
Abstract
The polymer complex [Hg(μ-mptrz)2] n and two binuclear complexes of {[H2en][Hg2(mptrz)4(μ-Br)2]} and {[H2en][Hg2(mptrz)4(μ-I)2]} were prepared from the reaction of 4-methyl-1,2,4-triazole-3-thiol (Hmptrz) and ethylene diamine (en) with HgCl2, HgBr2, and HgI2 in CH3OH, respectively. Complex [Hg(μ-mptrz)2] n was also prepared from the reaction of 4-methyl-1,2,4-triazole-3-thiol and ethylene diamine with Hg(OAc)2 and Hg(SCN)2 in CH3OH and Hg(NO3)2·H2O in a mixture of CH3OH/H2O. Analysis of these complexes was done by CHN elemental analysis, IR, UV–Vis, 1H and 13C NMR, and luminescence spectroscopy, as well as single-crystal X-ray diffraction. Thermal stabilities of these complexes were also studied by TGA/DTA analyses.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mercury(II) coordination compounds including nitrogen-, oxygen-, or sulfur-heterocyclic molecules, or a combination of these and an exocyclic thione (thioketo) group can be used for modeling of biosystematic interactions of mercury–cysteine thionate and also studying toxicological behavior [1–6]. 4-Methyl-1,2,4-triazole-3-thiol (Hmptrz) as a three dentate ligand can be coordinated to metal center through N and S atoms from triazole ring and thiol group. Under different experimental conditions, discrete and polymeric complexes with different coordination modes of this ligand can be formed, such as [Hg(mptrz)2Cl2] and [Hg(mptrz)2Br2] [7], [Pb(Hmptrz)4(NO3)2] and [Pb(μ-mptrz)2(H2O)] n [8], [Cu(μ4-mptrz)] n , [Cu(μ-Hmptrz)(μ-I)] n , [Cu12(μ4-mptrz)4(μ4-I)3(μ3-I)4(μ-I)] n ), and [Cu(μ-Hmptrz)(μ-DmptrzSS)I] n (DmptrzSS = 4,4′-dimethyl-3,3′-dithiodi-1,2,4-triazole) [9], [Cd(mptrz)2] n , [Cd(mptrz)X] n (X = I and Br), [Cd(mptrz)(μ-X)] n (X = Cl and Br) and [Cd3(μ3-OH)2(mptrz)4] n [10], [Ag2(mptrz)(µ3-X)] n (X = I and Br) and [Pb4(µ4-O)(mptrz)4(µ-X)2] (X = I and Cl) [11], [Ag2(NCS)2(μ-Hmptrz)2] n [12], and [Me3Sn(mptrz)] n and [Ph3Sn(mptrz)] n [13]. Recently some of us reported preparation and characterization of discrete [Hg(mptrz)2Cl2] and [Hg(mptrz)2Br2] complexes [7]. These compounds were synthesized by the reaction of 4-methyl-4H-1,2,4-triazole-3-thiol and HgCl2 and HgBr2 in methanol, respectively. Herein, HgX2 adducts of 4-methyl-4H-1,2,4-triazole-3-thiol in the presence of ethylene diamine have been synthesized and characterized as [Hg(μ-mptrz)2] n (1), {[H2en][Hg2(mptrz)4(μ-Br)2]} (2), and {[H2en][Hg2(mptrz)4(μ-I)2]} (3) complexes. Complex 1 is a polymeric compound while 2 and 3 have binuclear structures.
Results and discussion
Synthesis of 1, 2, and 3
Compound 1 was obtained from the reaction mixture of two equivalents of 4-methyl-1,2,4-triazole-3-thiol and one equivalent of ethylene diamine in CH3OH with one equivalent of HgX2 (X = Cl, OAc, and SCN in CH3OH or NO3 in a mixture of CH3OH/H2O) at room temperature. Compounds 2 and 3 were also obtained from the reaction mixture of two equivalents of 4-methyl-1,2,4-triazole-3-thiol and one equivalent of ethylene diamine in CH3OH with one equivalent of HgX2 (X is Br in 2 and I in 3) in CH3OH at room temperature. Suitable crystals of 1, 2, and 3 were obtained for X-ray diffraction measurement by methanol diffusion into a DMSO solution. The synthetic routes of these complexes are shown in Scheme 1.

