
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) causes porcine reproductive and respiratory syndrome (PRRS) worldwide, especially in domestic pigs, with an enormous economic impact, estimated at $664 million in losses every year to the pig industry. Current vaccines confer limited protection, and no direct-acting anti-PRRS treatment is available. Non-structural protein (NSP) 1β, a cysteine-like protease (CLPro) of PRRSV plays an essential role in viral polyprotein processing, subgenomic RNA synthesis, and evasion of host innate immunity. Therefore, agents that interfere with the bioactivity of NSP1β would be expected to inhibit virus replication. In this study, a porcine single-chain antibody (scFv)-phage display library was constructed and used as a tool for production of NSP1β-specific porcine scFvs (pscFvs). The pscFvs to NSP1β were linked to a cell-penetrating peptide to form cell-penetrating pscFvs (transbodies), which could be internalized and inhibit PRRSV replication in infected cells. A computer simulation indicated that the effective pscFvs used several residues in multiple complementarity determining regions (CDRs) to interact with multiple residues in the CLPro and C-terminal motifs, which might explain the mechanism of pscFv-mediated inhibition of virus replication. Although experiments are needed to determine the antiviral mechanism of the transbodies, the current data indicate that transbodies can potentially be applied for treatment and prevention of PRRSV infection.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) is a highly contagious virus causing porcine reproductive and respiratory syndrome (PRRS) (blue ear or mystery swine disease) worldwide, especially in domestic pigs, with an enormous economic impact, estimated at $664 million in losses every year to the pig industry [1]. PRRSV infection of pigs causes high morbidity, characterized by two overlapping clinical presentations, including reproductive impairment or failure of breeding of sows and gilts (late-term abortions, increased number of mummified fetuses, stillbirth, and/or weak-born neonates) and respiratory distress (interstitial pneumonitis with lymphocyte and mononuclear cell infiltration) of pigs at all ages, particularly young and growing piglets [2, 3]. Currently, there is no specific treatment for PRRS. The success of vaccination in controlling PRRSV infection has been limited, as the virus continues to evolve and cause new outbreaks with a high prevalence of infection in swine herds [4]. Development of direct-acting anti-PRRSV agents is therefore a worthy endeavor.
PRRSV is an enveloped single-stranded, positive-sense RNA virus of the phylum Pisuviricota, order Nidovirales, family Arteriviridae, and genus Betaarterivirus [5]. The mature virion (50-72 nm in diameter) is composed of an envelope with an isometric core (20-30 nm) that encapsidates a linear genomic RNA (~15 kb). The genome has a 5′-nontranslated region (NTR) with a cap structure and a 3′-NTR with a polyadenylated tail [6] and contains at least 10 open reading frames (ORFs). At the 5′ end of the genome, two ORFs – ORF1a and ORF1b – encode polyproteins that are cleaved by proteases into 13 or 14 non-structural proteins, and the 3′ end contains genes encoding the major and minor structural proteins, i.e., the envelope protein (E), glycoproteins (GPs), the membrane protein (M), and the nucleoprotein (N) [7]. Based on their genetic and immunogenic diversity, PRRSV strains are divided into two genotypes: European genotype 1 and North American genotype 2 [8].
During virus infection, host intracellular pathogen recognition receptors (PRRs), e.g., Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), and NOD-like receptors (NLRs), recognize viral pathogen-associated molecular patterns (PAMPs), such as ssRNA, dsRNA, and CpG, leading to stimulation of innate interferon (IFN) signaling pathways and production of several antiviral factors. PRRSV infection, however, produces several nonstructural proteins (NSPs), including NSP1, NSP2, NSP4, and NSP11, that are involved in suppression of the innate antiviral immunity of the host [9]. Of these four proteins, NSP1, or NSP1α/1β, had the strongest antagonistic effect on the innate interferon response. NSP1 is derived from the 383 N-terminal amino acids of the ORF1a polyprotein and can be self-cleaved into two active papain-like cysteine proteases: NSP1α (a 180-amino-acid zinc-finger protein also known as accessory papain-like cysteine protease α [PCPα]) and NSP1β (a 203-amino-acid-protein [PCPβ]) [10, 11]. NSP1β suppresses expression of IFN regulatory factor 3 (IRF3)- and NF-κB-dependent genes [9, 12], which consequently results in inhibition of the IFN-IFNRI signal transduction pathway and hence a lack of expression of innate antiviral factors, such as the 2’,5’-oligoadenylate synthetase (OAS)/RNase L system, MxA, and protein kinase R (PKR). It is therefore expected that an agent that inhibits NSP1β activity should be able to restore the antiviral immunity of the host, which would in turn inhibit the replication of the infecting virus. In this study, a porcine single-chain antibody (pscFv)-phage display library was constructed for use as a biological tool for production of NSP1β-specific pscFvs. The pscFvs were linked to a cell-penetrating peptide to target intracellular NSP1β and interfere with the activities of this viral protein. The results of this study suggest that NSP1β-specific pscFvs can be used effectively for passive immunization to treat PRRSV infections.
