5 (s, C=O), 150.6 (s, C=O), 86.3 (s, C-6), 79.5 (s, C-5), 62.5 (d, J = 6.4 Hz, POC), 62.2 (d, J = 6.8 Hz, POC), 47.8 (d, J = 13.5 Hz, CCCP), 22.1 (d, J = 142.4 Hz, CP), 21.1 (d, J = 4.3 Hz, CCP), 16.4 (d, J = 6.6 Hz, POCC), 16.3 (d, J = 5.3 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 33.88 ppm.
Diethyl 4-[5-chloro-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]butylphosphonate (6d, C12H20ClN2O5P)
White solid; yield 65 % (after column chromatography (hexane/ethyl acetate 2:1, 1:2 v/v) and crystallization from ethyl acetate-hexane mixture); m.p.: 139–142 °C; IR (KBr): \( \bar{\nu } \) = 3150, 2982, 2947, 2818, 1688, 1627, 1451, 1429, 1355, 1245, 1210, 1187, 1045, 1020, 971 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.38 (br s, 1H, NH), 7.45 (s, 1H, HC-6), 4.17–4.04 (m, 4H, 2 × POCH 2CH3), 3.76 (t, J = 7.3 Hz, 2H, CH 2CH2CH2CH2P), 1.88–1.61 (m, 6H, CH 2CH 2CH 2P), 1.33 (t, J = 7.1 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 159.6 (s, C=O), 150.1 (s, C=O), 141.2 (s, C-6), 108.8 (s, C-5), 61.9 (d, J = 6.6 Hz, POC), 48.8 (s, CCCCP), 29.7 (d, J = 14.3 Hz, CCCP), 25.2 (d, J = 141.7 Hz, CP), 19.7 (d, J = 4.9 Hz, CCP), 16.7 and 16.6 (2 × d, J = 5.9 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 31.46 ppm.
Diethyl 4-[6-chloro-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]butylphosphonate (7d, C12H20ClN2O5P)
White solid; yield 12 % [after column chromatography (hexane/ethyl acetate 2:1, 1:2 v/v)]; m.p.: 138–141 °C; IR (KBr): \( \bar{\nu } \) = 3412, 3231, 2961, 2924, 2854, 1745, 1698, 1482, 1439, 1254, 1027 cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.60 (br s, 1H, NH), 5.11 (s, 1H, HC-5), 4.16–3.98 (m, 4H, 2 × POCH 2CH3), 3.67 (dt, J = 13.7, 7.0 Hz, 1H, CH aHbCH2CH2CH2P), 3.49 (dt, J = 13.7, 7.1 Hz, 1H, CHa H bCH2CH2CH2P), 1.94–1.65 (m, 6H, CH 2CH 2CH 2P), 1.32 and 1.31 (2 × t, J = 7.0 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (151 MHz, CDCl3): δ = 161.6 (s, C=O), 150.6 (s, C=O), 85.6 (s, C-6), 79.5 (s, C-5), 62.2 (d, J = 6.5 Hz, POC), 62.0 (d, J = 7.1 Hz, POC), 46.3 (s, CCCCP), 28.2 (d, J = 11.7 Hz, CCCP), 24.3 (d, J = 141.3 Hz, CP), 19.1 (d, J = 4.8 Hz, CCP), 16.4 (d, J = 6.6 Hz, POCC), 16.3 (d, J = 6.6 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 33.19 ppm.
Diethyl [5-bromo-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]methylphosphonate (8a, C9H14BrN2O5P)
White solid; yield 69 % (after column chromatography (hexane/ethyl acetate 1:2 and ethyl acetate v/v) and crystallization from ethyl acetate-petroleum ether mixture); m.p.: 120–122 °C; IR (KBr): \( \bar{\nu } \) = 3415, 3164, 3040, 2943, 2851, 1707, 1626, 1422, 1338, 1224, 1012, 985 cm−1; 1H NMR (300 MHz, CDCl3): δ = 10.06 (br s, 1H, NH), 7.72 (s, 1H, HC-6), 4.21 (dq, J = 8.1, 7.0 Hz, 4H, 2 × POCH 2CH3), 4.20 (d, J = 11.1 Hz, 2H, CH2P), 1.35 (t, J = 7.0 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 159.5 (s, C=O), 150.1 (d, J = 2.0 Hz, C=O), 143.5 (s, C-6), 97.2 (s, C-5), 63.6 (d, J = 6.6 Hz, POC), 42.2 (d, J = 156.0 Hz, CP), 16.6 (d, J = 6.0 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 18.82 ppm.