Spectroscopic characterization of 1, 2, and 3
Infrared spectra in Table 1 show the vibration frequencies for free and coordinated Hmptrz and en ligands in compounds 1, 2, and 3. The infrared spectrum for compound 1 shows several bands in the region of 3,114–2,908 cm−1, which are assigned to the C–H stretching of the triazole ring and methyl group. The vibrational bands in the region of 3,104–2,572 cm−1 for compounds 2 and 3 are assigned to the N–H and C–H stretching vibrations of the ethylene diammonium and C–H stretching vibration of the triazole ring and methyl group.
The band observed at 2,642 cm−1 in the IR spectrum of free Hmptrz ligand is assigned to –SH stretching vibration. This band disappears for the complexes of 1, 2, and 3, showing the deprotonation of the Hmptrz ligand and formation of Hg–S bond [13].The bands observed in the range of 1,650–1,200 cm−1 are assigned to C–N, N–N, and C=N stretching vibrations and/or N–H deformation vibrations. The medium to strong vibrations in the region of 1,165–500 cm−1 are assigned to the C=S stretching and C=S, N–N, and C–N deformation vibrations [8–10, 14, 15]. The Hg–N stretching vibration for complex 1 is seen at 360 cm−1. The Hg–S stretching vibrations are found at 326 cm−1 for 1, 353 and 332 cm−1 for 2, and 351 and 335 cm−1 for 3. In addition, the bands observed at 258 and 252 cm−1, which are absent in the IR spectrum of 1, are assigned to Hg–X stretching vibrations for 2 and 3, respectively [16–21].
The electronic absorption spectra of dimethyl sulfoxide solutions of 1, 2, and 3 have broad bands in the region of 261-268 nm, which are assigned to the intra-ligand π–π* transitions [8], and a weaker band at 418 nm for 3 is assigned to the I → Hg LMCT (ligand to metal charge transfer) [22, 23].
The 1H NMR spectra of 1, 2, and 3 exhibited a singlet around 3.5 ppm for the methyl group and a singlet around 8.3 ppm for the =C–H of triazole ring. The 1H NMR spectra of 2 and 3 also exhibited a singlet around 2.9 ppm for the –C–H of ethylene diammonium. For complexes 2 and 3, a broad singlet signal which was exchangeable with D2O around 4 ppm is due to the two –NH3 + groups. The 1H NMR data showed that the signal of the –SH proton (13.65 ppm) in the spectrum of the ligand is absent in the spectra of title compounds, indicating the removal of the –SH proton and the formation of Hg–S bonds [13]. The 13C NMR spectra of 1, 2, and 3 showed a singlet at around 33 ppm for the methyl group and two singlets at 144–161 ppm for the triazole ring. The 13C NMR spectra of 2 and 3 also exhibited a singlet at around 36 ppm for –CH2 of ethylene diammonium.
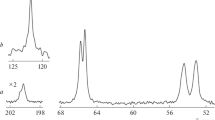
The luminescence emission spectra of Hmptrz, en, 1, 2, and 3 were obtained in DMSO at room temperature and the results are displayed in Fig. 1. As shown in Fig. 1a (λ ex = 261 nm), Hmptrz exhibits a broad luminescent emission centered at 322 nm and 1 displays a broad luminescent emission at 325 nm. The luminescent emission of complex 1 is stronger than that of the free Hmptrz ligand. As shown in Fig. 1b (λ ex = 264 nm), the en and Hmptrz exhibit a broad luminescent emission centered at 310 and 328 nm, respectively, and 2 displays a broad luminescent emission at 313 nm. There are blueshifts of the emission energies of Hmptrz after coordination to Hg(II) for 2 (15 nm blueshifted compared to the related emission band). The luminescent emission of complex 2 is stronger than that of the free en and Hmptrz ligands. As shown in Fig. 1c (λ ex = 268 nm), the en and Hmptrz exhibit a broad luminescent emission centered at 313 and 334 nm, respectively, and 3 displays a broad luminescent emission at 318 nm. There are blueshifts of the emission energies of Hmptrz after coordination to Hg(II) for 3 (16 nm blueshifted compared to the related emission band). The luminescent emission of complex 3 is stronger than that of the free en and Hmptrz ligands. The shapes of the luminescence emission spectra for Hmptrz, en, 1, 2, and 3 are similar, so the emission properties of these compounds are believed to have originated from π* → π or π* → n transitions in en and Hmptrz ligands [24–28].

