
Abstract
A series of ten novel isatin analogs have been synthesized and screened for their in vitro anti-breast cancer activity against MCF-7 cell line using sulforhodamine-B assay method. All the tested compounds showed highly potent activity against MCF-7 cell line with especially four compounds exhibited demonstrative antiproliferative effects on MCF-7 breast cancer cell line compared to reference adramycin (doxorubicin) and GI 50 <0.02 μM. Docking the synthesized compounds into the epidermal growth factor receptor, which is highly expressed in breast cancer, was employed to explore the possible interactions of these compounds with the receptor. Structure activity relationship as well as virtual ADME studies were carried out and a connection between activities, electronic and physicochemical properties of the target compounds was determined.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Globally, cancer is one of the leading causes of death. The death rate from various types of cancer continues to grow worldwide with an estimation of 12 million deaths in 2030 [1, 2]. One in ten of all new types of cancer diagnosed around the world each year is breast cancer and is the most common cancer in women, both in developing and developed countries [3]. Estrogen receptor (ER)-negative breast cancer accounts for approximately 30 % of all breast cancer diagnosed [4–6]. Chemotherapy has been the most frequently used treatment for breast cancer and other cancers till date. However, some normal proliferating cells are also destroyed by this method of treatment [7]. Hence, there is an intense worldwide search for identifying new drugs that are more effective and safe for the prevention and treatment of cancer.
A survey of the literature in this area showed that many heterocyclic scaffolds have been investigated as anti-breast cancer agents [8]. Among them isatin is a useful structural motif for the development of molecules of pharmaceutical or biological interests. Isatin is an indole derivative widely present in human and mammalian tissues. The synthetic versatility of isatin derived at C-2, C-3, and N positions has led to a wide variety of pharmacological responses including anticancer, antibacterial, antiviral, anti-HIV, anticholinesterase, antiinflammatory, antihypertensive, antihypoxic, antiulcer, anticonvulsant, COX-2 and carboxylesterase inhibitor activities [9–15]. Among these activities, anticancer activity studies on isatin derivatives have been accelerated after the FDA approval of C-3 derivative of isatin, sunitinib (SU11248) (Fig. 1). On the other hand, a literature survey on anticancer activity studies of N-alkylisatin derivatives reveals the importance of N-substitution. In addition, structure activity relationship (SAR) studies demonstrated that the introduction of an aromatic ring with 1–3 carbon atom linkers at the N atom enhanced the anticancer activity [16–18].

Structure of sunitinib (SU11248)
Thus, we describe herein the synthesis and anticancer activity of a series of isatin analogs 3a–3j and evaluation of their antitumor activity against breast cancer (MCF-7) cell line [19].
Substituted anilides have also been studied for their cytotoxic activity and the results suggested that the activity depends on the nature and the positions of the substituents on the N-phenyl ring [20]. In this context, a group of N-phenylisatin-1-acetamide derivatives bearing diverse substitutions with different electronic and hydrophobic natures on the phenyl ring were designed and synthesized. Chemical structures of the title compounds were confirmed by IR, CHNS, 13C NMR, 1H NMR, and ESI–MS spectra. The cytotoxic activity of the final compounds was screened against MCF-7 cell line.
Results and discussion
Chemistry
A small library of ten isatin analogs 3a–3j was synthesized following the reaction outlined in Scheme 1. The synthesis of the title compounds was realized in three steps.

Firstly, 1,3-dihydroindol-2-one derivatives 1a–1d were synthesized, following the method reported for the synthesis of isatin [21, 22]. For this, isatin (1) and substituted anilines were dissolved in warm ethanol in the presence of 1–2 drops of glacial acetic acid and refluxed for 2–3 h. After standing for approximately 24 h at room temperature, product was obtained. Next, the substituted anilines and 2-chloroacetylchloride (2) were reacted in the presence of glacial acetic acid in ice cold condition leading to the formation of chloroanilides 2a–2f [23]. Then, to the appropriate Schiff bases of isatin 1a–1d in 8–10 cm3 of anhydrous DMF, K2CO3 was added and stirred at room temperature for 1 h. After completion of 1 h, the solution turned red brown in color. Appropriate cholroanilides 2a–2f and KI were then added to this solution drop wise and heated at 60 °C for 5.5–9 h. The reaction was monitored by TLC and after completion of the reaction gives the entitled compounds 3a–3j [24–26].
