
Abstract
Iron zirconium phosphate nanoparticles have been used as an efficient catalyst for the selective oxidation of a wide range of alcohols to their corresponding ketones or aldehydes using H2O2 as an oxidizing agent without any organic solvent or additive. The steric factors associated with the substrates had a significant influence on the reaction conditions. The results showed that this method can be applied for chemoselective oxidation of benzyl alcohols in the presence of aliphatic alcohols. The catalyst used in the current study was characterized by ICP-OES, XRD, N2 adsorption–desorption, NH3-TPD, Py-FTIR, SEM, and TEM.
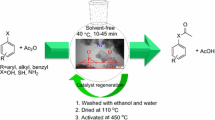
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
α‐Zirconium phosphate (ZP) is one of the most important compounds in inorganic chemistry, and the layered structure of this material has led to its use in a variety of different fields [1–3]. ZP behaves as a unique ion exchanger because of its exceptionally poor aqueous solubility, high thermal stability, resistance to radiation, and abrasive properties [4, 5]. The H+ of the P–OH moiety in ZP can be exchanged for various other ions, resulting in an enlargement of the interlayer distance [6–10]. Several studies pertaining to the successful exchange of this proton with various divalent and trivalent cations have been presented in the literature [10–14]. It has also been reported that ZP possesses excellent selectivity towards Pb2+, Zn2+, and Fe3+ as an ion exchanger [15–17]. Furthermore, ZP has been reported to exhibit antibacterial activity when it was loaded with Cu2+, Zn2+, or Ce3+ [5, 12–14]. There have also been several reports concerning the catalytic activities of ion-exchanged materials of this type, including the use of zinc zirconium phosphate (ZPZn) and copper zirconium phosphate (ZPCu) as catalysts in the acetylation of alcohols and phenols and the use of potassium iron zirconium phosphate as a catalysts in Friedel–Crafts benzoylation reaction [9, 18–24].
The selective oxidation of alcohols into the corresponding carbonyl compounds is an important research field since the corresponding aldehyde, ketone, or carboxylic derivatives serve as important and versatile intermediates for the synthesis of various chemicals, vitamins, drugs, and fragrances [25–27]. As a typical product of alcohol oxidation and a starting material, benzaldehyde (BzH) is an important chemical for the preparation of intermediates in the industry of dyestuff, agrochemicals, perfumery, and pharmaceuticals [27–29]. Thus, from economic and environmental viewpoints, there have been many recent publications emphasizing environmentally benign methods for the oxidation of alcohols, using molecular O2 or aqueous H2O2 as the oxidant, in the presence and/or absence of the solvent such as CuSO4 [25], CuBr2 [26], AMPA [28], MPA/V2O5/Al2O3 [29], TEMPO-IL/CuCl [30], Ni3[Fe(CN)6]2 [31], Pd/Fe@C [32], silicagel-TEMPO-NOx [33], CoTM4PyP-MT [34], PdO/SBA-15 [35] (TEAH)H2PW12O40 [36], Zn-Co-LDH [37], RuCl3·3H2O [38], VPO/HMS [39], AuRu/AC [40], oxidovanadium(V) complexes [41], Ag/SBA‐15 [42], DHPDMDO [43], H2WO4/[C8mim][NTf2] [44], SF-3-APTS-Fe(TClPP) [45], NaBrO3/[bmim]Br [46], DCMBF [47], WO4@PMO-IL [48], Cu-NHC-TEMPO complexes [49], Cu/AlO(OH) [50], SSA-SiO2/KMnO4 [51], copper/imidazolium/TEMPO [52], Ca(ClO)2/Al2O3 [53], PVPTB [54], Au/UiO-66 [55], PSFC [56], Au/Al2O3 [57], PMo11 M (M = Co, Mn, Ni) [58], Mn(salen)OAc [59], BPFC [60], KMnO4-aluminum silicate [61], KBr/oxone [62], PVP-H2O2 [63], TMP/KMnO4 [64], KMnO4-aluminum silicate [65], BQB [66], PS-TEMPO@Cu-IL [67], MPPC-alumina [68], CuBr2/TEMPO/bipyridine [69], KNa4[Ag(HIO6)2] [70], CPCC/alumina [71], and Fe3O4/MPS/PIL [72]. Homogeneous catalysts have drawbacks in terms of their corrosive nature, pollution of the product with catalyst, tedious catalyst separation and post synthesis disposals, and recovery from the effluents. On the other hand, the design of a new catalyst which gives excellent conversion with maximum selectivity for organic transformation is one of the challenges in the field of catalysis. H2O2 is a green and very clean oxidant for liquid phase oxidations, because it provides a high content of active oxygen species and water is the only by-product. This oxidant is much cheaper and safer than most other organic and inorganic oxidants and it is also readily available. With growing environmental concerns, one of the most promising ways to achieve these goals seems to be the use of green and insoluble catalysts or of ecofriendly solvent-free conditions. When an insoluble catalyst is used, it can be easily recovered from the reaction mixture by simple filtration and recycled and can be reused several times, making the process more economically and environmentally viable. Furthermore, the reported examples have demonstrated that heterogeneous catalysts typically require easier work-up procedures. With this in mind, and as part of ongoing work towards the development of efficient green catalysts for organic transformations [73, 74] with particular emphasis on the oxidation of alcohols [23], we report herein the use of iron zirconium phosphate (ZPFe) as an efficient catalyst for the mild and convenient selective oxidation of alcohols which was characterized by ICP-OES, XRD, BET, NH3-TPD, Py-FTIR, SEM, and TEM.
Results and discussion
Characterization
The ICP-OES analyses of ZP and ZPFe are shown in Table 1. The results obtained in the current study for ZPFe were compared with those reported previously in the literature [8–10]. Our results revealed that there was a negligible leach of iron ions into the reaction media after the reaction (i.e., following the first use of the catalyst).
Figure 1 shows the powder XRD patterns of the ZP and ZPFe materials. The results show some characteristic reflections in the 2θ range of 5°–40°. The diffraction peak in ZP at 2θ–12° was assigned to a d002 basal spacing of 7.5 Å between the planes, which was consistent with the patterns previously reported for ZP and its derivatives with a hexagonal crystal system [2]. It shows that the d-spacing of the (002) plane of ZPFe had increased, which indicated that the Fe3+ ions had intercalated into the interlayer of ZP and increased the d002 basal interlamellar spacing of ZP from 7.5 to 9.3 Å. It is well known that the ion radii of Fe3+ (0.64 Å) and hydrated Fe3+ (3.9 Å) are smaller than the basal spacing of ZP (7.5 Å) [75, 76]. These results therefore indicated that Fe3+ ions had inserted into the interlayer of ZP and increased the basal spacing of the modified ZP after the exchange [4, 9, 10, 17]. Taken together, these data indicated that ZPFe had been formed successfully. The XRD pattern of the ZPFe catalyst after the 8th run showed that the basal spacing of ZP was about 10.5 Å, which was only a little larger than that of the fresh ZPFe catalyst. This increase may have occurred because of the presence of less Fe3+ on the surface of ZP, and an increase in the number of water molecules between the layers following the seventh run (i.e., Fe3+ ions may have been washed off during the regeneration of the catalyst, Table 1).

