
Abstract
A mercury meniscus modified silver solid amalgam electrode was used for the first time for the determination of submicromolar concentrations of fenitrothion (FNT). The medium of ethanol–Britton–Robinson buffer of pH 7.0 (1:9) was chosen as the optimal one. The newly developed direct current and differential pulse (DP) voltammetric methods are fast, reliable, and robust. Moreover, the applicability of the DP voltammetric method was verified for the determination of FNT in spiked samples of drinking and river water, with the limits of quantification in the concentration order of 10−7 mol dm−3. Furthermore, the interaction of FNT with double-stranded (ds) DNA was investigated directly in a solution. From changes in the FNT electrochemical signals, we assume a formation of a FNT–dsDNA complex in which FNT bounds to dsDNA by electrostatic forces. DP voltammetry was employed to probe this interaction, while cyclic voltammetry was used for the investigation of voltammetric data of free FNT and FNT bound to dsDNA.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
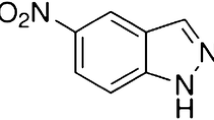
The evergrowing world population puts great demands on food production and its availability. That is inevitably associated with a more intensive use of agrochemicals. However, such trend causes negative impact to the environment and further deterioration of food and water quality. Agrochemicals include two large groups of compounds: chemical fertilizers and pesticides [1]. In this work, fenitrothion [O,O-dimethyl O-(3-methyl-4-nitrophenyl) phosphorothioate, FNT, see Fig. 1] has been chosen as a model representative of organophosphorous pesticides (OPPs) whose synthesis is relatively inexpensive and simple [2]. FNT is worldwide used under different tradenames, e.g., Agrothion, Sumithion. It effectively acts against various insects which damage agricultural crops, such as cereals, cotton, or rice [3]. FNT is toxic to birds, aquatic system and highly toxic to honeybees [4, 5]. It is either applied directly to the soil or sprayed over a variety of crops for pre- or postharvest protection [6]. Although FNT degrades rapidly in the environment, its small amount can be detected in food and drinking water [7, 8]. About 95 % of human intake appears to be from food [9]. It irreversibly inactivates acetylcholinesterase, which can result in accumulation of acetylcholine and overstimulation of its receptors in synapses and eventually damage the nervous system [10–13]. However, oxidative stress has been recognized as the main mechanism underlying the OPP toxicity from repeated exposure [14, 15]. Effects vary from mild to serious depending on exposure route, dose, and type of pesticide residue. Chronic effects were caused by long-term exposure to low levels of OPPs both in farmers and in consumers [8].

Structural formula of FNT
These facts do not imply that agrochemicals are completely useless or harmful, but current problems call upon much better control of their registration and use. As consequence, the European Union put pesticides, including OPPs plus their metabolites, in its list of priority environmental pollutants [6, 16]. Similarly, upon the Food Quality Protection Act of 1996, the US Environmental Protection Agency is required to conduct combined risk assessments for pesticides showing a “common mechanism of toxicity” [10, 17].
Therefore, there is a great demand for the rapid and sensitive detection of OPPs. Recently, many analytical methods of the FNT detection in technical products or biological and environmental samples have been developed, including gas chromatography [8, 18, 19] coupled with mass spectrometry [5, 20, 21] or liquid chromatography [22] hyphenated with mass spectrometry [20, 23]. The colorimetric methods have been described as well [24]. These traditional methods are suitable for large scale food quality inspection or to provide proof for law enforcement agencies [12]. However, voltammetric methods present a fast and inexpensive alternative to conventional methods [25, 26]. Their great advantage is that they do not require any sample preparation which is mainly accompanied by the extraction process, cleanup, and preconcentration. So far, several electrochemical methods for the FNT determination have been proposed, such as adsorptive stripping voltammetry [27], square wave voltammetry [24], or differential pulse stripping voltammetry [28], all at a hanging mercury drop electrode (HMDE). Square wave anodic stripping and cyclic voltammetry at a carbon nanotube paste electrode with mercury immobilized on its surface have been described by Lee and Ly [29]. Cyclic voltammetry, square wave adsorptive stripping voltammetry, and impedance techniques were used for the FNT determination using a non-traditional graphene and silver-zirconia nanosensor [30]. Moreover, the voltammetric methods enable to estimate and predict the conformational changes of DNA caused by pesticides [31–33]. The interaction of FNT with DNA has been satisfactory studied using cyclic voltammetry at the HMDE [34].