The luminescence spectra of a Hmptrz (4.63 × 10−4 M) and 1 (4.62 × 10−4 M) in DMSO at room temperature; excitation wavelength = 261 nm; b Hmptrz (4.63 × 10−4 M), en (4.66 × 10−4 M), and 2 (4.64 × 10−4 M) in DMSO at room temperature; excitation wavelength = 264 nm; c Hmptrz (4.63 × 10−4 M), en (4.66 × 10−4 M), and 3 (4.64 × 10−4 M) in DMSO at room temperature; excitation wavelength = 268 nm
Thermal studies of 1, 2, and 3
The thermal stability of [Hg(μ-mptrz)2] n (1), {[H2en][Hg2(mptrz)4(μ-Br)2]} (2), and {[H2en][Hg2(mptrz)4(μ-I)2]} (3) has been determined on single-crystalline samples between 30 and 780 °C in an air atmosphere during 75 min with a heating rate of 10 °C min−1 by thermogravimetric (TG) and differential thermal analyses (DTA) (Fig. 2). The TGA curve of 1 (Fig. 2a) exhibits five distinct weight loss steps. The two steps between 275 and 395 °C with a mass loss of 52.1 % correspond to the loss of two mptrz− anions and the framework decomposes (calcd. 49.5 %). The DTA curve of 1 displays one distinct endothermic peak at 285 °C and four distinct exothermic peaks at 375, 415, 542, and 682 °C. For complex 2 (Fig. 2b), TGA shows that chemical decomposition starts at about 195 °C and ends around 343 °C with the weight loss of 62.1 % corresponds to the removing of one ethylene diammonium cation, two bromide anions, and four mptrz− anions (calcd. 59.8 %). The DTA curve of 2 displays one distinct endothermic peak at 205 °C and four distinct exothermic peaks at 312, 405, 611, and 642 °C. Also, the TGA curve of 3 (Fig. 2c) shows that chemical decomposition starts at about 220 °C and ends around 360 °C with the weight loss of 64.8 % corresponds to the removing of one ethylene diammonium cation, two iode anions, and four mptrz− anions (calcd. 63.0 %). The DTA curve of 3 displays two distinct endothermic peaks at 232 and 318 °C and two distinct exothermic peaks at 389 and 625 °C. By comparison with the data from JCPDS file No. 37–1469, the solid residue formed at around 400 °C for 1, 343 °C for 2, and 360 °C for 3, is suggested to be orthorhombic mercury(II) oxide (HgO), which under higher temperature is evaporated [29–32]. The X-ray powder diffraction patterns of HgO as the final product of thermal analysis for title complexes are shown in Fig. 3.