The IR (KBr) spectra of all the synthesized compounds exhibited much related frequencies indicating the incidence of amide structures for the title compounds. The IR spectra of the synthesized compounds exhibited the acetamide carbonyl stretching (C=O) band at 1731–1607 cm−1. Similarly, N–H stretching band of amide was seen between 3400 and 3300 cm−1. The C=N band in IR spectra of all the compounds appeared at 1605–1550 cm−1 which is similar as that of the ordinary C=N absorption. The 1H NMR spectra of the title compounds 3a–3j were recorded in DMSO-d 6 solution which entirely concorded with the predictable resonance signals in terms of chemical shifts and integrations. 1H NMR spectra of the title compounds 3a–3j showed a broad singlet of one proton assigned to NH proton at δ = 10.96–10.82 ppm. Depending on the nature of the substituents and substitution patterns on the N-phenyl ring, the aromatic protons of certain compounds 3a–3j were observed in distinct chemical shifts with expected splitting patterns as doublets, triplets, or multiplets integrating more than one proton due to the close chemical shifts ranging from 6.36 to 7.58 ppm. In the aliphatic region, a broad singlet of two protons assigned to the methylenic proton of N–CH2–CO at range 4.61–4.67 ppm was observed for the compounds 3a–3j. A broad singlet of three protons assigned to methoxy OCH3 at 3.71 and 3.72 ppm was observed for the substituents at the N -phenyl ring of the compounds 3a and 3g, respectively. A broad singlet of three protons assigned to methyl protons of −CH3 at 1.99–2.28 ppm was observed for the substituents at the N-phenyl ring of the compounds 3b, 3c, 3d, 3f, 3g, 3h, 3i, and 3j. The NMR data of the title compounds 3a–3j are summarized in experimental sections. In 13C NMR spectra, aromatic carbons were observed in the region of 112.88–161.28 ppm and aliphatic carbon was observed (−CH3) in the region of 17.37–21.11 ppm and methoxy (−OCH3) was observed in the region of 55.31–55.47 ppm. The characteristics of carbon for C=O were observed in the region of 164.81–164.00 ppm and −CH2 was observed 45.17–45.67 ppm. The structural confirmations of these compounds were determined using ESI–MS. The physico-chemical data are summarized in Table 1.

Biological screening
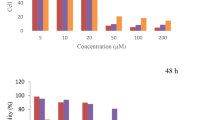
In this work, anticancer activity of the newly synthesized isatin analogs 3a–3j was evaluated against breast (MCF-7) cancer cell line, using sulforhodamine-B (SRB) assay method [27]. The GI 50 concentration for each compound was calculated with reference to a control sample, which represents the concentration that results in 50 % decrease of cell growth/proliferation after 48 h incubation in the presence of the drug. For each compound, GI 50 was calculated from dose–response curves that were generated with data obtained from two independent experiments carried out each in triplicate values. GI 50, LC 50, and TGI values were calculated in μM and presented in Table 2. The data of adramycin (doxrubiacin) were included as reference standard. The resultant data showed that, among ten synthesized compounds 3a–3j, four compounds exhibited demonstrative antiproliferative effects on MCF-7 breast cancer cell line (compounds 3b, 3c, 3e, and 3f showed the best activities compared to reference adramycin (doxorubicin) and GI 50 <0.02 uM). Rest of the compounds 3d, 3g, and 3h also showed moderate activity compared to reference standard adramycin (GI 50 = 0.13 uM, GI 50 = 0.03 uM, and GI 50 = 0.09 uM, respectively) but the compounds 3a, 3i, and 3j have showed less anticancer activity against MCF-7 cell line. The results also indicated that the hydrophobic group (−CH3) with 2nd phenyl ring substituted with 3-Cl or 4-Cl showed greater growth inhibitory potency. The plot of percentage control growth versus drug concentration (μg/cm3) shows the effective drug concentration on the MCF-7 human breast cancer cell line in Fig. 2.

The plot of percentage control growth versus drug concentration (μg/cm3) shows the effective drug concentration on the MCF-7 human breast cancer cell line
It was evident that incorporation of one or two hydrophobic methyl group/groups in the phenyl ring attached to the isatin moiety by the linker CH2CONH resulted in very high activity comparable to the standard. It was interesting to note that a combination of the chloro group (substituted at the para position of the second phenyl ring) along with one methyl group in the first phenyl ring showed a similar pattern of activity. It was also observed that absence of any substitution in the first phenyl ring with chloro group substituted in the second phenyl ring showed activity comparable to standard, as in case of compound 3e. However, when hydrophobicity of the total molecule was increased by incorporation of two methyl groups in the first phenyl ring and one chloro group at para position of the second phenyl ring, activity decreased significantly. SAR studies suggest that, an electron withdrawing halogen group is an essential feature for activity. It was observed that a combination of an electron withdrawing group with a hydrophobic electron releasing methyl group also played an important part in contribution to activity.