The main purpose of this work was to develop a voltammetric method for the determination of FNT at a mercury meniscus modified silver solid amalgam electrode (m-AgSAE) which represents a non-toxic alternative to traditional mercury electrodes regarding sensitivity and limit of quantification (LOQ). Its production is inexpensive and simple, operation costs are low, it is mechanical stable, robust, and suitable for flowing systems [35, 36]. Furthermore, high hydrogen overvoltage on solid amalgam allows to work in aqueous solutions up to −2 V [37, 38]. The second aim was to verify the applicability of the new method for model environmental samples. The last aim was to study an interaction of FNT with double-stranded (ds) DNA.
Results and discussion
Direct current and differential pulse voltammetric determination of FNT at the m-AgSAE
The electrochemical behavior of FNT in a mixture of ethanol and Britton–Robinson (BR) buffers (1:9) was investigated by means of direct current (DC) and differential pulse (DP) voltammetry at the m-AgSAE, which is possible due to the easily reducible nitro group in its structure [9]. First of all, the pH influence of BR buffers from 2.0 to 13.0 on the voltammetric FNT response was studied in cathodic potential region (see Fig. 2a for DC voltammetry and Fig. 2b for DP voltammetry, respectively). Using DC and DP voltammetry in acid medium and using DC voltammetry also in neutral medium, FNT yielded only one well-shaped wave/peak, corresponding to the four-electron reduction of the nitro group to the hydroxylamino moiety. Peak potential (E p) was monotonously shifted toward less negative values with increasing pH. In alkaline medium using both voltammetric techniques and in neutral medium using DP voltammetry, an additional signal appeared due to the lower concentration of H+. Thus, the nitro group did not undergo to the reduction as easily as at lower pH and reduction pathway was divided into two steps. After acception of one electron, a negatively charged radical was formed. At even more negative potential, this intermediate could accept other three electrons, resulting in the final reduction product, the same one as at lower pH. In the pH range from 8.0 to 13.0, the E p values practically did not vary. The suggested redox mechanism of FNT is analogous to that earlier proposed for analytes containing the nitro group in their structure at mercury electrodes or m-AgSAEs [39–41]. With respect to the substance decomposition in alkaline medium and further investigation of an interaction with DNA, ethanol–BR buffer of pH 7.0 (1:9) was chosen as the optimal medium.

DC (a) and DP (b) voltammograms of FNT (c = 1 × 10−4 mol dm−3) in the ethanol–BR buffer (1:9) medium recorded at the m-AgSAE, the BR buffer pH: 3.0 (1), 5.0 (2), 7.0 (3), 9.0 (4), 11.0 (5), and 13.0 (6)
Passivation of the working electrode surface is a common problem of solid electrodes. Therefore, appropriate regeneration potentials were sought. The repeatability of the DC and DP voltammetric peak height (I p) of 1 × 10−4 mol dm−3 FNT in ethanol–BR buffer of pH 7.0 (1:9) was tested. Various combinations of regeneration potentials, together with values obtained without any regenerating treatment, are depicted in Fig. 3. After application of 0 mV as an initial regeneration potential (E 1reg) and −1450 mV as a final regeneration potential (E 2reg), a relative standard deviation (RSD), which was calculated from twenty consecutive measurements, decreased from 4.0 to 2.5 %.

The dependence of the peak height of FNT (c = 1 × 10−4 mol dm−3) in ethanol–BR buffer of pH 7.0 (1:9) on a serial number of successive measurement with an application of different regeneration potentials at the m-AgSAE. The combinations of regeneration potentials were: 0, −1300 mV (1); −200, −1400 mV (2); without regeneration (3); 0, −1450 mV (4); and −400, −1300 mV (5). A couple of 0, −1450 mV was chosen as suitable initial and final regeneration potentials
The found optimal conditions were used for measuring calibration dependences in the concentration range from 0.6 to 100 μmol dm−3 using both DC and DP voltammetry. The parameters of corresponding calibration straight lines are summarized in Table 1. At the same place, the LOQ of FNT (whose computation is described in detail in the section “Experimental”) reached under the above-mentioned optimal conditions is evaluated for both techniques. The calculated LOQ for DC voltammetry corresponded well to the lowest measured concentration in reality. However, very good repeatability of the consecutive measurements of DP voltammetry caused that the calculated LOQ is much lower than it was possible to measure and evaluate in reality.