Diethyl 2-[5-bromo-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]ethylphosphonate (8b, C10H16BrN2O5P)
White solid; yield 75 % (after column chromatography (chloroform/methanol 200:1 v/v) and crystallization from ethyl acetate-petroleum ether mixture); m.p.: 134–135 °C; IR (KBr): \( \bar{\nu } \) = 3162, 3038, 2990, 2834, 1704, 1660, 1623, 1251, 1054, 1016, 962 cm−1; 1H NMR (300 MHz, CDCl3): δ = 10.06 (br s, 1H, NH), 7.73 (s, 1H, HC-6), 4.19–4.03 (m, 4H, 2 × POCH 2CH3), 4.02 (dt, J = 16.2, 6.9 Hz, 2H, CH 2CH2P), 2.23 (dt, J = 18.0, 6.9 Hz, 2H, CH2P), 1.30 (t, J = 6.8 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 159.9 (s, C=O), 150.3 (s, C=O), 144.8 (s, C-6), 96.1 (s, C-5), 62.5 (d, J = 6.6 Hz, POC), 44.6 (d, J = 3.4 Hz, CCP), 25.2 (d, J = 140.3 Hz, CP), 16.6 (d, J = 6.0 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 27.46 ppm.
Diethyl 3-[5-bromo-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]propylphosphonate (8c, C11H18BrN2O5P)
Creamy solid; yield 85 % [after column chromatography (chloroform/methanol 200:1 v/v)]; m.p.: 48–50 °C; IR (KBr): \( \bar{\nu } \) = 3549, 3504, 3162, 3052, 2986, 2824, 1702, 1618, 1452, 1346, 1272, 1242, 1180, 1014, 962 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.84 (br s, 1H, NH), 7.72 (s, 1H, HC-6), 4.22–4.04 (m, 4H, 2 × POCH 2CH3), 3.88 (t, J = 7.2 Hz, 2H, CH 2CH2CH2P), 2.04 (dqu, J = 15.0, 7.2 Hz, 2H, CH 2CH2P), 1.78 (dt, J = 18.0, 7.2 Hz, 2H, CH2P), 1.34 (t, J = 6.8 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 159.9 (s, C=O), 150.5 (s, C=O), 144.2 (s, C-6), 96.5 (s, C-5), 62.2 (d, J = 6.6 Hz, POC), 49.0 (d, J = 15.2 Hz, CCCP), 22.4 (d, J = 142.6 Hz, CP), 22.3 (d, J = 4.9 Hz, CCP), 16.6 (d, J = 6.0 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 31.36 ppm.
Diethyl 4-[5-bromo-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]butylphosphonate (8d, C12H20BrN2O5P)
White solid; yield 85 % (after column chromatography (chloroform/methanol 200:1 v/v) and crystallization from ethyl acetate-petroleum ether mixture); m.p.: 112–114 °C; IR (KBr): \( \bar{\nu } \) = 3141, 3074, 2980, 2928, 2817, 1705, 1686, 1621, 1424, 1353, 1247, 1211, 1040, 959 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.36 (br s, 1H, NH), 7.56 (s, 1H, HC-6), 4.17–4.02 (m, 4H, 2 × POCH 2CH3), 3.77 (t, J = 7.2 Hz, 2H, CH 2CH2CH2CH2P), 1.88–1.61 (m, 6H, CH 2CH 2CH 2P), 1.33 (t, J = 7.0 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 159.6 (s, C=O), 150.3 (s, C=O), 143.8 (s, C-6), 96.6 (s, C-5), 61.9 (d, J = 6.6 Hz, POC), 48.8 (s, CCCCP), 29.8 (d, J = 14.3 Hz, CCCP), 25.2 (d, J = 141.7 Hz, CP), 19.7 (d, J = 4.9 Hz, CCP), 16.7 and 16.6 (2 × d, J = 6.0 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 31.46 ppm.