Docking study: binding mode analysis
The epidermal growth factor receptor (EGFR) is highly expressed in many types of cancers, especially breast, colon, and bladder cancers [28]. Targeting this receptor represents a good strategy for the design of new anticancer drugs [29]. Thus, molecular docking studies for all the synthesized compounds 3a–3j into ATP-binding site of EGFR–TKs were performed to investigate the ability of these novel derivatives to inhibit these tumorogenic agents and explore their binding mode in the active site of EGFR–TKs. Considering the well-obtained in vitro results, a profound docking study was carried out to consider the possible binding mode of the highest active compound 3f inside the active site of EGFR–TKs using Schrodinger Suite software [30, 31]. In case of PDB ID 1M17 complexed with the cocrystallized ligand erlotinib (AQ4), the ligand erlotinib shows H-bond interactions with Met 769 as depicted in Fig. 3a, b. The interactions with Leu 820, Leu 768, Leu 694, and Gly 772 are very important for stable binding of AQ4 in the active site of 1M17 with a docking score of −8.74. Firstly, to validate the Glide software, we modeled the interaction between AQ4 and PDB ID 1M17. Superimposition of the experimental bound (co-crystallized) conformation of AQ4 and that predicted by Glide is shown in Fig. 3a. Glide successfully reproduced the experimental binding conformations of AQ4 in the binding pocket of 1M17 with an acceptable root-mean-square deviation (RMSD) of 1.737 Å (>3Å). Compound 3f with the highest docking score in the active site of 1M17 is visualized in its three-dimensional mode in Fig. 4a and the residues involved in inter-atomic contact has been shown in Fig. 4b. Analysis of the docking pose revealed that in compound 3f the isatin scaffold is similarly oriented in the binding site as the quinazoline moiety of erlotinib in the active site of 1M17. Here in, the isatin moiety interacts with multiple amino acid residues Met769, Leu820, Leu 764, Ala719, Lys721, Thr 766, Thr 830, and Gly722. The compound also forms a strong H-bond interaction between C=O group of the isatin ring and the C=O group present in residue Met 793 (COIsatin − C=O Met793 = 1. 889 Å). Thus, it is evident that these interactions and hydrogen bond may be responsible for the high docking score of compound 3f (−8.35) which was found to be comparable than that of the co-crystallized ligand AQ4 (−8.74). The docking scores of the synthesized compounds are given in Table 3.

a Redocked conformer of AQ4 (erlotinib) in the active site of the protein EGFR (PDB ID 1M17); b 2D representation of the ligand erlotinib (AQ4)

a Molecular model of most active compound 3f in the protein EGFR (protein data bank ID 1M17) (Glide XP Score −8.35); b schematic (2D) representation of interactions of compound 3f in the binding pocket of the protein EGFR
Virtual ADME study
We have analyzed 16 physical descriptors and pharmaceutically significant properties of isatin analogs using Qikprop v3.0 tool of Schrodinger software, among which major descriptors reported here are required for predicting the drug-like properties of molecules. These properties are
-
1.
Molecular weight (MW) (150–650).
-
2.
Octanol/water partition coefficient (Log Po/w) (−2 to 6.5).
-
3.
Aqueous solubility (QPlogS) (−6.5 to 0.5).
-
4.
Apparent MDCK cell permeability (QPPMDCK) (<25 poor, >500 great).
-
5.
Brain/blood partition coefficient (QPlogBB) (−3.0 to 1.2).
-
6.
Percent human oral absorption (≥80 % is high, ≤25 % is poor).
All the structures showed significant values for the properties analyzed and exhibited drug-like characteristics based on Lipinski’s rule of 5 [32–34]. The ADME values of newly designed compounds are given in Table 4. The first three properties are based on Lipinski’s rule of five, molecular weight (MW) less than 650, partition coefficient between octanol and water (logPo/w) between −2 and 6.5 and solubility (QPlogS) greater than −7. Brain/blood partition coefficient (QPlogBB) parameter indicated about the ability of the drug to pass through the blood–brain barrier which is mandatory for inhibition of 1M17. The QPPMDCK predicted apparent MDCK cell permeability in nm/s. MDCK cells are considered to be a good mimic for the blood–brain barrier. The higher the value of MDCK cell, the higher is the cell permeability. All the designed compounds showed ADME properties in acceptable range.