Materials and methods
Virus propagation
The highly pathogenic PRRSV-2 strain HP/Thailand/19500LL/2010 (TH19500LL/10) [13] was propagated in African green monkey kidney cells, i.e., the MARC-145 cell line (ATTC) cultured in Iscove’s modified Dulbecco’s medium (IMDM) (Gibco, USA) supplemented with 1% L-glutamine (Invitrogen, Canada), 10% fetal bovine serum (FBS) (Invitrogen), and 1% penicillin/streptomycin (Sigma Aldrich, USA) (complete IMDM). The median tissue culture infective dose (TCID50) of the virus stock was determined. The virus stock (1 × 105.8 TCID50/mL) was kept at -80°C until use.
Construction of a porcine scFv (pscFv) phage display library
Nucleotide sequences coding for porcine immunoglobulins were obtained from the GenBank database (Supplementary Table S1) and aligned using the web-based multiple sequence alignment program Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/). Oligonucleotide primers for amplification of the porcine variable heavy chain (VH) genes (vh) were designed to bind to the region coding for framework region (FR1) of the VH domain and JH segment, while the primers for amplification of porcine VLκ genes were designed to anneal to the regions encoding FR1 and FR4 of the variable light chain gene (vl). The individual forward and reverse primers contained restriction sites for SfiI and NotI, respectively, at their 5′ ends (Table 1).
EDTA blood samples were collected from six 7-week-old pigs (5 mL/pig) that had been vaccinated with circovirus, mycoplasma + classical swine fever, and classical swine fever vaccines at 2, 3, and 6 weeks of age. All animal experiments were approved by animal ethical committee of Kasetsart University, Thailand, and were performed by a qualified veterinarian. Peripheral blood mononuclear cells (PBMCs) were isolated from the blood samples using Ficoll-Paque (GE Life Science, USA). Total RNA was extracted from each PBMC preparation using TRIzol Reagent (Invitrogen) and reverse transcribed to cDNA using SuperScriptIII, (Invitrogen) for use as PCR templates. Porcine vh and vl sequences were amplified by PCR using degenerate forward and reverse primers (Table 1) and Taq DNA polymerase (Thermo Scientific, USA). The PCR conditions for vh and vl amplification were as follows: initial denaturation at 94ºC for 5 minutes; 30 cycles of denaturation at 94ºC for 1 minute, annealing at 50ºC for 1 minute, and extension at 72ºC for 1 minute; and final extension at 72ºC for 10 minutes. The vh and vl amplicons were purified from agarose gel slabs using an Agarose Gel Extraction Kit (Jena Bioscience, Germany) and pooled. The vh and vl sequences were linked randomly via a nucleotide linker coding for (Gly4Ser)3 by splicing by overlap extension PCR (SOE-PCR) into the vh-linker-vl or pscfv sequence [15]. The pscfv repertoire was cloned into pCANTAB5E phagemid vectors (GE Life Science, USA) at the SfiI and NotI restriction sites. The insert sequences were verified by DNA sequencing at Macrogen, Korea. Then, the recombinant pscfv phagemids were introduced by transformation into competent E. coli TG1 cells. Mature phage particles that displayed phage P3-pscFvs fusion proteins on the surface and carried the respective pscfv genes in their genomes were recovered from the E. coli culture supernatant after coinfecting the growing bacteria with the helper phage M13KO7. The diversity of the MvaI-digested-pscfv sequences from the pscfv-phage transformed E. coli TG1 clones was assessed by analysis of restriction fragment length polymorphism (RFLP) [14].
Preparation of recombinant NSP1β protein
Total RNA was extracted from the PRRSV stock using TRIzol Reagent (Invitrogen) and reverse transcribed to cDNA. Specific oligonucleotide primers for amplification of the nsp1β gene were designed from an nsp1 sequence available in the GenBank database (AY262352). EcoRI and XhoI restriction sites were incorporated into the 5′ ends of the forward and the reverse primers, respectively. The PCR reaction conditions for nsp1β amplification were as follows: initial denaturation at 94ºC for 5 minutes; followed by 30 cycles of denaturation at 94ºC for 1 minute, annealing at 50ºC for 1 minute, and extension at 72ºC for 1 minute; and final extension at 72ºC for 10 minutes. The product was digested with EcoRI and XhoI PCR and ligated to the pGEX-5X-3 expression vectors cut with the same enzymes (GE Life Sciences, USA), and the recombinant constructs were introduced by transformation into E. coli DH5α. The transformed E. coli clone carrying the nsp1β plasmid was cultured in LB with ampicillin (LB-A broth) in the presence of 0.2 mM IPTG to induce expression of the cloned gene. The recombinant protein was then purified from an E. coli homogenate using a GST column (GE Life Sciences) and verified by SDS-PAGE and protein staining, Western blot analysis, and liquid chromatography tandem mass spectrometry (LC-MS/MS).