XRD patterns of powder ZP (down), ZPFe (up)
Figure 2 shows the N2 adsorption–desorption isotherm of ZPFe, as a representative example, in the relative pressure range (p/p 0) of 0.1–1.0. The surface area of ZPFe was determined to be 120.5 m2/g. The isotherm for ZPFe shows three adsorption stages. The first of these stages was observed at p/p 0 < 0.36, whereas the second stage was observed in the range of 0.36 < p/p 0 < 0.92, and the third stage was observed at higher relative pressures (p/p 0 > 0.92). The N2 adsorption–desorption isotherm of ZPFe exhibited a typical type IV isotherm shape with a distinct hysteresis loop, which is characteristic of a mesoporous material [77]. The hysteresis loop (type H3) is associated with the occurrence of capillary condensation in the mesopores, which indicates the presence of a mesoporous structure in the ZPFe catalyst. The observed increase in adsorption at the higher p/p 0 value indicated the presence of larger mesopores in the sample [9, 10]. The surface area of ZPFe after the 8th run was found to be 64.2 m2/g.

N2 adsorption–desorption isotherm of ZPFe
Pyridine adsorption was used to determine the acidic sites using FTIR. Prior to the measurements, 20 mg of a catalyst was pressed in self-supporting disc and activated in the IR cell attached to a vacuum line at 350 °C for 4 h. The adsorption of pyridine was performed at 150 °C for 30 min. The excess of probe molecules was further evacuated at 150 °C for 0.5 h. The adsorption–evacuation was repeated several times until no changes in the spectra were observed (Fig. 3). The main bands observed over the samples are assigned according to the literature data [78, 79]. Pyridine-desorbed FTIR spectra of the ZPFe show the strong bands at 1632 and 1541 cm−1, indicating typical pyridinium ion. The band at 1488 cm−1 is a combination band between those at 1541 and 1444 cm−1, corresponding to Brønsted and Lewis acid sites, respectively [9, 10].

Pyridine‐desorbed FTIR spectra of the calcined ZPFe
Total acidity of the samples was determined by temperature-programmed desorption of ammonia (TPD-NH3) with a Quantachrome ChemBET 3000. Before the adsorption of ammonia, the samples were pre-treated in He at 250 °C for 30 min and then, 1 h at 350 °C and cooled to 100 °C. Then, ammonia was adsorbed on the samples for 1 h. The TPD-NH3 was carried out between 150 and 550 °C, at 10 °C/min, and analyzed by a thermal conductivity detector (TCD) for continuous monitoring of the desorbed ammonia. TPD-NH3 provides a quantitative estimation of the total number of acid sites and the distribution of acid strengths. Because of the strong basicity of NH3 gas, it was expected that all acid sites on the catalysts interact with NH3. The total amount of NH3 desorbed after saturation permits the quantification of the number of acid sites on the surface, while the position of the peak, desorption temperature, indicates the strength of the catalyst, i.e., the higher temperature of desorption, the stronger the acid strength [73]. The TPD-NH3 curves of ZPFe are shown in Fig. 4. ZPFe desorbed ammonia in a wide range of temperatures from 212 to 538 °C, which mostly corresponds to the medium and the strong acidic sites. The NH3 desorption peak at temperatures below 250 °C belongs to the physisorption/chemisorptions of NH3 molecules on weak acidic sites. The peak at about 250–450 °C shows the existence of intermediate strength acidic sites and finally the peak at 450–538 °C demonstrates the presence of strong acidic sites on the surface of ZPFe. Figure 4 shows that the desorption of ammonia starts at almost 212 °C, centered at 317. The NH3-TPD curves subsequently decreased with further increase in temperature and almost complete at 538 °C.

NH3-TPD profile of ZPFe
This indicates that ZPFe contains a considerable number of acid sites which is attributed to the presence of Fe3+ groups on the surface of zirconium phosphate layers and makes it suitable as a solid acid catalyst. The extent of desorptions is found to be ca. 1.9 mmol NH3/g of catalyst. A TPD experiment was carried out after the 8th cycle by recovering the catalyst, to magnify the difference from the fresh catalysts (Table 1).
The SEM image of ZP (Fig. 5a) revealed the presence of hexagonal plates with well-defined shapes and very smooth surfaces. Figure 5b, c shows the SEM images of ZPFe. These images revealed that the structure of ZPFe was much less ordered than that of ZP, and that the ZPFe particles had aggregated to form both sheets and spheres of different shapes and sizes [4, 10].