Conclusion
A series of new isatin analogs have been synthesized and characterized by IR, 13C NMR, 1H NMR, and mass spectroscopy. All the newly synthesized compounds were evaluated for their potential as antitumor lead compounds in vitro on human breast cancer cell line MCF-7. Most of the tested compounds showed potent antitumor activity. Molecular docking studies for the synthesized compounds were performed and molecular interactions were explored. From the modeling studies, it can be concluded that incorporation of hydrophobic and electron withdrawing/electron releasing groups in both the aryl moieties attached to the isatin pharmacophore is essential; however, hydrophobicity should be optimal for activity. These results also support the clinical promise of these compounds as a component of therapeutic strategies for cancer, for which high concentrations of chemotherapeutic agents are always a major limitation. Moreover, the uncomplicated methodology used for the preparation of these potent compounds allows for obtaining sufficient amounts for more in-depth clinical studies. The simplicity of the prepared compounds with their potent anti-breast cancer activity highlights the potential of elegant organic synthesis for the discovery of new drug leads.
Experimental
All the chemicals were purchased from the following companies: Sigma-Aldrich Chemicals, Rankem, and Spectrochem PVT LTD. Precursors 1a–1d [21, 22] and 2a–2f [23] were synthesized according to published methods. The purity and homogeneity of the compounds were assessed by TLC performed on Merck silica gel 60 F254 aluminum sheets using ethyl acetate:n-hexane (3:7) as eluents. Iodine chamber and Shimadzu (UV-254) spectrometer were used for visualization of TLC spots. Ashless Whatmann No. 1 filter paper was used for vacuum filtration. Melting points were determined on an OPTIMELT automated system apparatus. Infrared spectra were determined as KBr solid disks or pellets, recorded on SHIAMADZU FT/IR 8400 and were reported in cm−1. 13C NMR spectra were recorded on a Bruker Avance 100 MHz spectrometer using CDCl3 as solvent. Chemical shifts were reported in ppm using TMS as the internal standard. 1H NMR spectra were recorded on a Varian spectrometer 400 MHz using DMSO-d 6 as solvent. Chemical shifts are expressed in δ values (ppm) relative to tetramethylsilane (TMS) as an internal standard. The peak multiplicity is reported as singlet (s), doublet (d), triplet (t), and multiplet (m). Mass spectra were recorded using (ESI+) MS. Elemental analysis was performed on a Vario Micro cube CHNS analyzer of Elementar and values were within the acceptable ±0.3 % limit of the calculated values.
All the novel isatin analogs were screened on MCF-7 breast cancer cell line by the Advanced Centre for Treatment Research and Education in Cancer (ACTREC) Mumbai, India. The cell viability was measured using SRB assay. All the environmental conditions were maintained throughout the experiment for all the groups. The assay was performed in triplicate for each of the compounds. The growth curve was plotted against μg/cm3 drug concentration of compounds and % control growth.
General procedure for the synthesis of 2-[2-oxo-3-(arylimino)indolin-1-yl]-N-arylacetamides 3a–3j
To the appropriate Schiff bases of isatin (10 mmol) in 8–10 cm3 of anhydrous DMF, K2CO3 (15 mmol) was added and stirred at room temperature for 1 h. After completion of 1 h, the solution turned red brown in color. Appropriate chloroanilides (10 mmol) and KI (2 mmol) were then added to this solution drop wise and heated at 60 °C for 5.5–9 h. After conforming the end of reaction by TLC (ethyl acetate: n-hexane 30:70), the mixture was poured into ice cold water. Precipitated crude product was filtered and washed thoroughly with cold water (3 × 200 cm3). Compounds were recrystallized from ethanol/water mixture (1:1). Reaction times, melting points, and yields are depicted in Table 1.
N-(4-Methoxyphenyl)-2-[2-oxo-3-(phenylimino)indolin-1-yl]acetamide (3a, C23H19N3O3)
Brownish yellow powder; IR (KBr): \(\overline{\nu }\) = 3286.81 (N–H stretching), 3074, 2939.61 (Ar–CH stretching), 1733.10, 1722 (C=O stretching), 1603.86 (C=N stretching) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ = 3.717 (s, 3H, −OCH3), 4.613 (s, 2H, −CH2−), 6.42 (d, 1H, J = 7.6 Hz, Ar–H), 6.79–6.83 (t, 1H, J = 7.6 Hz, Ar–H), 6.908–6.865 (dd, 2H, J = 8.4, 8.8 Hz, Ar–H), 7.02–7.00 (d, 2H, J = 7.6 Hz, Ar–H), 7.16–7.09 (m, 1H, Ar–H), 7.27 (d, J = 7.6 Hz, 1H, Ar–H), 7.521–7.403 (m, 5H, Ar–H), 10.228 (s, 1H, −NH−) ppm; 13C NMR (100 MHz, CDCl3): δ = 45.08, 55.47, 110.35, 114.14, 115.88, 117.78, 121.99, 123.45, 125.64, 126.29, 129.52, 130.31, 134.46, 146.58, 149.97, 153.77, 156.74, 163.91, 164.42 ppm; MS (ESI): m/z = 386.0 ([M + 1)+], calc for C23H19N3O3 385.42.