The mixture of deionized water and BR buffer (9:1) was used to provide a step between the determination of FNT in the ethanol–BR buffer medium and in model water samples. As we supposed, it was possible to evaluate the voltammetric signals of FNT at lower concentrations in the supporting electrolyte in the absence of ethanol than in the presence of ethanol. This became due to the fact that ethanol, whose addition was necessary because of a limited solubility of FNT in water at its higher concentrations, reduced voltammetric signals. Therefore, the calibration dependences of FNT in deionized water–BR buffer of pH 7.0 (9:1) were measured at the FNT concentrations not higher than 10 μmol dm−3.
Overall, the attained LOQ was slightly higher than in the case of the ethanolic-aqueous medium. This is caused by a way of LOQ calculation. The corresponding DP voltammograms and calibration straight lines are shown in Fig. 4, their parameters are summarized in Table 1. Afterwards, an attempt to increase the sensitivity of the FNT determination at the m-AgSAE using adsorptive stripping DC voltammetry was made, unfortunately, with no reasonable positive result at any accumulation time or accumulation potential investigated.

DP voltammograms of FNT in spiked deionized water–BR buffer of pH 7.0 (9:1) at the m-AgSAE; E 1reg = 0 mV, E 2reg = −1450 mV. Final concentrations of FNT in the measured solution for a 0 (1), 0.2 (2), 0.4 (3), 0.6 (4), 0.8 (5), and 1.0 (6) µmol dm−3; and for b 2.0 (7), 4.0 (8), 6.0 (9), 8.0 (10), and 10 (11) µmol dm−3
Voltammetric determination of FNT in drinking and river water at the m-AgSAE
As was previously mentioned, one of the aims of this work was to verify usability of the newly developed and above-described method for voltammetric determination of FNT in model water samples. For that purpose, only more sensitive DP voltammetry was used. It is necessary to emphasize that FNT was not present in drinking and river water used for the following determination. Therefore, the model water samples were spiked by FNT from its stock solution. The precise procedure of the sample preparation is described in the section “Experimental.” Calibration dependences were measured in the FNT concentration range from 0.2 to 10 μmol dm−3. The voltammograms obtained in the concentration range from 0.2 to 1.0 μmol dm−3 are depicted in Fig. 5. The parameters of the calibration straight lines are summarized in Table 1; the LOQs for both water samples are comparable. Overall, the results confirm the possible application of the proposed DP voltammetric method for both drinking and river water, with no significant matrix effect appeared.

DP voltammograms of FNT in spiked drinking (a)/river (b) water–BR buffer of pH 7.0 (9:1) at the m-AgSAE; E 1reg = 0 mV, E 2reg = −1450 mV. Final concentrations of FNT in the measured solution: 0 (1), 0.2 (2), 0.4 (3), 0.6 (4), 0.8 (5), and 1.0 (6) µmol dm−3
Electrochemical study of DNA-binding agents
In recent years, there has been a growing interest in the electrochemical investigation of DNA, a carrier of genetic information, and its interaction with nucleic acid-binding molecules [42–45] because damage to DNA, upon the interaction with various chemical and physical agents occurring in the environment, often results in mutation that may subsequently lead to cancer development [46]. Electrochemical investigations can provide a useful complement to the techniques usually used to study these interactions (mass spectrometry, capillary electrophoresis, surface plasmon resonance, spectrometric methods). The binding of small molecules, like FNT, to dsDNA could involve the electrostatic interaction with negatively charged sugar-phosphate backbone, intercalation, which is typical for planar organic molecules containing several aromatic condensed rings, and minor or major DNA groove binding [47]. Investigated xenobiotic compound itself can serve as redox indicator because its E p is shifted in the positive direction when the analyte binds to DNA by intercalation between the stacked base pairs of dsDNA. The E p is shifted in the negative direction when the interaction with DNA occurs by electrostatic attraction [48]. The interaction of FNT with DNA has been already studied using cyclic voltammetry at a HMDE by Ahmadi and Jafari [34]. They have suggested a partial-intercalative mode as a result of the mutual interaction. Moreover, they have calculated the binding constant, binding site size and diffusion coefficient of free FNT and its complex with DNA.