SEM images of regular morphology of prepared ZP (a), ZPFe fresh (b, c) and after 8th run (d)
Figure 6 shows the TEM images of ZPFe. It shows that ZPFe catalyst retained the original morphology of ZP (layered structure) and that the particles were approximately 150 nm in size. These images also showed nanoparticles of different sizes on the smooth surface of the ZP.

TEM images of regular morphology of prepared ZPFe fresh (a, b) (different magnification) and after the 8th run (c)
The presence of metallic crystal nanoparticles on the surface of ZP indicated that the iron deposited on the surface of the ZP had agglomerated. Similar observations have also been reported for zinc and cerium with ZP [12, 14, 24]. Figures 5d and 6c show the SEM and TEM images of the catalyst following its 8th run, respectively. Both of these images showed that the sheets and particles had conglomerated to a much greater extent following the 8th run because of the process used to regenerate the catalyst.
Oxidation reaction and expected mechanism
Initially, the benzyl alcohol (BzOH) was selected as the model substrate to determine the optimal conditions and the results are summarized in Table 2. Although the major product was BzH, benzoic acid was also formed as a side product (Table 2, entries 4, 5, 8, 11, 14, 15). It was found that by changing the reaction parameters such as time (Table 2, entries 1–5), mole ratio (Table 2, entries 6–8), temperature (Table 2, entries 9–11) or catalyst mol % (Table 2, entries 12–15), the selectivity and/or yield were changed.

As can be seen from Table 2, an increase in the reaction time (up to 15 min) causes an increase in yield of BzH as the only product but after that, benzoic acid was produced as a by-product and BzH yield was reduced to 65 %. When the mole ratio of BzOH: H2O2 reached at 1:3, highest yield was obtained and then, by further increasing of BzOH: H2O2 mol ratio, the yield was slightly increased, but selectivity toward BzH decreased sharply (Table 2, entries 2, 6–8). For finding the optimum reaction temperature for oxidation of BzOH, the reaction was performed at different temperatures. The yield of BzH was very low at room temperature even after 90 min (Table 2, entry 9). Table 2 shows that the temperature rising can accelerate the reaction (up to 50 °C), but increasing the temperature further results in a decrease in the yield, which might be due to the overoxidation of BzH to benzoic acid or self-decomposition of H2O2 at higher temperatures, leading to insufficient oxidation of BzOH (entries 2, 9–11). Hence, when the reaction was performed at 50 °C, BzOH: H2O2 mol ratio 1:3 and 15 min, best result was achieved (Table 2, entry 2). To evaluate the role of our catalyst, the oxidation reaction was performed when the ZP was used as the catalyst (Table 2, entry 17). Also, the oxidation reaction was performed in the absence of the any catalyst (Table 2, entry 16). No significant amount of BzH was detected indicating that H2O2 alone is unable to oxidize BzOH to BzH.
As shown in Table 3, a wide range of alcohols bearing either electron-donating or electron-drawing groups were successfully oxidized into their corresponding carbonyl compounds within short reaction times. Notably, the efficient transformations of primary alcohols into the desired aldehydes were observed without any overoxidation to the corresponding carboxylic acid (Table 3, entries 1–14). Both electron-donating and electron-drawing groups accelerated the oxidation reaction, but ortho-substituted substrates (Table 3, entries 2, 5, 7, 9) gave relatively lower yields compared to the corresponding para-isomer due to steric hindrance [26, 50, 52]. Compared to benzylic alcohols, aliphatic alcohols showed relatively low reactivity toward oxidation (Table 3, entries 22–25). It should be noted that under this reaction condition, various secondary alcohols were oxidized into their corresponding ketone in fair yields (Table 3, entries 15–21, 23, 25). To investigate the selectivity of this method, a mixture of 1-hexanol and BzOH was subjected to oxidation, BzH was produced in 85 % yield and only 5 % of 1-hexanal was detected. Such selectivity can be considered as a useful practical achievement in oxidation of alcohols.