Thermal behavior of a complex 1, b complex 2, and c complex 3

X-ray powder diffraction patterns of HgO, as the final product of thermal analysis of 1 (a), 2 (b), and 3 (c)
Description of the molecular structure of 1, 2, and 3
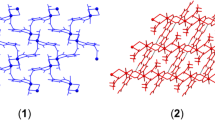
Crystallographic data for 1, 2, and 3 are given in Table 2 and selected bond lengths and angles are presented in Table 3. An ortep view of 1 is shown in Fig. 4. As it is clear in this figure, the asymmetric unit of 1 is constructed by one mptrz− anion and a half-occupied Hg(II) cation. Crystal packing diagram for 1 is shown in Fig. 5. As it is depicted in this figure, complex 1 has a one-dimensional polymeric looped chain structure. Coordination environment in this complex is made by two S atoms and two N atoms from four mptrz− anions. Coordination geometry can be considered as distorted tetrahedral geometry. As shown in Table 3, the Hg–S and Hg–N bond lengths, 2.440(2) Å and 2.370(7)Å, respectively, are within normal range and are comparable with reported similar structures [1, 2]. In the crystal packing of this complex, as shown in Fig. 5, two neighboring mercury centers are linked to each other through two mptrz− ligands to generation of 8-membered [Hg2S2C2N2] ring. The Hg···Hg interatomic distance in the polymeric chains of 1 is 4.3511(6) Å. In the crystal structure of 1 (Fig. 5) there is no π···π interaction between the triazole rings, and only intermolecular C–H···N hydrogen bonds (Table 4) are effective in the stabilization of the crystal structure and formation of the 2-D supramolecular assembly.

The molecular structure of [Hg(μ-mptrz)2] n (1), with the atom-numbering scheme and 50 % probability displacement ellipsoids; symmetry codes: (i) 3/2 − x, y, 1 − z; (ii) 2 − x, 1 − y, 1 − z; (iii) −1/2 + x, 1 – y, z

Crystal packing diagram for [Hg(μ-mptrz)2] n (1). Intermolecular interactions are shown as dashed lines
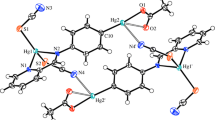
Figures 6 and 7 show the molecular structures of two centrosymmetric binuclear complexes of 2 and 3 linked through halogen bridges. Both complexes crystallize in the triclinic space group Pī where the center of each molecule is located over an inversion center of this space group. The asymmetric units of 2 and 3, {[H2en][Hg2(mptrz)4(μ-X)2]} (X is Br for 2 and I for 3), contain one half-molecule. The structure of these complexes consists of [Hg2(mptrz)4(μ-X)2]2− anions and protonated ethylene diamine cations. The mercury cation is four-coordinated by two S atoms from two mptrz− anions and two (µ-X)− anions in a distorted tetrahedral geometry. The Hg–S average bond distance is 2.427(2) Å and 2.438(2) Å in complexes 2 and 3, respectively. The Hg–S bond distances are within the ranges of those for other analogical mercury(II) complexes [2, 33]. The two mercury and two halogen atoms constitute a perfect plane, with no deviation from the least-diamond Hg2X2 plane in 2 and 3. The Hg1-Br1, Hg1-Br1i, Hg1-Hg1ii, Hg1-Br1-Hg1ii, and Br-Hg1-Bri bridge bond distances and bond angles are 2.8173(11) Å, 2.9059(11) Å, 4.1208(8) Å, 92.10(3)° and 87.90(3)°, respectively (i = −x + 2, −y + 1, −z + 2; ii = −x + 1, −y + 2, −z + 1) and are within normal range [2, 33–35]. The Hg1-I1, Hg1-I1ii, Hg1-Hg1i, Hg1-I1-Hg1ii, and I-Hg1-Iii bridge bond distances and bond angles are 2.9899(6) Å, 3.0448(6) Å, 4.1931(7) Å, 88.022(17)° and 91.978(17)°, respectively (i = −x, −y + 1, −z + 1; ii = −x + 1, −y, −z + 2) and are within normal range [2, 36]. The additional Hg···Si (i is 2 − x, 2 − y, 1 − z in 2 and −x, −y, 2 − z in 3) interaction in complexes of 2 and 3 are 3.276(2) Å and 3.3287(19) Å, respectively, and links all binuclear complexes into one-dimensional chain polymer (Figs. 8, 9). Thus, in the generated chain it is possible to note the short distance Hg···Hgi (i is −x + 2, −y + 1, −z + 2 in 2 and −x, −y + 1, −z + 1 in 3) is 4.3082(8) Å and 4.3515(7) Å for 2 and 3, respectively.