The DNA–xenobiotic interactions can be investigated in three different ways: (1) DNA-modified electrode, (2) xenobiotic-modified electrode, and (3) interaction of both in bulk solution. In this paper, the third type of arrangement was chosen, i.e., FNT and DNA were placed in the same solution and the changes in the electrochemical signals of FNT–DNA complex were compared with the signals obtained with FNT alone in the solution [47].
Investigation of FNT interaction with DNA by differential pulse voltammetry
First, to make the investigation simpler, BR buffer was replaced by 0.1 mol dm−3 phosphate buffer of pH 6.8 in which all measurements took place. Also DNA was dissolved in phosphate buffer to obtain its stock solution. The pretreatment of the working electrode, including the regeneration using regeneration potentials 0 and −1450 mV, remained unchanged. A signal of FNT (c = 1 × 10−6 mol dm−3, 1 × 10−5 mol dm−3) in phosphate buffer at the m-AgSAE was recorded using DP voltammetry. After that, dsDNA in the concentration range from 2 to 100 µg cm−3 was added to the same solution, i.e., to phosphate buffer containing FNT. The voltammograms displayed in Fig. 6 illustrate the shift of the FNT E p toward more negative values, thus, the reduction of FNT becomes more difficult. The decrease of the FNT peak current was also obvious. We assume the change of the electrochemical behavior of both DNA and FNT molecules, which indicates a formation of a mutual complex. As the FNT was bonded to DNA, its electron transfer process was affected and its electron transport ability decreased with increasing concentration of DNA until no free FNT remained in the solution [49]. In the case of the FNT concentration of 1 × 10−5 mol dm−3, the E p shift stabilized at 12 µg cm−3 and the I p at 20 µg cm−3 of added dsDNA. While in the case of 1 × 10−6 mol dm−3 FNT, both the E p and the I p stabilized at 8 µg cm−3 of added dsDNA. Moreover, the FNT signal was not possible to be evaluated at the concentration of dsDNA in the solution of 16 µg cm−3. Upon the information in [47, 48], the E p shift indicated the electrostatic binding of FNT to DNA. Nevertheless, it would require other techniques to prove the type of the interaction. In principle, we have observed the same voltammetric behavior of free FNT, as well as after the addition of dsDNA, at the m-AgSAE as other authors using the HMDE [34].

a DP voltammograms of FNT (c = 1 × 10−5 mol dm−3) in phosphate buffer with the dsDNA additions. The final concentration of dsDNA in the FNT solution: 0 (1), 2 (2), 4 (3), 6 (4), 8 (5), 12 (6), 16 (7), 20 (8), and 100 (9) µg cm−3. The curve number 10 corresponds to pure phosphate buffer. b The corresponding dependence of the E p and I p on the added dsDNA concentration to the FNT solution; the error bars are constructed for n = 5
Electrochemical behavior of FNT–DNA complex investigated by cyclic voltammetry
In order to comprehend the mutual FNT and dsDNA interaction more deeply, further investigation using cyclic voltammetry was carried out. Although the electroactivity of DNA itself, respectively of nucleic bases, has been reported many times, even at the amalgam electrodes [42, 50–54], we did not observed any signal of dsDNA over the concentration range of 0.002–2 µg cm−3 in 0.1 mol dm−3 phosphate buffer of pH 6.8 under the used conditions, i.e., using cyclic voltammetry at the scan rate of 20 mV s−1 at the m-AgSAE. It could be explained by lower sensitivity of the m-AgSAE compared to the HMDE and by low scan rate (in [55], the authors did not obtained any signal of cytosine, adenine, or guanine until the scan rate reached at least 1 V s−1).