Based on the observation, the typical mechanism proposed for ZPFe-catalyzed oxidation of alcohols with H2O2 is given in Scheme 1. At first, the Fe Lewis acid site (on the surface of ZPFe) interacts with H2O2 to form (I) and it reacts with alcohol to give an intermediate (II) which gives the corresponding carbonyl compound and regenerated active sites by two steps of dehydration. Hence, any steric hindrance around hydroxyl group of alcohols such as presence of substitutes at the ortho position (Table 3, entries 2, 5, 7, 9) or more hindered secondary alcohols (Table 3, entries 15–21) causes longer reaction time and lower yields. Almost similar behavior was reported before [26, 50, 52].

The reusability of the ZPFe catalyst was investigated under the optimum reaction conditions for the oxidation of alcohols, and the results are shown in Table 4. The elemental composition of the catalyst remained largely unchanged following its 8th run, although the amount of iron in the catalyst was reduced by almost 75 % compared with the first run (Table 1). The recycled ZPFe catalyst gave a similar product yield to the freshly prepared catalyst up until the sixth cycle. All products were identified by their GC–MS (GC-Mass, Agilent 5975C) and 1H NMR spectra (Bruker-Avance AQS 400 MHz spectrometer; in CDCl3) with authentic samples [23, 28, 47, 49, 51].
A comparison of the catalytic efficiency of ZPFe with selected previously known catalysts is collected in Table 5. BzOH was oxidized to BzH in less than 15 min at 50 °C in 90 % isolated yield using the present protocol (Table 5, entry 17). Of course, the reaction conditions are different, but the catalyst used in this study (ZPFe) might be one of the best catalysts with regard to lower reaction time and temperature, better BzH yield and selectivity toward BzH as the major product. The catalysts can also be easily regenerated for further applications.

Conclusion
In summary, we have reported the catalytic performance of water-insoluble ZPFe in the oxidation of alcohols using H2O2. The catalyst was characterized by various methods and results showed good agreement with the literature. ZPFe showed outstanding catalytic performance with excellent conversion of BzOH and selectivity to BzH at 50 °C for 15 min. The selectivity remained unchanged for all the reactions, although the yields were found to be affected by the steric hindrance. Alcohols having bulky groups required a longer reaction time compared with others. The results clearly reveal that this method can be applied for chemoselective oxidation of BzOHs in the presence of aliphatic alcohols. This procedure is environmentally benign, general, efficient, high yielding, safe, and operationally simple.
Experimental
All chemicals and solvents were purchased from Sigma-Aldrich and Merck Chemical Companies and used without further purification. The chemical composition of the ZPFe catalyst was evaluated at different stages of the reaction (i.e., before and after the catalytic reaction) by ICP-OES using an Optima 7300 V ICP-OES spectrometer (PerkinElmer). The samples were ground into a fine powder and analyzed by XRD on a Philips X’pert X-ray diffractometer. The specific surface areas of the samples were determined from their N2 adsorption–desorption isotherms using the Brunauer–Emmett–Teller (BET) method on a Quantachrome ChemBET 3000 instrument. Each sample was degassed at 400 °C for 2 h before being analyzed to remove any adsorbed species from their surfaces. The BET surface areas of the materials were estimated from their N2 adsorption–desorption isotherms. The surface morphologies of the ZP and ZPFe materials were studied by SEM on a Philips XL scabbing electron microscope (Philips). TEM images of ZPFe were obtained on a CENTRA 100 TEM system (Zeiss). All yields refer to isolated products after purification. All products were characterized by comparison with authentic samples and by spectroscopy data FT-IR and 1H NMR analysis. 1H NMR spectra were recorded on a Bruker-Avance AQS 400 MHz spectrometer. The spectra were measured in CDCl3 unless otherwise stated, relative to TMS (0.00 ppm). FT-IR spectra were recorded on a Jasco-680 spectrophotometer.
