
Abstract
Here, we report a novel ourmia-like mycovirus, named “Phomopsis asparagi magoulivirus 1” (PaMV1), derived from the phytopathogenic fungus Phomopsis asparagi. The genome of PaMV1 consists of a positive-sense single-stranded RNA (+ ssRNA) that is 2,639 nucleotides in length, with a GC content of 57.13%. It contains a single open reading frame (ORF) encoding a putative RNA-dependent RNA polymerase (RdRp) consisting of 686 amino acids with a molecular mass of 78.57 kDa. Phylogenetic analysis based on RdRp sequences revealed that PaMV1 grouped together with Diaporthe gulyae magoulivirus 1 (DgMV1) in a distinct clade. Sequence comparisons and phylogenetic analysis suggest that PaMV1 is a novel member of the genus Magoulivirus, family Botourmiaviridae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Mycovirus infections are common in most fungi [1]. The first mycovirus was identified in the 1960s in the basidiomycete Agaricus bisporus [2]. Since then, an increasing number of mycoviruses have been identified in various fungi. According to the International Committee on Taxonomy of Viruses (ICTV) (https://talk.ictvonline.org/taxonomy), mycoviruses encompass a wide range of families, including Barnaviridae, Endornaviridae, Alphaflexiviridae, Deltaflexiviridae, Gammaflexiviridae, Narnaviridae, Mitoviridae, Botourmiaviridae, Hypoviridae, Mymonaviridae, Phenuiviridae, Chrysoviridae, Megabirnaviridae, Quadriviridae, Orthototiviridae, Curvulaviridae, Partitiviridae, Botybirnaviridae, Polymycoviridae, Spinareoviridae, Genomoviridae, Metaviridae, Pseudoviridae, Discoviridae, Tulasviridae, Alternaviridae, Hadakaviridae, Yadokariviridae, Amalgaviridae, and Fusariviridae. Mycoviruses can have a double-stranded RNA (dsRNA), positive-sense single-stranded RNA (+ ssRNA), negative-sense single-stranded RNA (-ssRNA), or single-stranded DNA (ssDNA) genome [3].
The majority of mycoviruses cause symptomless infection in their host; however, advancements in fungal virology have revealed that some mycoviruses can induce changes in their host fungi. Sclerotinia sclerotiorum hypovirulence-associated DNA virus 1 (SsHADV-1) infects Sclerotinia sclerotiorum, leading to a reduced growth rate and reduced virulence while transforming it into a beneficial endophyte for protection and yield enhancement of Brassica sp. [4,5,6]. Pestalotiopsis theae chrysovirus 1 (PtCV1) has the ability to convert its host fungus from a phytopathogen to a nonpathogenic endophytic fungus and induce plant resistance [7]. Fusarium oxysporum ourmia-like virus 1 (FoOuLV1), a member of the family Botourmiaviridae, induces hypovirulence in Fusarium oxysporum f. sp. momordicae (FoM) [8]. Currently, plant fungal diseases are controlled predominantly through the use of chemical agents. However, excessive pesticide usage poses various risks, such as pesticide contamination, environmental pollution, and pesticide resistance, which necessitates research efforts towards finding alternative approaches for replacing chemical pesticides. Therefore, fungal viruses hold potential as environmentally friendly biological control agents.
Members of the fungal genus Phomopsis (anamorph of Diaporthe) exhibit a wide distribution in nature, occurring as endophytes, saprobes, or plant pathogens that cause diseases on a diverse range of host plants, including economically significant crops. The symptoms caused by Phomopsis spp., such as canker, withering, rotting, and even plant death, result in substantial economic losses [9]. Phomopsis asparagi has been identified as the causal agent responsible for asparagus stem blight, which is prevalent in countries cultivating asparagus across Asia, Europe, and North America. Asparagus stem blight exhibits its most severe impact in hot and humid regions of China, Japan, Thailand, Indonesia, and other Asian countries [10].
Here, we present the identification of a novel mycovirus from Phomopsis asparagi strain XJ5, which exhibits a close phylogenetic relationship to members of the genus Magoulivirus in the family Botourmiaviridae. Therefore, we designate it as “Phomopsis asparagi magoulivirus 1” (PaMV1), representing a newly discovered member of the genus Magoulivirus.