Production of porcine scFvs (pscFvs) to NSP1β
Purified recombinant NSP1β (5 μg), purified recombinant GST (5 μg), and a lysate of the original E. coli strain HB2151 were immobilized separately in three wells of a microtiter plate. The pscFv-phage display library was added to the well containing the immobilized E. coli lysate and incubated for 20 minutes. Then, the fluid containing unbound phage (subtracted library) was collected from the well and added to the well containing immobilized GST (subtracted GST). Finally, the unbound phages were collected and added to the well containing immobilized NSP1β. After incubation, the fluid was discarded and the well was washed thoroughly with PBS, pH 7.4, containing 0.05% Tween-20 (PBST). Log-phase-grown E. coli HB2151 was added to the well containing the NSP1β-bound phages, and phage infection was allowed to proceed for 20 minutes. The fluid in the well was collected and spread onto 2× YT-AG plates, and the plates were incubated at 37°C overnight. The presence of pscfvs in the phagemids was detected by direct colony PCR using phagemid-specific primers [15]. The pscfv-positive clones were grown in the presence of 0.2 mM IPTG to induce expression of the cloned gene, and the bacterial cells were harvested, homogenized by sonication, and centrifuged. The supernatants of the E. coli lysates were tested for the presence of pscFvs by Western blot analysis using an anti-E-tag antibody to detect the E-tagged-pscFvs, after which the pscFvs were tested for their ability to bind to the recombinant NSP1β by indirect ELISA [15].
Preparation of cell-penetrating pscFvs (PEN-pscFv)
The pscfv gene of the selected E. coli clone was subcloned into the recombinant plasmid pET23b+ immediately downstream of the inserted DNA coding for the cell-penetrating peptide (CPP) penetratin (PEN), a 16-mer peptide derived from the third helix of the Drosophila Antennapedia homeodomain [16]. The pen-pscfv-pET23b+ construct was introduced into competent E. coli BL21 (DE3), and transformants carrying the recombinant plasmid were grown in the presence of 0.2 mM IPTG for expression of the cloned gene. The 6× His-tagged PEN-pscFv fusion proteins specific for NSP1β were then purified from cell lysates using an affinity resin.
Determination of cell internalization efficiency of the PEN-pscFvs
A MARC-145 cell monolayer was treated with 10 µg of the PEN-pscFv preparation for 1 h. The cell culture supernatants were collected, and the cells were washed with plain IMDM, after which a fixed volume of PBS was added, the cells were homogenized, and the lysates were cleared by centrifugation. PEN-pscFvs in the cell lysates were quantified by indirect ELISA as described previously [17]. The intracellular localization of the PEN-pscFvs was determined by laser scanning confocal microscopy. MARC-145 cells were grown on glass coverslips in tissue culture wells. After incubation with 10 µg of PEN-pscFvs at 37°C in a 5% CO2 incubator for 1 h, the cells were washed with PBS, fixed, permeabilized with cold methanol for 20 min, and blocked with 3% bovine serum albumin (BSA). After washing, mouse anti-6 His tag (1: 3,000) and goat anti-mouse immunoglobulin (Alexa Fluor 488; Invitrogen) (1:200) were added sequentially to the cells with washing between the steps. DAPI (Invitrogen) was used to stain cell nuclei. The stained cells were viewed using 2 µm laser scanning confocal microscopy to localize of the PEN-pscFvs in different cellular layers.