Catalyst synthesis
All of the reagents and solvents used in the current study were purchased from Merck Chemical Company and used without further purification. The catalyst was prepared according to previously published procedures, with minor modifications [2, 8–10]. ZP was prepared according to the following procedure. ZrOCl2·8H2O (5 g) was heated at reflux in a solution of 50 cm3 H3PO4 (12 mol/dm3) for 24 h. The resulting mixture was cooled to ambient temperature to give a suspension, which was filtered, and the filter cake was then washed with a solution of H3PO4 (0.1 mol/dm3) until the filtrate was free of chloride ions. The filter cake was then washed several times with distilled water until the pH of the filtrate was neutral. The solid was then collected and dried in an oven at 110 °C for 24 h [2]. ZPFe was prepared through an ion exchange reaction [8–10]. Briefly, 3 g ZP was dispersed in 50 cm3 deionized water at 50 °C, and the resulting suspension was treated with 100 cm3 of a solution of Fe(OAc)3 (0.1 mol/dm3) in water (excess amount of Fe3+). This mixture was then heated at reflux for 4 d. It is noteworthy that the acetate ion performed effectively as a base to keep the hydrogen ion concentration in solution sufficiently low to achieve high loadings of the catalyst [8]. A complete exchange between the cations and the hydrogen ions of the P–OH groups could not be achieved in less than 3 d or at temperatures below 80 °C [13]. The resulting slurry was filtered hot to give a light yellow solid, which was washed with distilled water until no Fe3+ ions could be detected in the filtrate (i.e., until the filtrate was colorless). The solid product was then dried at 100 °C for 24 h before being calcined at 600 °C for 4 h to give the final product, Fe1/3[Zr2(PO4)3], as a pale yellow solid (Scheme 2).

General experimental procedure for the oxidation reaction
Substrate (5 mmol) and ZPFe (0.5 mol %) were added into a 25 cm3 two-necked flask. It was heated in an oil bath to 50 °C and then, 30 % H2O2 (0.015 mol) was added slowly with continuous stirring for the specified time. The reaction progress was monitored by GC. At the end, the reaction mixture was cooled to room temperature and then, the catalyst was removed from the reaction mixture by centrifuge. After that, the organic layer was separated from the aqueous phase by extraction with n-hexane and dried over anhydrous CaCl2. The identity of reaction products was confirmed by FT-IR, GC–MS, and 1H NMR.
Recyclability studies of catalyst
To examine the recyclability of the catalyst, the used ZPFe was recovered from the reaction media and re-used. For recycling, after the first use, the catalyst was separated from the reaction mixture by centrifugation, washed sequentially with ethanol and water before being dried at 110 °C for 2 h, and then activated at 450 °C for 2 h.