The molecular structure of {[H2en][Hg2(mptrz)4(μ-Br)2]} (2), with the atom-numbering scheme and 50 % probability displacement ellipsoids; symmetry codes: (1) 1 − x, 2 − y, 1 − z; (2) 2 − x, 1 − y, 2 − z

The molecular structure of {[H2en][Hg2(mptrz)4(μ-I)2]} (3), with the atom-numbering scheme and 50 % probability displacement ellipsoids; symmetry codes: (1) 1 − x, −y, 2 − z; (2) −x, 1 − y, 1 − z

Polymer organization in 2 due to Hg…S interactions. Symmetry code: 2 − x, 2 − y, 1 − z

Polymer organization in 3 due to Hg…S interactions. Symmetry code: −x, −y, 2 − z
In the crystal structure of complex 2 (Figs. 8, 10), the Hg···S interaction and π···π interaction between the triazole rings, Cg2···Cg2i (distance = 3.437(6) Å, symmetry code: 2 − x, 1 − y, 1 − z, where Cg2 is centroid of the ring (N1/N2/C2/N3/C1)) and intra- and intermolecular N–H···N and C–H···N hydrogen bonds (Table 4) are effective in the stabilization of the crystal structure and the formation of the 3D supramolecular complex.

Crystal packing diagram for {[H2en][Hg2(mptrz)4(μ-Br)2]} (2). Intra and intermolecular N–H···N and C–H···N hydrogen bonds and π–π contacts are shown as dashed lines
Figures 9 and 11 illustrate the crystal structure of 3, where Hg···S interaction, π···π interaction between the triazole rings, Cg2···Cg2i (distance = 3.465(5) Å, symmetry code: −x, 1 − y, 2 − z, where Cg2 is centroid of the ring (N1/N2/C2/N3/C1)) and intra- and intermolecular N–H···N and C–H···N hydrogen bonds (Table 4) result in a 3-D supramolecular complex, in the crystal structure of 3.

Crystal packing diagram for {[H2en][Hg2(mptrz)4(μ-I)2]} (3). Intra and intermolecular N–H···N and C–H···N hydrogen bonds and π–π contacts are shown as dashed lines
Conclusion
A new coordination polymer of complex [Hg(μ-mptrz)2] n (1) has been synthesized by the reaction of the Hmptrz ligand and HgCl2 presence of ethylene diamine in methanol. This complex was also prepared from the reaction of the Hmptrz ligand in the presence of ethylene diamine with Hg(OAc)2 and Hg(SCN)2 in methanol and Hg(NO3)2·H2O in a mixture of methanol/water. Two new centrosymmetric binuclear complexes of {[H2en][Hg2(mptrz)4(μ-Br)2]} (2) and {[H2en][Hg2(mptrz)4(μ-I)2]} (3) have been synthesized by the reaction of the Hmptrz ligand and HgBr2 and HgI2 in methanol, respectively, in the presence of ethylene diamine. All of these complexes were fully characterized.
Experimental
4-Methyl-1,2,4-triazole-3-thiol was purchased from Aldrich, and used as received. Other materials were purchased from Merck and used without further purification. Infrared spectra (4,000–250 cm−1) of solid samples were taken as 1 % dispersion in CsI pellets using a Shimadzu-470 spectrometer. NMR spectra were recorded on a Bruker AC-300 spectrometer for protons at 300.13 MHz and for 13C at 75.45 MHz in DMSO-d 6 . Melting points were obtained on a Kofler Heizbank Rechart type 7,841 melting point apparatus. Elemental analysis was performed using a Heraeus CHN-O Rapid analyzer. Thermal behavior was measured with a STA 503 Bähr apparatus. UV–Vis spectra were recorded on a Shimadzu 2100 spectrometer using a 1 cm path length cell in DMSO at room temperature, and luminescence spectra were recorded on a Perkin Elmer LS 45 using a 1 cm path length cell. The X-ray powder diffraction (XRD) measurements were performed using a θ/θ STADIP diffractometer of Stöe company with monochromatizes Cu Kα radiation.