General procedure for the iodination reactions
A solution of the respective phosphonate 5a–5d (0.30 mmol), iodine (0.18 mmol) and (NH4)2Ce(NO3)6 (0.15 mmol) in 10 cm3 anhydrous acetonitrile was refluxed for 2 h. After the solvent was removed, the residue was washed with diethyl ether (3 × 15 cm3) and purified by column chromatography with 2-propanol-hexane mixture (1:3, v/v) to give phosphonates 9a–9d.
Diethyl [5-iodo-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]methylphosphonate (9a, C9H14IN2O5P)
Colorless solid; yield 72 % (after crystallization from ethyl alcohol-petroleum ether); m.p.: 116–118 °C; IR (KBr): \( \bar{\nu } \) = 3156, 3008, 2984, 2929, 2827, 1694, 1613, 1418, 1352, 1226, 1055, 1017, 967 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.24 (br s, 1H, NH), 7.78 (s, 1H, HC-6), 4.20 (dq, J = 8.1, 7.0 Hz, 4H, 2 × POCH 2CH3), 4.17 (d, J = 11.3 Hz, 2H, CH2P), 1.35 (t, J = 7.0 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 160.4 (s, C=O), 150.3 (d, J = 2.0 Hz, C=O), 148.5 (s, C-6), 68.8 (s, C-5), 63.6 (d, J = 6.6 Hz, POC), 42.2 (d, J = 155.7 Hz, CP), 16.6 and 16.5 (2 × d, J = 6.0 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 18.02 ppm.
Diethyl 2-[5-iodo-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]ethylphosphonate (9b, C10H16IN2O5P)
Colorless oil; yield 77 %; IR (film): \( \bar{\nu } \) = 3473, 3167, 3051, 2984, 2817, 1691, 1613, 1443, 1420, 1359, 1284, 1226, 1097, 1051, 1024, 973, 794 cm−1; 1H NMR (300 MHz, CDCl3): δ = 10.04 (br s, 1H, NH), 7.84 (s, 1H, HC-6), 4.21–4.04 (m, 4H, 2 × POCH 2CH3), 4.04 (dt, J = 15.7, 6.9 Hz, 2H, CH 2CH2P), 2.25 (dt, J = 18.2, 6.9 Hz, 2H, CH2P), 1.32 (t, J = 7.0 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 161.0 (s, C=O), 150.6 (s, C=O), 149.7 (s, C-6), 67.7 (s, C-5), 62.5 (d, J = 6.3 Hz, POC), 44.5 (d, J = 2.6 Hz, CCP), 25.2 (d, J = 140.3 Hz, CP), 16.6 (d, J = 5.7 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 27.49 ppm.
Diethyl 3-[5-iodo-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]propylphosphonate (9c) [42]
White solid; yield 71 % (after crystallization from chloroform–diethyl ether mixture); m.p.: 82–84 °C.
Diethyl 4-[5-iodo-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]butylphosphonate (9d, C12H20IN2O5P)
Colorless oil; yield 91 %; IR (film): \( \bar{\nu } \) = 3445, 3165, 3050, 2984, 2816, 1690, 1612, 1441, 1420, 1228, 1051, 1026, 966 cm−1; 1H NMR (300 MHz, CDCl3): δ = 9.63 (br s, 1H, NH), 7.67 (s, 1H, HC-6), 4.18–4.02 (m, 4H, 2 × POCH 2CH3), 3.77 (t, J = 7.1 Hz, 2H, CH 2CH2CH2CH2P), 1.88–1.61 (m, 6H, CH 2CH 2CH 2P), 1.33 (t, J = 7.1 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (75.5 MHz, CDCl3): δ = 160.9 (s, C=O), 150.7 (s, C=O), 148.8 (s, C-6), 68.1 (s, C-5), 61.9 (d, J = 6.6 Hz, POC), 48.6 (s, CCCCP), 29.8 (d, J = 14.9 Hz, CCCP), 25.1 (d, J = 142.0 Hz, CP), 19.6 (d, J = 3.7 Hz, CCP), 16.8 and 16.7 (2 × d, J = 5.9 Hz, POCC) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 31.55 ppm.