N-(2,5-Dimethylphenyl)-2-[2-oxo-3-(phenylimino)indolin-1-yl]acetamide (3b, C24H21N3O2)
Pale yellow powder; IR (KBr): \(\overline{\nu }\) = 3282.95 (N–H stretching), 3019.66, 2922.25 (Ar–CH stretching), 1728.10, 1688.12 (C=O stretching), 1605.79 (C=N stretching) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ = 2.153 (s, 3H, CH3), 2.236 (s, 3H, CH3), 4.659 (s, 2H, −CH2−), 6.426 (d, 1H, J = 7.6 Hz, Ar–H), 6.800–6.838 (dd, 1H, J = 8.0, 7.2 Hz, Ar–H), 6.924 (d, 1H, J = 7.2 Hz, Ar–H), 7.089–7.120 (m, 2H, Ar–H), 7.007 (d, 2H, J = 7.6 Hz, Ar–H), 7.187 (s, 1H, Ar–H), 7.265–7.337 (m, 1H, Ar–H), 7.426–7.520 (m, 3H, Ar–H), 9.668 (s, 1H, −NH−) ppm; 13C NMR (100 MHz, CDCl3): δ = 17.37, 21.11, 45.17, 110.38, 116.37, 117.04, 120.26, 122.99, 123.80, 124.85, 126.10, 126.32, 129.09, 131.01, 134.46, 136.68, 137.45, 146.45, 148.91, 153.82, 164.07, 164.81 ppm; MS (ESI): m/z = 384.0 ([M + 1)+], calc for C24H21N3O2 383.44.
2-[3-(2,5-Dimethylphenylimino)-2-oxoindolin-1-yl]-N-(o-tolyl)acetamide (3i, C25H23N3O2)
Yellow powder; IR (KBr): \(\overline{\nu }\) = 3212.35 (N–H), 3048.84, 2920.32 (Ar–CH), 1602.90 (C=N), 1708.10, 1658 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ = 1.993 (s, 3H, −CH3), 2.218 (s, 3H, −CH3), 2.282 (s, 3H, −CH3), 4.686 (s, 2H, −CH2−), 6.365 (d, 1H, J = 7.6 Hz, Ar–H), 6.666 (s, 1H, Ar–H), 6.843 (dd, 1H, J = 7.2, 7.6 Hz, Ar–H), 6.999 (d, 1H, J = 7.6 Hz, Ar–H), 7.12 (d, 2H, J = 7.6 Hz, Ar–H), 7.158 (dd, 1H, J = 7.2, 1.6 Hz, Ar–H), 7.239 (d, 2H, J = 7.6 Hz, Ar–H), 7.375 (d, 1H, J = 7.6 Hz, Ar–H), 7.451 (dd, 1H, J = 8.0, 7.2 Hz, Ar–H), 9.759 (s, 1H, −NH−) ppm; 13C NMR (100 MHz, CDCl3): δ = 17.23, 17.72, 21.03, 45.63, 110.10, 116.29, 116.83, 122.55, 122.74, 123.93, 125.57, 126.13, 126.25, 126.80, 128.92, 130.62, 130.91, 134.37, 135.05, 136.58, 145.86, 148.84, 153.40, 163.82, 164.68 ppm; MS (ESI): m/z = 398.1 ([M + 1)+], calc for C25H23N3O2 397.47.