References
Gan H, Zhao X, Song B, Guo L, Zhang R, Chen C, Chen J, Zhu W, Hou Z (2014) Chin J Catal 35:1148
Sun L, Boo WJ, Sue HJ, Clearfield A (2007) New J Chem 31:39
Hajipour AR, Karimi H (2014) Mater Lett 116:356
Sun Z, Liu Z, Xu L, Yang Y, He Y (2004) Stud Surf Sci Catal 154:1060
Shi QS, Tan SZ, Ouyang YS, Yang QH, Chen AM, Li WR, Shu XL, Feng J, Feng J, Chen YB (2011) Adv Mater Res 150:852
Khare S, Chokhare R (2012) J Mol Catal A: Chem 353–354:138
Wang Q, Yu J, Liu J, Guo Z, Umar A, Sun L (2013) Sci Adv Mater 5:469
Clearfield A, Kalnins JM (1978) J Inorg Nucl Chem 40:1933
Gawande M, Deshpande S, Sonavane S, Jayaram R (2005) J Mol Catal A: Chem 241:151
Khare S, Chokhare R (2011) J Mol Catal A: Chem 344:83
Giannoccaro P, Gargano M, Fanizzi A, Ferragina C, Aresta M (2005) Appl Catal A 284:77
Cai X, Dai G-J, Tan S-Z, Ouyang Y, Ouyang Y-S, Shi Q-S (2012) Mater Lett 67:199
Yang Y, Dai G, Tan S, Liu Y, Shi Q, Ouyang Y (2011) J Rare Earths 29:308
Dai G, Yu A, Cai X, Shi Q, Ouyang Y, Tan S (2012) J Rare Earths 30:820
Zhang QR, Du W, Pan BC, Pan BJ, Zhang WM, Zhang QJ, Xu ZW, Zhang QX (2008) J Hazard Mater 152:469
Costantino U, Szirtes L, Kuzmann E, Megyeri J, Lázár K (2001) Solid State Ionics 141–142:359
Gobechiya ER, Kabalov YK, Orlova AI, Trubach IG, Bykov DM, Kurazhkovskaya VS (2004) Crystallogr Rep 49:390
Wang XY, Hua WM, Yue YH, Gao Z (2013) Chem J Chin Univ 34:1913
Barhon Z, Saffaj N, Albizane A, Azzi M, Mamouni R, El Haddad M (2012) J Mater Environ Sci 3:879
Pylinina A, Mikhalenko I (2013) Russ J Phys Chem 87:372
Pylinina A, Mikhalenko I (2011) Russ J Phys Chem 85:2109
Hajipour A, Karimi H, Karimzadeh M (2014) Monatsh Chem 145:1461
Hajipour AR, Karimi H (2014) Chin J Catal 35:1529
Hajipour AR, Karimi H (2014) Chin J Catal 35:1982
Ahmad JU, Räisänen MT, Leskelä M, Repo T (2012) Appl Catal A 411–412:180
Hu Z, Kerton FM (2012) Appl Catal A 413–414:332
Wang P, Cai J, Yang J, Sun C, Li L, Hu H, Ji M (2013) Tetrahedron Lett 54:533
Hosseinzadeh R, Tajbakhsh M, Khaledi H (2008) J Chin Chem Soc 55:239
Rao PSN, Rao KTV, Sai Prasad PS, Lingaiah N (2011) Chin J Catal 32:1719
Liu L, Ji LY, Wei YY (2008) Monatsh Chem 139:901
Ali SR, Chandra P, Latwal M, Jain SK, Bansal VK, Singh SP (2011) Chin J Catal 32:1844
Zhang H, Liu Y, Zhang X (2011) Chin J Catal 32:1693
Zhang H, Fu L, Zhong H (2013) Chin J Catal 34:1848
Zhou X, Ji H (2012) Chin J Catal 33:1906
Atashin H, Malakooti R (2014) J Chin Chem Soc 61:1039
Su H, Yang C (2014) Chin J Catal 35:1224
Zou X, Goswami A, Asefa T (2013) J Am Chem Soc 135:17242
Zhou C, Liu Y (2010) Chin J Catal 31:656
Mahdavi V, Hasheminasab HR, Abdollahi S (2010) J Chin Chem Soc 57:189
Villa A, E-chan-thaw C, Schiavoni M, Campisi S, Wang D, Prati L (2014) Chin J Catal 35:945
Ghorbani-Vaghei R, Veisi H, Amiri M (2007) J Chin Chem Soc 54:1257
Ma L, Jia L, Guo X, Xiang L (2014) Chin J Catal 35:108