Provenance of the virus material
The phytopathogenic fungus Phomopsis asparagi strain XJ5 was provided by Dr. Yingqing Yang (Jiangxi Academy of Agricultural Sciences). It was originally isolated from asparagus displaying typical symptoms of asparagus stem blight. The Phomopsis asparagi strain XJ5 was cultured on PDA plates layered with a cellophane membrane for hyphal growth at 25°C for 7 days. Then, the hyphae were collected and used for RNA extraction. dsRNA was extracted through selective absorption to columns of CF-11 cellulose powder in the presence of 16% ethanol, followed by DNase I and S1 nuclease digestion for purification of dsRNA and ssRNA segments, respectively [11]. Agarose gel electrophoresis results indicated the presence of viruses in strain XJ5. A cDNA library was constructed using a Revert Aid First Strand cDNA Synthesis Kit (Thermo) with the tagged oligonucleotide 5’-CGATCGATCATGATGCAATGCNNNNNN-3’ [11]. The mycovirus cDNA sample was sequenced and analyzed using next-generation sequencing technology by Guangzhou Gene Denovo Biotechnology. Specific primers were designed based on the high-throughput sequencing results. To determine the terminal sequences of PaMV1, rapid amplification of cDNA ends (RACE) was used [12]. Amplified cDNA products were purified using a gel extraction kit and then cloned to the pMD18-T vector for Sanger sequencing [13]. The resulting PaMV1 sequence has been deposited in the GenBank database under the accession number OR578827.
The viral genome sequence was analyzed using the NCBI online prediction software ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/). Conserved domain prediction was performed using the Conserved Domain Database (CDD) prediction tool (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Potential stem-loop structures of 5’ and 3’ untranslated regions (UTRs) were predicted using the UNAfold online website (http://www.unafold.org/). Multiple sequence alignments of the RdRp sequences were performed using the MEGA7 and GeneDoc programs [14, 15]. Phylogenetic trees were constructed by the maximum-likelihood (ML) method, using the Molecular Evolutionary Genetics Analysis (MEGA7) program with 1,000 bootstrap replicates [14, 16].
Sequence properties
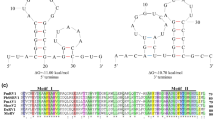
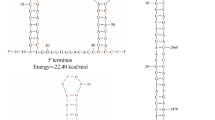
The complete genome sequence of PaMV1 is 2,639 nucleotides (nt) in length with a GC content of 55.13%. The 5’ and 3’ UTRs of PaMV1 are 36 and 542 nt in length, respectively (Fig. 1A and B). Notably, the analysis predicted two potential stem-loop structures within the 5’ and 3’ UTRs, with initial ΔG values of -20.10 kcal/mol and − 10.40 kcal/mol, respectively (Fig. 1C).

(A) Agarose gel electrophoresis of the PaMV1 genome, with M representing the molecular markers of DNA Ladder 4500. (B) The genome organization of the novel mycovirus PaMV1. The open reading frame (ORF) and untranslated regions (UTRs) are indicated by boxes and black lines, respectively. The initiation and termination codons of the RdRp ORF are indicated above the solid lines, along with the molecular weight prediction. (C) Predicted RNA secondary structures of the 5’ and 3’ UTRs of PaMV1. Stem-loop structures were identified at the 5’ and 3’ termini of the PaMV1 genome, with predicted ΔG values of -20.10 and − 10.40 kcal/mol, respectively. Hydrogen bonds between different base pairs are indicated by short red or black lines: red for G-C pairs and black for A-U pairs
The PaMV1 genome contains a single open reading frame (ORF) (Fig. 1B). The ORF of PaMV1 is 2,061 nt in length and encodes a protein of 686 aa with a predicted molecular weight of 78.57 kDa. A BLASTp analysis revealed that the ORF of PaMV1 exhibits sequence similarity to the RdRp proteins found in members of the family Botourmiaviridae. Specifically, the RdRp of PaMV1 shares 76.53%, 54.32%, and 43.56% sequence identity with those of Diaporthe gulyae magoulivirus 1, Fusarium mangiferae botourmiavirus 3, and Penicillium citrinum ourmia-like virus 1, respectively.
A conserved domain database (CDD) search and multiple protein sequences alignment revealed that the RdRp domain contains eight conserved motifs that are characteristic of members within the family Botourmiaviridae (Fig. 2A). Phylogenetic analysis demonstrated a close relationship between PaMV1 and other magouliviruses, forming a well-supported clade (Fig. 2B). Therefore, based on these findings, we conclude that PaMV1 should be classified as a novel member of the genus Magoulivirus in the family Botourmiaviridae.