N-(2,5-Dimethylphenyl)-2-[3-(2,5-Dimethylphenylimino)-2-oxoindolin-1-yl]acetamide (3j, C26H25N3O2)
Shiny yellow crystals; IR (KBr): \(\overline{\nu }\) = 3281.99 (N–H), 3031.23, 2912.29 (Ar–CH), 1606.79 (C=N), 1730.10, 1665.90 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ = 1.991 (s, 3H, −CH3), 2.164 (s, 3H, −CH3), 2.237 (s, 3H, −CH3), 2.281 (s, 3H, −CH3), 4.671 (s, 2H, −CH2−), 6.362 (d, 1H, J = 7.2 Hz, Ar–H), 6.662 (s, 1H, Ar–H), 6.821–6.859 (t, 1H, J = 7.6 Hz, Ar–H), 6.925 (d, 1H, J = 7.6 Hz, Ar–H), 6.996 (d, 1H, J = 7.6 Hz, Ar–H), 7.095–7.120 (m, 2H, Ar–H), 7.205–7.249 (dd, 2H, J = 9.6, 8.0 Hz, Ar–H), 7.432–7.468 (dd, 1H, J = 8.0, 6.4 Hz Ar–H), 9.6904 (s, 1H, −NH−) ppm; 13C NMR (100 MHz, CDCl3): δ = 17.42, 19.34, 19.96, 21.16, 45.67, 110.33, 116.44, 117.03, 117.75, 121.59, 123.01, 123.91, 126.20, 126.34, 130.15, 131.02, 133.48, 134.47, 134.91, 136.69, 137.51, 146.26, 153.65, 164.00, 164.52 ppm; MS (ESI): m/z = 412.2 ([M + 1)+], calc for C26H25N3O2 411.19.
In vitro anticancer activity evaluation by SRB assay
Anticancer activity of the newly synthesized isatin analogs was evaluated against MCF-7 breast cancer cell line using sulforhodamine-B (SRB) assay method [35–38]. Human breast cancer cell line (MCF-7) evaluation was performed at the Advanced Centre for Treatment, Research and Education in Cancer (ACTREC), Mumbai, India and adriamycin (doxorubicin) was used as a reference drug. Reagents and chemicals were purchased from Sigma-Aldrich Chemical Company. The cell line was grown in RPMI 1640 medium containing 10 % fetal bovine serum and 2 mM l-glutamine. For present screening experiment, cells were inoculated into 96-well microtiter plates in 90 mm3 at plating densities, depending on the doubling time of the individual cell line [37, 38]. The microtiter plates were incubated at 37 °C in a carbon dioxide (CO2) incubator at 5 % CO2, 95 % air, and 100 % relative humidity for 24 h prior to the addition of experimental drugs. After 24 h, one plate of every cell line was fixed in situ with TCA, to represent a measurement of the cell population for every cell line at the time of drug addiction. Experimental drugs were solubilized in suitable solvent at 400-fold the preferred final maximum test concentration and stored frozen prior to use. At the time of drug addition, an aliquot of frozen concentrate was thawed and diluted to ten times the preferred final maximum test concentration with complete medium containing test article at a concentration of 10−3. Additional three, tenfold serial dilutions were made to provide a total of four drug concentrations plus control. Aliquots of 10 mm3 of these different drug dilutions were added to the appropriate microtiter wells already containing 90 mm3 of medium, resulting in the necessary final drug concentrations.
After compound addition, the cells were then incubated for 48 h with the test compounds. The experiment was completed by adding 30 % chilled TCA to the wells. Then, cells were fixed in situ by the gentle addition of 50 mm3 of cold 30 % (w/v) TCA (final concentration, 10 % TCA) and incubated for 60 min at 4 °C. The supernatant was discarded, the plates were washed five times with tap water and air dried. Sulforhodamine B (SRB) solution (50 mm3) at 0.4 % (w/v) in 1 % acetic acid was added to each of the wells, and plates were incubated for 20 min at room temperature. After staining, unbound dye was recovered and the residual dye was removed by washing five times with 1 % acetic acid. The plates were air dried. Bound stain was afterward eluted with 10 mM trizma base, and the absorbance was read on an Elisa plate reader at a wavelength of 540 with 690 nm reference wavelength [38].
Percent growth was calculated on a plate-by-plate basis for test wells relative to control wells. Percent growth was expressed as the ratio of average absorbance of the test well to the average absorbance of the control wells × 100: [(Ti − Tz)/(C − Tz)] × 100.
Using the six absorbance measurements [time zero (Tz), control growth (C), and test growth in the presence of drug at the four concentration levels (Ti)], the percentage growth was calculated at each of the drug concentration levels [39].
For concentrations for which Ti ≥ Tz (Ti − Tz) positive or zero = [(Ti − Tz)/(C − Tz)] × 100.
For concentrations for which Ti < Tz (Ti − Tz) negative = [(Ti − Tz)/Tz] × 100.
Growth inhibition of 50 %, GI 50 = [(Ti − Tz)/(C − Tz)] × 100.