General procedure for the benzoyl derivatives 10a–10d
To a solution of the respective phosphonate 5a–5d (0.20 mmol) in 6 cm3 dichloromethane, 0.022 g triethylamine (0.22 mmol) and 0.030 g benzoyl chloride (0.21 mmol) were added at 0 °C and the mixture was stirred at room temperature for 72 h. The solution was washed with water (3 × 15 cm3), dried over MgSO4, and purified by column chromatography with chloroform–methanol mixtures (200:1, 20:1, v/v) to give pure phosphonates 10a–10d.
Diethyl [3-benzoyl-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]methylphosphonate (10a, C16H19N2O6P)
Colorless oil; yield 63 %; IR (film): \( \bar{\nu } \) = 3482, 3090, 2988, 2939, 1750, 1706, 1668, 1598, 1437, 1381, 1341, 1241, 1047, 1023, 976 cm−1; 1H NMR (600 MHz, CDCl3): δ = 7.95–7.94 (m, 2H), 7.68–7.66 (m, 1H), 7.53–7.50 (m, 2H), 7.44 (d, J = 8.0 Hz, 1H, HC-6), 5.87 (d, J = 8.0 Hz, 1H, HC-5), 4.16–4.09 (m, 4H, 2 × POCH 2CH3), 4.18 (d, J = 11.3 Hz, 2H, CH2P), 1.34 (t, J = 7.1 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (151 MHz, CDCl3): δ = 168.2 (s, C=O), 162.0 (s, C=O), 149.5 (d, J = 2.3 Hz, C=O), 143.8 (s, C-6), 135.2, 131.3, 130.5, 129.2, 102.7 (s, C-5), 63.4 (d, J = 6.5 Hz, POC), 42.2 (d, J = 155.7 Hz, CP), 16.3 (d, J = 5.7 Hz, POCC) ppm; 31P NMR (243 MHz, CDCl3): δ = 17.86 ppm.
Diethyl 2-[3-benzoyl-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]ethylphosphonate (10b, C17H21N2O6P)
Colorless oil; yield 64 %; IR (film): \( \bar{\nu } \) = 3494, 3088, 2984, 2931, 2910, 1746, 1705, 1664, 1441, 1391, 1353, 1242, 1051, 1026, 987 cm−1; 1H NMR (600 MHz, CDCl3): δ = 7.96–7.95 (m, 2H), 7.68–7.66 (m, 1H), 7.53–7.50 (m, 2H), 7.41 (d, J = 8.0 Hz, 1H, HC-6), 5.81 (d, J = 8.0 Hz, 1H, HC-5), 4.18–4.09 (m, 4H, 2 × POCH 2CH3), 4.05 (dt, J = 15.9, 7.0 Hz, 2H, CH 2CH2P), 2.26 (dt, J = 18.2, 7.0 Hz, 2H, CH2P), 1.36 (t, J = 7.0 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (151 MHz, CDCl3): δ = 168.6 (s, C=O), 162.4 (s, C=O), 149.6 (s, C=O), 144.9 (s, C-6), 135.1, 131.5, 130.5, 129.2, 101.9 (s, C-5), 62.2 (d, J = 6.5 Hz, POC), 44.8 (d, J = 4.0 Hz, CCP), 24.9 (d, J = 140.7 Hz, CP), 16.4 (d, J = 6.0 Hz, POCC) ppm; 31P NMR (243 MHz, CDCl3): δ = 26.76 ppm.