(A) Multiple alignment of the amino acid sequences of viral RNA-dependent RNA polymerases (RdRps). Shaded areas indicate identical amino acids residues. (B) Phylogenetic analysis based on the RdRp amino acids sequences of PaMV1 (indicated by a red star) and other related viruses. The tree was constructed by the maximum-likelihood (ML) method, using MEGA X software. Bootstrap scores (1,000 replicates) 50% are shown at the nodes. Scale bars represent a genetic distance of 0.10 aa substitutions per site
References
Ghabrial SA, Castón JR, Jiang D, Nibert ML, Suzuki N (2015) 50-plus years of fungal viruses. Virology 479–480:356–68
Ghabrial SA (1988) Origin, adaptation and evolutionary pathways of fungal viruses. Virus Genes 16(1):119–131
Kondo H, Botella L, Suzuki N (2022) Mycovirus diversity and evolution revealed/inferred from recent studies. Annu Rev Phytopathol 60:307–336
Nuss DL (1992) Biological control of chestnut blight: an example of virus-mediated attenuation of fungal pathogenesis. Microbiol Rev 56(4):561–576
Yu X, Li B, Fu Y, Jiang D, Ghabrial SA, Li G, Peng Y, Xie J, Cheng J, Huang J, Yi X (2010) A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc Natl Acad Sci U S A 107(18):8387–8392
Zhang H, Xie J, Fu Y, Cheng J, Qu Z, Zhao Z, Cheng S, Chen T, Li B, Wang Q, Liu X, Tian B, Collinge DB, Jiang D (2020) A 2-kb mycovirus converts a pathogenic fungus into a beneficial endophyte for brassica protection and yield enhancement. Mol Plant 13(10):1420–1433
Zhou L, Li X, Kotta-Loizou I, Dong K, Li S, Ni D, Hong N, Wang G, Xu W (2021) A mycovirus modulates the endophytic and pathogenic traits of a plant associated fungus. ISME J 15(7):1893–1906
Zhao Y, Zhang Y, Wan X, She Y, Li M, Xi H, Xie J, Wen C (2020) A novel ourmia-like mycovirus confers hypovirulence-associated traits on Fusarium oxysporum. Front Microbiol 11:569869
Udayanga D, Liu X, McKenzie EHC et al (2011) The genus Phomopsis: biology, applications, species concepts and names of common phytopathogens. Fungal Divers 50(1):189–225
Yang YQ, Sun Q, Li CM, Chen HF, Zhao F, Huang JH, Zhou JS, Li XM, Lan B (2020) Biological characteristics and genetic diversity of Phomopsis asparagi, causal agent of asparagus stem blight. Plant Dis 104(11):2898–2904
Zheng L, Liu H, Zhang M, Cao X, Zhou E (2013) The complete genomic sequence of a novel mycovirus from Rhizoctonia solani AG-1 IA strain B275. Arch Virol 158(7):1609–1612
Darissa O, Willingmann P, Adam G (2010) Optimized approaches for the sequence determination of double-stranded RNA templates. J Virol Methods 169(2):397–403
Liu R, Cheng J, Fu Y, Jiang D, Xie J (2015) Molecular characterization of a novel positive-sense, single-stranded RNA mycovirus infecting the plant pathogenic fungus Sclerotinia sclerotiorum. Viruses 7(5):2470–2484
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874
Chen Y, Ao WL, Zhang T, Wu QF, Dang ZS, Chen JH, Hu W (2015) Cloning, expression and bioinformatics analysis of pyruvate dehydrogenase of Echinococcus granulosus. Chin J Prev Med 27(4):376–380
King KM, Van Doorslaer K (2016) Building (viral) phylogenetic trees using a maximum likelihood approach. Curr Protoc Microbiol 51(1):e63
Funding
This study was financially supported by Hainan Province Key R&D Project (no. ZDYF2022XDNY242), the National Natural Science Foundation of China (no. 32260648), the National Natural Science Foundation of China (no. 32370180), and the Project of Collaborative Innovation Center Research from Hainan University (no. XTCX2022NYB10).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with animals or human participants performed by any of the authors.
Conflict of interest
All authors declare no conflict of interest.
Additional information
Communicated by Robert H.A. Coutts
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhou, J., Liu, S., Xu, Y. et al. Complete genome sequence of a novel botourmiavirus infecting the fungus Phomopsis asparagi. Arch Virol 169, 161 (2024). https://doi.org/10.1007/s00705-024-06084-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-024-06084-6