FNT yielded one irreversible cathodic peak, corresponding to the reduction of the nitro group. According to the dependences of the I p of FNT (c = 1 × 10−5 mol dm−3) on the scan rate and scan rate square root, which are shown in Fig. 7, we can presume a determining process for the electrochemical reaction. With the increasing scan rate, the height of the FNT signal increased and its potential shifted toward more negative values. Simultaneously, both relations were not linear and implied a process including both adsorption and diffusion. Diffusion prevailed mainly at the lower scan rates, which was confirmed by plotting the dependence of the logarithm of the I p on the logarithm of the scan rate (not shown), resulting in the slope of 0.55–0.6. When FNT formed a complex with dsDNA, the electrochemical behavior changed. Using cyclic voltammetry, we have recognized the same changes as those resulting from the FNT–dsDNA interaction obtained by DP voltammetry, i.e., the I p declined and the E p shifted toward the same direction. Equally, the slope of the dependence of the logarithm of the I p on the logarithm of the scan rate dropped under 0.5.

The dependences of the FNT (c = 1 × 10−5 mol dm−3) I p on the scan rate (a) and the square root of scan rate (b) without (1) and with (2) dsDNA (γ = 2 µg cm−3) obtained by cyclic voltammetry at the m-AgSAE
Conclusion
The mercury meniscus modified silver solid amalgam electrode (m-AgSAE) represents a suitable alternative to the mercury electrodes, offering a variety of applications [36]. In the present work, the m-AgSAE was employed for the voltammetric determination of fenitrothion (FNT). Electrochemical reduction of FNT was found to be irreversible and pH-dependent using both direct current (DC) and differential pulse (DP) voltammetry. The supporting electrolyte composition, pH, and regeneration potentials were optimized with respect to the maximal and well-defined peak. A wide linear range was observed between the peak currents and the concentration of FNT over three orders of magnitude, with the limits of quantification (LOQs) of 0.3 and 0.04 µmol dm−3 for DC voltammetry and DP voltammetry, respectively, in ethanol–Britton–Robinson (BR) buffer of pH 7.0 (1:9), and over two orders of magnitude, with the LOQ of 0.06 µmol dm−3 for DP voltammetry in spiked deionized water–BR buffer of pH 7.0 (9:1). The newly developed DP voltammetric method was applied for the determination of FNT in model water samples.
Moreover, DP and cyclic voltammetry were used for the investigation of the interaction between FNT and double-stranded (ds) DNA in the solution. On the basis of the measured results, we propose an electrostatic binding as the main type of the mutual interaction between the studied analyte FNT and dsDNA.
Experimental
Chemicals and reagents
The FNT stock solution (c = 1 × 10−3 mol dm−3) was prepared by dissolving an accurately weighed amount of FNT (Sigma-Aldrich, St. Louis, USA) in ethanol and stored in a dark place at 4 °C. Working solutions were prepared freshly before use by exact dilution of the stock solution with ethanol. BR (pH 2.0–12.0) and phosphate (pH 6.8) buffer solutions were prepared in a usual way. Sodium hydroxide of 0.1 mol dm−3 was used to obtain a solution of pH 13.0. A solution of 0.2 mol dm−3 potassium chloride was used for the m-AgSAE activation. The DNA stock solution was prepared by dissolving 10 mg of low molecular weight dsDNA from salmon sperm (Sigma-Aldrich, St. Louis, USA) in 10 cm3 of 0.1 mol dm−3 phosphate buffer of pH 6.8 and stored in germ-free plastic tubes at −18 °C. The dsDNA stock solutions were prepared at least 12 h before measurements to completely solubilize DNA. Unless stated otherwise, all reagents were p.a. grade (Lach-Ner, Neratovice, Czech Republic).
Instrumentation
All voltammetric measurements were performed using an Eco-Tribo Polarograph (running with the Polar Pro 5.1 software, Polaro-Sensors, Prague, Czech Republic) with an m-AgSAE working electrode, a platinum wire counter electrode, and a reference Ag|AgCl electrode (3 mol dm−3 KCl). The DP voltammetry parameters were as follows: pulse amplitude of −50 mV and pulse width of 100 ms, with current sampling for the last 20 ms. Scan rate of 20 mV s−1 was used for DC, DP, and cyclic voltammetry if not specifically mentioned otherwise.