GI 50 is that value of the drug concentration resulting in a 50 % reduction in the net protein increase (as measured by SRB staining) in control cells during the drug incubation. The drug concentration resulting in total growth inhibition (TGI) was calculated from Ti = Tz. The LC 50 is the drug concentration resulting in a 50 % reduction in the measured protein at the end of the drug treatment as compared to that at the beginning. During this, there is a net loss of 50 % cells following treatment and is calculated from [(Ti − Tz)/Tz] × 100 = −50 [40].
Statistical analysis
Values were calculated for each of these three parameters if the level of activity was reached; however, if the effect was not reached or was exceeded, the values for that parameter were expressed as greater or less than the maximum or minimum concentration tested. The experiment data were estimated using linear regression method of plots of the cell viability against the μg/cm3 drug concentration of tested compounds.
In silico molecular docking studies
Carcinogenesis is usually accompanied by over activation of receptor tyrosine kinase (RTK) signaling pathways, so inhibitors which block these receptors have a significant therapeutic potential in cancer treatment [41]. On this basis, RTK was selected as the target receptor for docking studies of the synthesized compounds. EGFR kinase domain in complex with 4-anilinoquinazoline inhibitor (AQ4) (PDB ID: IM17) [42] was used. For docking studies, all water molecules and ligands were removed from the protein for docking studies, and the protein was minimized by the protein preparation wizard. Partial atomic charges were assigned according to the OPLS_AA force field [43, 44]. A radius of 10 Å was selected for active site cavity during receptor grid generation. The docking of the ligands was performed with extra precision mode (XP) which is employed in GLIDE 5.0 module implemented in the Schrodinger LLC. The three-dimensional structures of ligands were drawn using the Maestro 8.5. The ligands were prepared using ligprep utility of Schrodinger suite with default parameters, the ligand energy minimized using OPLS 2005 (macromodel multiple minimization) and water as solvent. The ligands did not show the formation of any tautomers or isomers after ligprep and macromodel multiple energy minimizations. In the present study, extra precision (XP) glide docking procedure was validated by removing AQ4 from the crystallographic structures of the proteins with PDB ID: 1M17. We set up a very good concord between the localization of the inhibitors upon docking and from the crystal structures. The root mean square deviations (RMSDs) between the predicted conformation and the observed X-ray crystallographic conformation of compound AQ4 (erlotinib) equaled 1.737 Å (>3 Å). This indicates the reliability of the docking method in reproducing the experimentally observed binding mode for 1M17.
In silico ADME evaluation
In silico ADME evaluation of compounds was performed by ADME software Qikprop v 3.0 [33, 34]. Structures of the compounds were saved in the mol format using Chem Office software. Then, mol files of compounds were uploaded into the ADME predictor software for further evaluation. All descriptors were estimated at pH 7.4.
References
Solomon VR, Hua C, Lee H (2009) Med Chem 17:7585
Patel K, Karthikeyan C, Solomon VR, Narayana NSH, Lee H, Sahu K, Deora GS, Trivedi P (2011) Lett Drug Des Discov 8:308
Weber J, Antonietti M, Thomas A (2007) Macromolecules 40:1299
Somers-Edgar TJ, Taurin S, Larsen L, Chandramouli A, Nelson MA, Rosengren RJ (2011) Invest New Drugs 29:87
Yadav BD, Taurin S, Larsen L, Rosengren RJ (2011) Eur J Cancer 47:S355
Farag AM, Mayhoub AS, Eldebss TMA, Amr AGE, Ali KK, Abdel-Hafez NA, Abdulla MM (2010) Arch Pharm Chem Life Sci 343:384
Wang S, Liu Q, Zhang Y, Liu K, Yu P, Liu K, Luan J, Duan H, Lu Z, Wang, Wu E, Yagasaki K, Zhang G (2009) Mol Cancer 8:81