Diethyl 3-[3-benzoyl-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]propylphosphonate (10c, C18H23N2O6P)
Colorless oil; yield 56 %; IR (film): \( \bar{\nu } \) = 3448, 3083, 2984, 2932, 1747, 1704, 1663, 1439, 1242, 1028, 967 cm−1; 1H NMR (600 MHz, CDCl3): δ = 7.95–7.94 (m, 2H), 7.68–7.66 (m, 1H), 7.53–7.51 (m, 2H), 7.43 (d, J = 8.0 Hz, 1H, HC-6), 5.83 (d, J = 8.0 Hz, 1H, HC-5), 4.18–4.08 (m, 4H, 2 × POCH 2CH3), 3.91 (t, J = 7.5 Hz, 2H, CH 2CH2CH2P), 2.07 (dqu, J = 14.9, 7.5 Hz, 2H, CH 2CH2P), 1.79 (dt, J = 18.5, 7.5 Hz, 2H, CH2P), 1.35 (t, J = 7.1 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (150 MHz, CDCl3): δ = 168.7 (s, C=O), 162.4 (s, C=O), 149.8 (s, C=O), 144.4 (s, C-6), 135.1, 131.5, 130.4, 129.2, 102.2 (s, C-5), 61.9 (d, J = 6.6 Hz, POC), 49.0 (d, J = 13.7 Hz, CCCP), 22.3 (d, J = 143.0 Hz, CP), 22.2 (d, J = 4.6 Hz, CCP), 16.5 (d, J = 5.8 Hz, POCC) ppm; 31P NMR (243 MHz, CDCl3): δ = 29.96 ppm.
Diethyl 4-[3-benzoyl-3,4-dihydro-2,4-dioxopyrimidin-1(2H)-yl]butylphosphonate (10d, C19H25N2O6P)
Colorless oil; yield 73 %; IR (film): \( \bar{\nu } \) = 3451, 3083, 2983, 2939, 2872, 1747, 1704, 1663, 1439, 1241, 1027, 963 cm−1; 1H NMR (600 MHz, CDCl3): δ = 7.94–7.93 (m, 2H), 7.67–7.65 (m, 1H), 7.52–7.50 (m, 2H), 7.30 (d, J = 8.0 Hz, 1H, HC-6), 5.81 (d, J = 8.0 Hz, 1H, HC-5), 4.14–4.04 (m, 4H, 2 × POCH 2CH3), 3.78 (t, J = 7.3 Hz, 2H, CH 2CH2CH2CH2P), 1.88–1.83 (m, 2H, CH 2CH2CH2P), 1.81–1.76 (m, 2H, CH2P), 1.72–1.64 (m, 2H, CH 2CH2P), 1.32 (t, J = 7.0 Hz, 6H, 2 × POCH2CH 3) ppm; 13C NMR (150 MHz, CDCl3): δ = 168.8 (s, C=O), 162.4 (s, C=O), 149.8 (s, C=O), 144.1 (s, C-6), 135.0, 131.6, 130.4, 129.2, 102.2 (s, C-5), 61.6 (d, J = 6.6 Hz, POC), 48.5 (s, CCCCP), 29.5 (d, J = 14.6 Hz, CCCP), 25.0 (d, J = 141.9 Hz, CP), 19.5 (d, J = 4.7 Hz, CCP), 16.4 (d, J = 5.7 Hz, POCC) ppm; 31P NMR (243 MHz, CDCl3): δ = 30.88 ppm.