The m-AgSAE was prepared by immersing a glass capillary tip filled by silver powder into freshly filtered liquid mercury and leaving there for 2 h. Then, the electrode surface was polished with filter paper and, afterwards, it was immersed into liquid mercury for 15 s while stirring to modify the electrode surface by a mercury meniscus. After creating a new meniscus or each day before starting measurements, the working electrode had to be electrochemically activated in stirred 0.2 mol dm−3 KCl at constant potential of −2200 mV for 300 s [56, 57].
Procedures
Before starting voltammetric measurement, oxygen was removed from the measured solutions by purging with nitrogen (purity 4.0, Linde, Prague, Czech Republic) for 300 s. In order to reduce the influence of electrode passivation, an electrochemical regeneration of the working electrode, based on 150 times switching the electrode potential between E 1reg and E 2reg for 0.1 s, was carried out before recording each curve.
The LOQ was calculated as a quotient of tenfold standard deviation of the peak height at the lowest measurable concentration obtained from ten repetitive determinations and a slope of a corresponding calibration curve [58].
The Origin Pro 8.0 software (OriginLab Corporation, Northampton, USA) was used for calculation of all parameters of calibration curves and construction of graphs. All measurements were carried out at laboratory temperature.
Preparation of model water samples
Drinking water from the public water pipeline in the building of the Faculty of Science of the Charles University in Prague (Czech Republic) and river water from the Vltava in Prague (Czech Republic) were used for model water samples. Both types of waters were used without further pretreatment or purification, only a decantation and equalization to laboratory temperature were applied. The preparation procedure of the measured solution with the model water sample addition was as follows: 1 cm3 of BR buffer of pH 7.0 was added to 9 cm3 of water sample which have been already spiked with an appropriate amount of the FNT stock solution.
References
Carvalho FP (2006) Environ Sci Policy 9:685
Henych J, Stengl V, Slusna M, Grygar TM, Janos P, Kuran P, Stastny M (2015) Appl Surf Sci 344:9
Rougier NM, Vico RV, Rossi RH, Bujan EI (2014) J Phys Org Chem 27:935
Yuan Z, Yao J, Liu H, Han J, Trebse P (2014) Ecotox Environ Saf 108:84
Lopez DR, Ahumada DA, Diaz AC, Guerrero JA (2014) Food Control 37:33
Gonzalez-Curbelo MA, Herrera-Herrera AV, Hermandez-Borges J, Rodriguez-Delgado MA (2013) J Sep Sci 36:556
Lijuan Z, Faqing Z, Baizhao Z (2014) Biosens Bioelectron 62:19
Sapbamrer R, Hongsibsong S (2014) Arch Environ Contam Toxicol 67:60
WHO (2004) Fenitrothion in drinking-water. Geneva. http://www.who.int/water_sanitation_health/dwq/chemicals/fenitrothion.pdf. Accessed 16 July 2015
Costa LG (2006) Clin Chim Acta 366:1
Fukuto TR (1990) Environ Health Persp 87:245
Meng X, Schultz CW, Cui C, Li X, Yu H-Z (2015) Sensor Actuat B Chem 215:577
Deng N, Ni Y, Kokot S (2010) Chin J Chem 28:404
Taib IS, Budin SB, Ghazali AR, Jayusman PA, Louis SR, Mohamed J (2015) Toxicol Res 4:132
Lukaszewicz-Hussain A (2010) Pestic Biochem Phys 98:145
Council of the European Union (2000) Council directive 98/83/EC on the quality of water intended for human consumption. Brussel. http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:31998L0083. Accessed 16 July 2015