Measurement of antiviral activity of the PEN-pscFvs to NSP1β
We hypothesized that treatment of host cells infected with PRRSV with the PEN-pscFvs would inhibit NSP1β activity and thereby bring about restoration of the antiviral immunity of the host and inhibition of virus growth. For this experiment, 500 μL (1 × 105.8 TCID50/mL) of the PRRSV stock was added to a MARC-145 cell monolayer, which was incubated at 37°C in a 5% CO2 incubator for 1 hour to allow virus entry. Then, the cells were washed, and complete IMDM containing 10 µg of purified PEN-pscFvs was added. Infected cells treated with control PEN-pscFvs, convalescent pig serum obtained after PRRSV infection, poly(I:C), and medium alone were included as background virus inhibition, positive virus inhibition, innate interferon stimulator, and positive infection controls, respectively. MARC-145 cells in medium alone served as a negative infection control. At 3 days postinfection, the number of infected cells was determined using an immunoperoxidase monolayer assay (IPMA). Pig immune serum to PRRSV was used to detect the virus in the infected MARC-145 cells. Co-localization of PEN-pscFv with NSP1β was also examined using a fluorescence dye. Mouse anti-His-tag and goat anti-mouse immunoglobulin (Alexa Fluor 488) were used to locate PEN-pscFv, whereas rabbit anti-NSP1 and goat anti-rabbit immunoglobulin (Alexa Fluor 555) were used to locate PRRSV particles. Stained infected cells were visualized using fluorescence microscopy.
Immunoperoxidase monolayer assay
After the various treatments, the cells were washed with PBS, fixed with cold methanol for 30 minutes, and washed again. A solution of 1% hydrogen peroxide was then added, and the cells were kept in the dark for 30 minutes. After washing with PBST, a pig immune serum against PRRSV was used to detect the virus in the infected MARC-145 cells. HRP-conjugated goat anti-pig IgG (Sigma) and DAB substrate (1:1,000) were used to identify infected cells by bright field microscopy (200× magnification) (Olympus FSX-100 microscope, Japan). A total of 200 cells of each treatment were counted.
Focus assay
A focus assay was used to determine the number of PRRSV particles in the cell culture supernatants after each treatment. Culture fluids from the individual wells were diluted 1:10 with fresh culture medium and added to a MARC-145 cell monolayer. After incubation for 3 h, the fluid in each well was discarded, the cells were washed thoroughly with PBS and cultured for 3 days, and virus foci were identified and counted using the immunoperoxidase assay.
Prediction of the contact interface between pscFv and NSP1
For computer simulations of intermolecular docking between the NSP1 and pscFv, a 3D model of pscFv was generated by submission of its amino acid sequence to the I-TASSER online server [20]. This model was then refined using the Mod-Refiner algorithm, and the lowest-free-energy conformation was predicted using Fragment-Guided Molecular Dynamics (FG-MD) simulation [20]. This model was then verified using the Yet Another Scientific Artificial Reality Application (YASARA) online service [18]. The NSP1 crystal structure (3MTV) [19] and the pscFv model were then used for a docking simulation, using the ClusPro 2.0 protein-protein docking server in antibody mode. The largest cluster with the lowest local energy was selected to simulate the antigen-antibody interaction, and the final model was built and visualized using Pymol software [20].
Statistical analysis
Statistical comparison of different treatments were performed using one-way ANOVA. P < 0.05 was considered statistically significant.
Results
Porcine-scFv phage display library
The amplicons of porcine vh (450 bp) and vl (350 bp) are shown in Figure 1A and 1B, respectively. SOE-PCR was used to link the vh and vl sequences randomly via a (Gly4Ser1)3 linker (Supplementary Table S1). The pscfv amplicon (~800 bp) is shown in Figure 1C. The pscfv repertoire was inserted into pCANPAB5E, and the recombinant phagemids were introduced into competent E. coli TG1 bacteria. After phage rescue by coinfecting the growing E. coli TG1 with helper phage MOKO7, mature phage particles (pscFv-phage display library) were recovered from the TG1 culture supernatant. The rescued phage titer of this library was 2.5 × 107 cfu/mL. The pscfv sequence diversity of the pscFv-phage display library was confirmed by RFLP using the restriction enzyme MvaI (Fig. 1D). The clones were found to be especially diverse in the CD3 region (Supplementary Fig. S1).

Construction of a porcine scFv phage display library. (A) PCR amplicons of genes coding for porcine VH (vh, 450 bp, arrow). (B) PCR amplicons of genes coding for porcine VL (vlκ, 350 bp, arrow). (C) SOE-PCR amplicon of pscfv sequences (~800 bp, arrow) derived from vh and vl sequences that were randomly linked using a nucleotide linker. (D) Representative patterns of MvaI restriction fragments of pscfv sequences from pscfv-phage-transformed E. coli TG1 clones.
Recombinant NSP1β protein
The amplicon of nsp1β cDNA (~550 bp) is shown in Figure 2A. E. coli DH5α carrying nsp1β- pGEX-5X-3 was grown in the presence of IPTG to induce expression of the cloned gene. The recombinant protein was purified from the E. coli lysate using a GST column and verified by SDS-PAGE and Coomassie brilliant blue G-250 (CBB) staining (Fig. 2B) and by Western blot, using anti-GST antibody as a probe (Fig. 2C). The identity of the recombinant protein was also verified by LC-MS/MS (Supplementary Table S2).