Antiviral activity assays
The compounds were evaluated against the following viruses: herpes simplex virus type 1 (HSV-1) strain KOS, thymidine kinase-deficient (TK−) HSV-1 KOS strain resistant to ACV (ACVr), herpes simplex virus type 2 (HSV-2) strains Lyons and G, varicella-zoster virus (VZV) strain Oka, TK− VZV strain 07-1, human cytomegalovirus (HCMV) strains AD-169 and Davis, vaccinia virus Lederle strain, respiratory syncytial virus (RSV) strain Long, vesicular stomatitis virus (VSV), Coxsackie B4, Parainfluenza 3, Influenza virus A (subtypes H1N1, H3N2), influenza virus B, Reovirus-1, Sindbis, Reovirus-1, Punta Toro, human immunodeficiency virus type 1 strain IIIB, and human immunodeficiency virus type 2 strain ROD. The antiviral, other than anti-HIV, assays were based on inhibition of virus-induced cytopathicity or plaque formation in human embryonic lung (HEL) fibroblasts, African green monkey cells (Vero), human epithelial cells (HeLa), or Madin-Darby canine kidney cells (MDCK). Confluent cell cultures in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50 % of the cell cultures) or with 20 plaque forming units (PFU) (VZV) in the presence of varying concentrations of the test compounds. Viral cytopathicity or plaque formation was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50 or compound concentration required to reduce virus-induced cytopathogenicity or viral plaque formation by 50 %.
The methodology of the anti-HIV assays was as follows: human CEM (~3 × 105 cells/cm3) cells were infected with 100 CCID50 of HIV(IIIB) or HIV-2(ROD)/cm3 and seeded in 200 mm3 wells of a microtiter plate containing appropriate dilutions of the test compounds. After 4 days of incubation at 37 °C, HIV-induced CEM giant cell formation was examined microscopically.
Cytostatic activity assays
All assays were performed in 96-well microtiter plates. To each well were added (5–7.5) × 104 tumor cells and a given amount of the test compound. The cells were allowed to proliferate for 48 h (murine leukemia L1210 cells) or 72 h (human lymphocytic CEM and human cervix carcinoma HeLa cells) at 37 °C in a humidified CO2-controlled atmosphere. At the end of the incubation period, the cells were counted in a Coulter counter. The IC50 (50 % inhibitory concentration) was defined as the concentration of the compound that inhibited cell proliferation by 50 %.
References
De Clercq E (2011) Annu Rev Pharmacol Toxicol 51:1
De Clercq E (2007) Antiviral Res 75:1
De Clercq E, Holý A (2005) Nat Rev Drug Discov 4:928
Freeman S, Gardiner JM (1996) Mol Biotechnol 5:125
De Clercq E (2003) Clin Microbiol Rev 16:569
De Clercq E (2004) J Clin Virol 30:115
Jordheim LP, Durantel D, Zoulim F, Dumontet C (2013) Nat Rev Drug Discov 12:447
Galmarini CM, Mackey JR, Dumontet C (2001) Leukemia 15:875
Anderson PL, Kakuda TN, Lichtenstein KA (2004) Clin Infect Dis 38:743
Holý A (2003) Curr Pharm Des 9:2567
Franchetti P, Abu Sheikha G, Cappellacci L, Grifantini M, De Montis A, Piras G, Loi AG, La Colla P (1995) J Med Chem 38:4007
Nair V, Chun BK (2003) ARKIVOC i:9
Shealy YF, O’Dell CA (1976) J Heterocycl Chem 13:1015
Rao JR, Jha AK, Rawal RK, Sharon A, Day CW, Barnard DL, Smee DF, Chu CK (2010) Bioorg Med Chem Lett 20:2601
Russ P, Schelling P, Scapozza L, Folkers G, De Clercq E, Marquez VE (2003) J Med Chem 46:5045
Kálmán FK, Woods M, Caravan P, Jurek P, Spiller M, Tirsco G, Király R, Brucher E, Sherry AD (2007) Inorg Chem 46:5260