Catena-Poly[bis[μ-(4-methyl-1,2,4-triazole-3-thiolato-κ 2 N,S)]mercury(II)] [Hg(μ-mptrz) 2 ] n (1, C6H8HgN6S2)
4-Methyl-1,2,4-triazole-3-thiol (0.37 g, 3.20 mmol) was dissolved in a mixture of 10 cm3 methanol and 8 cm3 ethylene diamine (0.2 M in methanol solution, 1.60 mmol). The solution was then stirred for 5 min and added gradually to a solution of 0.43 g mercury(II) chloride (1.60 mmol) in 10 cm3 CH3OH at room temperature and the resulting white precipitant was dissolved in DMSO. Suitable crystals for X-ray diffraction measurement were obtained by methanol diffusion into the colorless solution of 1 in DMSO over 1 week. It is notable that using 0.51 g Hg(OAc)2, 0.51 g Hg(SCN)2 in 10 cm3 CH3OH or 0.55 g Hg(NO3)2·H2O in a mixture of 5 cm3 CH3OH and 5 cm3 H2O resulted in the same product as when using HgCl2 salt. Yield: 0.53 g (77.2 %) for HgCl2, 0.51 g (74.3 %) for Hg(OAc)2, 0.54 g (78.7 %) for Hg(SCN)2, 0.49 g (71.5 %) for Hg(NO3)2·H2O; m.p.: 280 °C; IR (CsI): \(\overline{\nu}\) = 3,114 m, 3005w, 2950w, 2908w, 1,507 s, 1,465 m, 1,417 s, 1,387 m, 1,355 s, 1,320 s, 1,202 s, 1,164 s, 1,064 s, 1,037 s, 968 m, 859 s, 691 s, 653 s, 512 m, 422 m, 360 m, 326 m cm−1; UV–Vis (DMSO): λ max = 261 nm; 1H NMR (DMSO-d 6 ): δ = 3.52 (s, 6H), 8.34 (s, 2H) ppm; 13C{1H} NMR (DMSO-d 6 ): δ = 32.5 (s), 145.1 (s), 159.2 (s) ppm.
Ethylenediammonium [di-μ-bromidotetrakis(4-methyl-1,2,4-triazole-3-thiolato-κS)dimercury(II)] {[H 2 en][Hg 2 (mptrz) 4 (μ-Br) 2 ]} (2, C14H26Br2Hg2N14S4)
4-Methyl-1,2,4-triazole-3-thiol (0.37 g, 3.20 mmol) was dissolved in a mixture of 10 cm3 methanol and 8 cm3 ethylene diamine (0.2 M in methanol solution, 1.60 mmol). The solution was then stirred for 5 min and added gradually to a solution of 0.58 g HgBr2 (1.60 mmol) in 10 cm3 CH3OH at room temperature and the resulting white precipitant was dissolved in DMSO. Suitable crystals for X-ray diffraction measurement were obtained by methanol diffusion into the colorless solution of 2 in DMSO over 2 weeks. Yield: 0.66 g (76.4 %); m.p.: 202 °C; IR (CsI): \(\overline{\nu}\) = 3,104 m, 3,020 m, 2,915 m, 2,845 m, 2,754 m, 2,572 m, 1,648 m, 1,520 s, 1,480 s, 1,419 s, 1,356 s, 1,222 s, 1,162 s, 1,067 m, 1,026 m, 970 m, 830 m, 795 m, 698 s, 652 s, 506 m, 445w, 353 m, 332 m, 258 s cm−1; UV–Vis (DMSO): λ max = 264 nm; 1H NMR (DMSO-d 6 ): δ = 2.92 (s, 4H), 3.52 (s, 12H), 4.28 (s, br, 6H, disappeared after D2O exchange), 8.35 (s, 4H) ppm; 13C{1H} NMR (DMSO-d 6 ): δ = 32.8 (s), 36.2 (s), 144.5 (s), 160.2 (s) ppm.