US Congress (1996) Food quality protection act. Washington. http://epa.gov/pesticides/regulating/laws/fqpa/backgrnd.htm. Accessed 16 July 2015
Zuin VG, Yariwake JH, Bicchi C (2003) J Chromatorgr A 985:159
Ko KY, Shin JY, Kim D, Kim MK, Son S (2014) J Anal Toxicol 38:667
Grimalt S, Harbeck S, Shegunova P, Seghers J, Sejerøe-Olsen B, Emteborg H, Dabrio M (2015) Anal Bioanal Chem 407:3083
Baroja O, Unceta N, Sampedro MC, Goicolea MA, Barrio RJ (2004) J Chromatogr A 1059:165
Sanchez-Ortega A, Sampedro MC, Unceta N, Goicolea MA, Barrio RJ (2005) J Chromatogr A 1094:70
Hernandez F, Sancho JV, Pozo OJ (2004) J Chromatogr B 808:229
Melgar LZ, Machado SAS (2005) J Braz Chem Soc 16:743
Barek J, Peckova K, Vyskocil V (2008) Curr Anal Chem 4:242
Krejcova Z, Barek J, Vyskocil V (2015) Electroanal 27:185
Galeano-Dıaz T, Guiberteau-Cabanillas A, Espinosa-Mansilla A, Lopez-Soto MD (2008) Anal Chim Acta 618:131
Ni Y, Qiu P, Kokot S (2004) Anal Chim Acta 516:7
Lee C, Ly S (2010) J Environ Sci 19:941
Sreedhar NY, Kumar MS, Krishnaveni K (2015) Monatsh Chem 146:1385
Ibrahim MS (2001) Anal Chim Acta 1:443
Ni Y, Wang P, Kokot S (2012) Biosens Bioelectron 38:245
Erdem A, Ozsoz M (2002) Electroanal 14:14
Ahmadi F, Jafari B (2011) Electroanal 23:675
Vyskocil V, Barek J (2009) Crit Rev Anal Chem 39:173
Danhel A, Barek J (2011) Curr Org Chem 15:2957
Yosypchuk B, Barek J (2009) Crit Rev Anal Chem 39:189
Yosypchuk B, Sestakova I (2008) Electroanal 20:426
Tocher JH (1997) Gen Pharmacol 28:485
Barek J, Fischer J, Navratil T, Peckova K, Yosypchuk B, Zima J (2007) Electroanal 19:2003
Yosypchuk O, Karasek J, Vyskocil V, Barek J, Peckova K (2012) Sci World J 2012:231986
Vyskocil V, Labuda J, Barek J (2010) Anal Bioanal Chem 397:233
Yola ML, Ozaltin N (2011) J Electroanal Chem 653:56
Abreu FC, Goulart MOF, Brett AMO (2002) Biosens Bioelectron 17:913
Palecek E, Fojta M, Tomschik M, Wang J (1998) Biosens Bioelectron 13:621
Fojta M (2002) Electroanal 14:1449
Rauf S, Gooding JJ, Akhtar K, Ghauri MA, Rahman M, Anwar MA, Khalid AM (2005) J Pharm Biomed 37:205
Carter MT, Rodriguez M, Bard AJ (1989) J Am Chem Soc 111:8901
Wang Y, Ni Y, Kokot S (2011) Anal Biochem 419:76
Labuda J, Oliveira-Brett AM, Evtugyn G, Fojta M, Mascini M, Ozsoz M, Palchetti I, Palecek E, Wang J (2010) Pure Appl Chem 82:1161
Neto BAD, Lapis AAM (2009) Molecules 14:1725
Vacek J, Havran L, Fojta M (2009) Collect Czech Chem Commun 74:1727
Fojta M (2004) Collect Czech Chem Commun 69:715
Kucharikova K, Novotny L, Yosypchuk B, Fojta M (2004) Electroanal 16:410
Danhel A, Raindlova V, Havran L, Pivonkova H, Hocek M, Fojta M (2014) Electrochim Acta 126:122
Barek J, Fischer J, Navratil T, Peckova K, Yosypchuk B (2006) Sensors 6:445
Yosypchuk B, Novotny L (2002) Electroanal 14:1733
Inczedy J, Lengyel T, Ure AM (1998) Compendium of analytical nomenclature: definitive rules 1997, 3rd edn. Blackwell Science, Malden
Acknowledgments
This research was carried out within the framework of the Specific University Research (SVV). Z. Krejcova thanks the Grant Agency of the Charles University in Prague (Project 140214/2014/B-CH/PrF) and J. Barek and V. Vyskocil thank the Grant Agency of the Czech Republic (Project P206/12/G151) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Krejcova, Z., Barek, J. & Vyskocil, V. Voltammetric determination of fenitrothion and study of its interaction with DNA at a mercury meniscus modified silver solid amalgam electrode. Monatsh Chem 147, 135–142 (2016). https://doi.org/10.1007/s00706-015-1595-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1595-4