Ohrlein R, Baisch G (2009) Metal oxide nanoparticles coated with specific N-acylaminomethylene phosphonates. US Patent 20090123507 A1, May 14, 2009; (2006) Chem Abstr 145:336796
Fernández F, García-Mera X, Morales M, Rodríguez-Borges JE (2001) Synthesis 239
Bakó P, Novák T, Ludányi K, Pete B, Tõke L, Keglevich G (1999) Tetrahedron Asymmetry 10:2373
Ali MM, Woods M, Caravan P, Opina ACL, Spiller M, Fettinger JC, Sherry AD (2008) Chem Eur J 14:7250
Moree WJ, Sears P, Kawashiro K, Witte K, Wong C-H (1997) J Am Chem Soc 119:3942
Neidlein R, Greulich P (1992) Helv Chim Acta 75:2545
Efimtseva EV, Mikhailov SN, Jasko MV, Malakov DV, Semizarcv DG, Fomicheva MV, Kern ER (1995) Nucleosides Nucleotides 14:373
Gali H, Prabhu KR, Karra SR, Katti KV (2000) J Org Chem 65:676
Sauer R, Froimowicz P, Schöller K, Cramer J-M, Ritz S, Mailänder V, Landfester K (2012) Chem Eur J 17:5201
Mercklé L, de Andrés-Gómez A, Dick B, Cox RJ, Godfrey CRA (2005) ChemBioChem 6:1866
Talukdar A, Morgunova E, Duan J, Meining W, Foloppe N, Nilsson L, Bacher A, Illarionov B, Fischer M, Ladenstein R, Cushman M (2010) Bioorg Med Chem 18:3518
Głowacka IE, Balzarini J, Wróblewski AE (2012) Nucleosides Nucleotides Nucleic Acids 31:293
Głowacka IE, Balzarini J, Wróblewski AE (2013) Eur J Med Chem 70C:703
Tucker-Schwartz AK, Garrell RL (2010) Chem Eur J 16:12718
Dijkstra HP, Sprong H, Aerts BNH, Kruithof CA, Egmond MR, Gebbink RJMK (2008) Org Biomol Chem 6:523
Artyushin OI, Vorob’eva DV, Vasil’eva TP, Osipov SN, Röschenthaler GV, Odinets IL (2008) Heteroat Chem 19:293
An I-H, Seong HR, Ahn KH (2004) Bull Korean Chem Soc 25:420
Lange M, Pettersen AL, Undheim K (1998) Tetrahedron 54:5745
Holý A (1993) Collect Czech Chem Commun 58:649
Rejman D, Kovačková S, Pohl R, Dračínský M, Fiedler P, Rosenberg I (2009) Tetrahedron 65:8513
Pudry DF, Zintek LB, Nair V (1994) Nucleosides Nucleotides 13:109
Pickering L, Nair V (1996) Nucleosides Nucleotides 15:1751
Tietze LF, Schneider C, Pretor M (1993) Synthesis 1079
Pohl R, Rulíšek L, Rejman D (2011) J Phys Org Chem 24:423
Lazrek HB, Taourirte M, Oulih T, Barascut JL, Imbach JL, Pannecouque C, Witrouw M, De Clercq E (2001) Nucleosides Nucleotides Nucleic Acids 20:1949
Parchina A, Froeyen M, Margamuljana L, Rozenski J, De Jonghe S, Briers Y, Lavigne R, Herdewijn P, Lescrinier E (2013) ChemMedChem 8:1373
Lee YS, Kim BH (2002) Bioorg Med Chem Lett 12:1395
Głowacka IE, Balzarini J, Schols D, Piotrowska DG (2014) Bioorg Med Chem 22:3629
Song X-P, Bouillon C, Lescrinier E, Herdewijn (2011) ChemBioChem 12:1868
Sauer R, El-Tayeb A, Kaulich M, Müller CE (2009) Bioorg Med Chem 17:5071
Brel VK (2012) Synthesis 44:2359
Acknowledgments
The authors wish to express their gratitude to Mrs. Leentje Persoons, Mrs. Frieda De Meyer, and Mrs. Lizette van Berckelaer for excellent technical assistance. This work was supported by the Medical University of Lodz internal funds (503/3-014-1/503-06-300 and 502-03-/3-014-01/502-34-038). The virological part of this work was supported by the KU Leuven (GOA No. 10/014).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rygielska-Tokarska, D., Andrei, G., Schols, D. et al. Synthesis, antiviral, cytotoxic and cytostatic evaluation of N 1-(phosphonoalkyl)uracil derivatives. Monatsh Chem 147, 1081–1090 (2016). https://doi.org/10.1007/s00706-016-1701-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-016-1701-2