Al-Said MS, Bashandy MS, Al-Qasoumi SI, Ghorab MM (2011) Eur J Med Chem 46:137
Medvedev A, Buneeva O, Glover V (2007) Biol: Targets Ther 1:151
Hou L, Ju C, Zhang J, Song J, Ge Y, Yue W (2008) Eur J Pharmacol 589:27
Vine KL, Matesic L, Locke JM, Ranson M, Skropeta D (2009) Anti-Cancer Agents Med Chem 9:397
Martin P, Bouhfid R, Joly N, Ohmani F, Essassi EM, Lequart V, Banoub J, Kheddid K, Charof R, Massoui M (2008) Lett Org Chem 5:3
Matheus ME, Violante FA, Garden SJ, Pinto AC, Fernandes PD (2007) Eur J Pharmacol 556:200
Pandeya SN, Smitha S, Jyoti M, Sridhar SK (2005) Acta Pharm 5:27
Hyatt JL, Moak T, Hatfield MJ, Tsurkan L, Edwards CC, Wierdl M, Danks MK, Wadkins RM, Potter PM (2007) J Med Chem 50:1876
Vine KL, Locke JM, Ranson M, Pyne SG, Bremner JB (2007) Bioorg Med Chem 15:931
Sabet R, Mohammadpour M, Sadeghi A, Fassihi A (2010) Eur J Med Chem 45:1113
Matesic L, Locke JM, Bremner JB, Pyne SG, Skropeta D, Ranson M, Vine KL (2008) Bioorg Med Chem 16:3118
Grosse S, Mathieu V, Pillard C, Massip S, Marchivie M, Jarry C, Guillaumet G (2014) Eur J Med Chem 84:718
Yang Y, Shang P, Cheng C, Wang D, Yang P, Zhang F, Li T, Lu A, Zhao Y (2010) Eur J Med Chem 45:4300
Sridhar SK, Saravanan M, Ramesh A (2001) Eur J Med Chem 36:615
Azizian J, Mohammadi MK, Firuzi O, Razzaghi-asl N, Miri R (2012) Med Chem Res 21:3730
Soyer Z, Kilic FS, Erol K, Pabuccuoglu V (2004) Il Farmaco 59:595
Akgul O, Tarikogullari AH, Kose FA, Ballar P, Pabuccuoglu V (2013) Turk J Chem 37:204
Modi NR, Shah RJ, Patel MJ, Suthar M, Chauhan BF, Patel LJ (2011) Med Chem Res 20:615
Debnath B, Ganguly S (2015) Toxicol Environ Chem 97:741
Hu C, Solomon VR, Cano P, Lee H (2010) Eur J Med Chem 45:705
Gullick WJ (1991) Br Med Bull 47:87
Abdelgawad MA, Belal A, Omar HA, Hegazy L, Rateb ME (2013) Arch Pharm 346:534
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS (2004) J Med Chem 47:1739
Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, Fagan P, Marvin J, Padilla D, Ravichandran V, Schneider B, Thanki N, Weissig H, Westbrook JD, Zardecki C (2002) Acta Crystallogr Sect D: Biol Crystallogr 58:899
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2012) Adv Drug Deliv Rev 64:4
Debnath B, Ganguly S (2014) Asian J Pharm Clin Res 7:186
Ganguly S, Debnath B (2014) Med Chem 4:558
Patel K, Karthikeyan C, Moorthy NHN, Deora GS, Solomon VR, Lee H, Trivedi P (2012) Med Chem Res 21:1780
Solomon VR, Hu C, Lee H (2010) Eur J Med Chem 45:3916
Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) J Natl Cancer Inst 82:1107
Vichai V, Kirtikara K (2006) Nat Protoc 1:1112
Pandey K, Sharma PK, Dudhe R (2012) Asian J Pharm Clin Res 5:28
Qian J, Zhou CH, Qian Z, Nan FJ, Ye QZ (2001) Acta Pharmacol Sin 22:821
Belal A (2015) Bioorg Chem 59:124
http://www.rcsb.org/pdb/explore.do?structureId¼1M17 (accessed Feb 2015)
Hopkins AL, Ren J, Esnouf RM, Willcox BE, Jones EY, Ross C, Miyasaka T, Walker RT, Tanaka H, Stammers DK, Stuart DI (1996) J Med Chem 39:1589
Jorgensen WL, Maxwell DS, Tirado-Rives J (1996) J Am Chem Soc 118:11225
Acknowledgments
The authors acknowledge the University Grants Commission for providing financial support in the form of a Major Research Project. The authors are also grateful to Dr. Reddy’s Institute of Life sciences, Hyderabad for spectral characterization and ACTREC, Mumbai for anticancer screening facility. One of the authors (BD) gratefully acknowledges the University Grants Commission-Basic Science Research (UGC-BSR) for the award of fellowship during the work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Debnath, B., Ganguly, S. Synthesis, biological evaluation, in silico docking, and virtual ADME studies of 2-[2-Oxo-3-(arylimino)indolin-1-yl]-N-arylacetamides as potent anti-breast cancer agents. Monatsh Chem 147, 565–574 (2016). https://doi.org/10.1007/s00706-015-1566-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1566-9