Ethylenediammonium [di-μ-iodidotetrakis(4-methyl-1,2,4-triazole-3-thiolato-κS)dimercury(II)] {[H 2 en][Hg 2 (mptrz) 4 (μ-I) 2 ]}(3, C14H26I2Hg2N14S4)
Complex 3 was prepared according to the procedure described for complex 2. 4-Methyl-1,2,4-triazole-3-thiol (0.37 g, 3.20 mmol) was dissolved in a mixture of 10 cm3 methanol and 8 cm3 ethylene diamine (0.2 M in methanol solution, 1.60 mmol). The solution was then stirred for 5 min and added gradually to a solution of 0.73 g HgI2 (1.60 mmol) in 10 cm3 CH3OH at room temperature. Suitable crystals for X-ray diffraction measurement were obtained by methanol diffusion into the colorless solution of 3 in DMSO over 2 weeks. Yield: 0.71 g (75.6 %); m.p.: 227 °C; IR (CsI): \(\overline{\nu}\) = 3,109 m, 3,015 m, 2,927 m, 2,850 m, 2,756 m, 2,591 m, 1,647 m, 1,518 s, 1,467 s, 1,417 s, 1,393 m, 1,354 s, 1,207 s, 1,161 s, 1,138 m, 1,031 m, 968 m, 830 m, 788 m, 696 s, 647 s, 501 m, 444w, 351 m, 335 m, 252 s cm−1; UV–Vis (DMSO): λ max = 268, 418 nm; 1H NMR (DMSO-d 6 ): δ = 2.85 (s, 4H), 3.50 (s, 12H), 3.98 (s, br, 6H, disappeared after D2O exchange), 8.33 (s, 4H) ppm; 13C{1H} NMR (DMSO-d 6 ): δ = 32.9 (s), 36.3 (s), 144.8 (s), 160.8 (s) ppm.
X-ray structure analysis
The X-ray diffraction measurements were made on a Bruker APEX II CCD area detector diffractometer at 298 K (Mo Kα radiation, graphite monochromator, λ = 0.71,073Å). The structures of 1, 2, and 3 were solved by SHELX-97 and absorption corrections were done using the SADABS programs [37, 38]. Softwares including Bruker APEX II (data collection and cell refinement) [39], Bruker SHELXTL (data reduction) [40], and WinGX (publication material) [41] were properly used. The molecular graphics programs used were ORTEP-3 for windows [42], PLATON, and MERCURY [43].
Full crystallographic details are deposited with the Cambridge Structural Database (CCDC Nos. 991304, 991305, and 991306 for 1, 2, and 3, respectively). Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB21EZ, UK.