Azarifar D, Najminejad Z, Khosravi K (2013) J Iran Chem Soc 10:979
Chatel G, Monnier C, Kardos N, Voiron C, Andrioletti B, Draye M (2014) Appl Catal A 478:157
Rahimi R, Ghoreishi SZ, Dekamin MG (2012) Monatsh Chem 143:1031
Shaabani A, Farhangi E, Rahmati A (2008) Monatsh Chem 139:905
Khazaei A, Raiatzadeh A, Rostami A (2007) J Chin Chem Soc 54:465
Karimi B, Rostami FB, Khorasani M, Elhamifar D, Vali H (2014) Tetrahedron 6114
Liu X, Xia Q, Zhang Y, Chen C, Chen W (2013) J Org Chem 78:8531
Babu SG, Priyadarsini PA, Karvembu R (2011) Appl Catal A 392:218
Mirjalili BF, Zolfigol MA, Bamoniri A, Zarei A (2004) J Chin Chem Soc 51:509
Chen C, Liu B, Chen W (2013) Synthesis 45:3387
Mojtahedi MM, Saidi MR, Bolourtchian M, Shirzi JS (2001) Monatsh Chem 132:655
Ali Zolfigol M, Hajjami M, Ghorbani-Choghamarani A (2012) J Iran Chem Soc 9:13
Zhu J, Wang PC, Lu M (2014) Appl Catal A 477:125
Bekhradnia AR, Zahir F, Arshadi S (2008) Monatsh Chem 139:521
Rautiainen S, Simakova O, Guo H, Leino AR, Kordás K, Murzin D, Leskelä M, Repo T (2014) Appl Catal A 485:202
Pathan S, Patel A (2013) Appl Catal A 459:59
Bahramian B, Mirkhani V, Moghadam M, Amin AH (2006) Appl Catal A 315:52
Özgün B, Yaylaoglu A, Şendil K (2007) Monatsh Chem 138:161
Lou JD, Gao CL, Li L, Fang ZG (2006) Monatsh Chem 137:1071
Moriyama K, Takemura M, Togo H (2014) J Org Chem 79:6094
Ghorbani-Choghamarani A, Azadi G (2011) J Iran Chem Soc 8:1082
Zhu C, Ji L, Wei Y (2010) Monatsh Chem 141:327
Lou J-D, Zhu L-Y, Wang L-Z (2004) Monatsh Chem 135:31
Laila A (2013) Monatsh Chem 144:307
Liu L, Liu D, Xia Z, Gao J, Zhang T, Ma J, Zhang D, Tong Z (2013) Monatsh Chem 144:251
Tajbakhsh M, Ghaemi M, Sarabi S, Ghassemzadeh M, Heravi MM (2000) Monatsh Chem 131:1213
Geißlmeir D, Jary WG, Falk H (2005) Monatsh Chem 136:1591
Bugarcěić Z, Novokmet S, Senić Z, Bugarcěić Z (2000) Monatsh Chem 131:799
Heravi MM, Kiakoojori R, Mirza-Aghayan M, Tabar-Hydar K, Bolourtchian M (1999) Monatsh Chem 130:481
Zhu J, Wang PC, Lu M (2013) Monatsh Chem 144:1671
Hajipour AR, Karimi H (2014) Appl Catal A 482:99
Hajipour AR, Karimi H (2014) Chin J Catal 35:1136
Bingham PA, Hannant OM, Reeves-Mclaren N, Stennett MC, Hand RJ (2014) J Non-Cryst Solids 387:47
Pandit AA, Shitre AR, Shengule DR, Jadhav KM (2005) J Mater Sci 40:423
Sing KS (1985) Pure Appl Chem 57:603
Corma A (1995) Chem Rev 95:559
Tyagi B, Chudasama CD, Jasra RV (2006) Appl Clay Sci 31:16
Acknowledgments
We gratefully acknowledge the funding support received for this project from the Isfahan University of Technology (IUT), IR, Iran.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hajipour, A.R., Karimi, H. & Masti, A. An efficient selective oxidation of alcohols with iron zirconium phosphate under solvent-free conditions. Monatsh Chem 147, 413–423 (2016). https://doi.org/10.1007/s00706-015-1499-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1499-3