References
Bell NA, Clegg W, Creighton JR, Raper ES (2000) Inorg Chim Acta 303:12
Popović Z, Matković-Ćalogović D, Soldin Ž, Pavlović G, Davidović N, Vikić-Topić D (1999) Inorg Chim Acta 294:35
Steele RA, Opella SJ (1997) Biochem 36:6885
Dance IG (1986) Polyhedron 5:1037
Cheesman BV, Arnold AP, Rabenstein DZ (1988) J Am Chem Soc 110:6359
Nielsen KB, Atkin CL, Winge DR (1985) J Biol Chem 260:5342
Samiei Paqhaleh DM, Hashemi L, Amani V, Morsali A, Aminjanov AA (2013) Inorg Chim Acta 407:1
Samiei Paqhaleh DM, Aminjanov AA, Amani V, Morsali A (2014) J Inorg Organomet Polym Mater 24:340
Wang YL, Zhang N, Liu QY, Shan ZM, Cao R, Wang MS, Luo JJ, Yang EL (2011) Cryst Growth Des 11:130
Jiang YL, Wang YL, Lin JX, Liu QY, Lu ZH, Zhang N, Wei JJ, Li LQ (2011) CrystEngComm 13:1697
Wang YL, Jiang YL, Liu QY, Wei JJ, Li LQ (2012) Aust J Chem 65:50
Kodcharat K, Pakawatchai C, Saithong S (2013) Acta Crystallogr E69:m265
Ma C, Tian G, Zhang R (2006) J Inorg Organomet Polym Mater 16:139
Jolley J, Cross WI, Pritchard RG, McAuliffe CA, Nolan KB (2001) Inorg Chim Acta 315:36
Bellamy LJ (1975) The infrared spectra of complex molecules, 3rd edn. Chapman and Hall, London
Brill TB, Wertz DW (1970) Inorg Chem 9:2692
Giusti A, Peyronel G (1982) Spectrochim Acta A 38:975
Barr RM, Goldstein M, Unsworth WD (1974) J Cryst Mol Struct 4:165
Allman T, Goel RG, Pilon P (1979) Spectrochim Acta A 35:923
Alizadeh R, Amani V, Farshady AA, Khavasi HR (2010) J Coord Chem 63:2122
Nakamoto K (2009) Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Application in Coordination, Organometallic and Bioinorganic Chemistry. John Wiley and Sons, New York
Mendizabal F, Burgos D, Azar CO (2008) Chem Phys Lett 463:272
Horváth O, Vogler A (1994) Inorg Chim Acta 221:79
Abedi A, Safari N, Amani V, Khavasi HR (2012) J Coord Chem 65:325
Al-Hashemi R, Safari N, Amani V, Amani S, Ng SW (2014) J Iran Chem Soc 11:341
Amani V, Abedi A, Safari N (2012) Monatsh Chem 143:589
Abedi A, Amani V, Safari N (2012) Main Group Chem 11:223
Amani V, Safari N, Notash B (2013) J Iran Chem Soc 10:751
Mahmoudi G, Morsali A, Zeller M (2009) Inorg Chim Acta 362:217
Mahmoudi G, Morsali A, Zhu LG (2007) Polyhedron 26:2885
Collins LW, Gibson EK, Wendelandt WW (1975) Thermochim Acta 11:177
Askarinejad A, Morsali A (2009) Chem Eng J 153:183
Bell NA, Branston TN, Clegg W, Creighton JR, Cucurull-Sánchez L, Elsegood MRJ, Raper ES (2000) Inorg Chim Acta 303:220
Bell NA, Branston TN, Clegg W, Parker L, Raper ES, Sammon C, Constable CP (2001) Inorg Chim Acta 319:30
Tirloni B, Back DF, Burrow RA, Oliveira GNM, Villetti MA, Lang ES (2010) J Braz Chem Soc 21:1230
Isaia F, Aragoni MC, Arca M, Caltagirone C, Castellano C, Demartin F, Garau A, Lippolis V, Pintus A (2011) Dalton Trans 40:4505
Sheldrick GM (1998) SADABS. Bruker AXS, Madison
Bruker (2005) APEX2 Software Package, vers 20-1. Bruker AXS Inc, Madison
Sheldrick GM (1998) SHELXTL (Version 51), Structure Determination Software Suite. Bruker AXS, Madison
Sheldrick GM (2008) Acta Crystallogr A64:112
Farrugia LJ (1999) J Appl Crystallogr 32:837
Farrugia LJ (1997) J Appl Crystallogr 30:565
Mercury 1.4.1 (2001–2005) Copyright Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK
Acknowledgments
We would like to thank the University of Kurdistan and Islamic Azad University, Yadegar-e-Imam Khomeini (RAH) Branch, for financial support.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Taheriha, M., Ghadermazi, M. & Amani, V. Dimeric and polymeric mercury(II) complexes containing 4-methyl-1,2,4-triazole-3-thiol ligand: X-ray studies, spectroscopic characterization, and thermal analyses. Monatsh Chem 146, 559–569 (2015). https://doi.org/10.1007/s00706-014-1333-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-014-1333-3