Production of recombinant NSP1β. (A) PCR amplicon of nsp1β cDNA at ~ 600 bp (lane 1, arrow); lane M, DNA marker. Numbers at the left are DNA marker sizes in bp. (B) SDS-PAGE and CBB staining of recombinant NSP1β-GST fusion protein at ~ 48 kDa (lane 2, upper); lane 1, GST (lower arrow); lane M, protein standard marker. (C) Western blot of the gel in panel B probed with anti-GST antibody. Numbers at the left in panels B and C are protein molecular masses in kDa.
Porcine scFvs and cell-penetrating pscFv to NSP1β
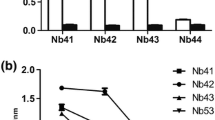
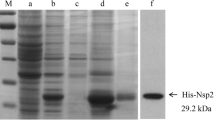
Phage clones that were found by bio-panning to bind to the recombinant NSP1β were introduced by transfection into E. coli HB2151. Out of 12 single colonies of phage-transformed E. coli grown on a selective agar plate, seven carried pscfvs, resulting in amplicons of ~1000 bp (Fig. 3A). These E. coli clones expressed pscFvs (expected size, ~25-30 kDa), as shown in Figure 3B. Indirect ELISA showed that pscFvs in the lysates of E. coli HB2151 clones 1, 2, 3, and 5 bound to recombinant NSP1β, giving an ELISA signal at 405 nm wavelength that was more than twice as high as the BSA control) (Fig. 3C). Clone 5 gave the strongest ELISA signal and was therefore selected for further experiments.

Amplicons of pscfv genes in pscfv-phagemid-transformed E. coli HB2151. (A) PCR amplicons of pscfv of phagemid-transformed E. coli HB2151 clones 1-3, 5, 7, 8, and 11 (1000 bp, arrow); clones 4, 6, 9, and 10 contained truncated pscfv genes. Numbers at the left are DNA marker sizes in bp. (B) Western blot of pscFvs expressed from clones 1-3, 5, 7, 8, and 11 (lanes 1-7, respectively). The protein doublets at ~25-30 kDa consist of an upper band representing immature pscFvs with signal peptides, and the lower bands represent mature pscFvs without signal peptides. Lane M, protein standard marker. Numbers at the left are protein molecular masses in kDa. (C) Binding of pscFvs of clones 1-3, 5, 7, 8, and 11 to recombinant NSP1β, measured by indirect ELISA. A lysate of the original E. coli strain HB2151 was used as a negative antibody (background binding) control, and BSA was used as a control antigen. Clone no. 5, which produced the pscFv with the strongest binding to recombinant NSP1β, was selected for further experiments.
Clone 5, which had 97% sequence identity to the V gene region of Sus scrofa IGHV1-15*01 F (accession no. AB513624), was subcloned into pPET23b, immediately downstream of the region encoding penetratin (pen), and the resulting construct, pen-pscfv5- pPET23b, was introduced by transformation into E. coli BL21. PEN-pscFv5 containing a His tag (transbody) was purified from the E. coli lysate using Ni-NTA affinity resin. PEN-pscFv from E. coli clone 8, which did not bind to NSP1β, was prepared similarly and used as a control antibody.
Cell internalization efficiency of cell-penetrating pscFv
After treatment of MARC-145 cells with 10 µg of PEN-pscFv, it was found that more than 80% of the PEN-pscFv had been internalized (data not shown). The localization of PEN-pscFv in MARC-145 cells is shown in Figure 4.

Intracellular localization of PEN-pscFv5 revealed by laser scanning confocal microscopy. The four upper panels show PEN-pscFv5-treated cells at 2, 4, 6, and 8 µm from the cell surface, respectively; the bottom panels show untreated cells at 4 µm from the cell surface. The left column of panels shows cells under bright field. The panels in the second column DAPI to locate the nuclei (blue). The upper four panels in the third column show the intracellular localization of PEN-pscFvs in cells probed with a mouse anti-His-tag antibody and stained with a goat anti-mouse-immunoglobulin Alexa Fluor 488 conjugate (green fluorescence). The negative control is shown at the bottom. Merged images of DAPI- and Alexa Fluor 488-stained cells are shown at the right. The PEN-pscFvs were scattered in the cytoplasm and nucleus of the cells at the four layers.
Inhibition of PRRSV replication by PEN-pscFv to NSP1β
MARC-145 cells infected with PRRSV were treated with 10 µg of purified PEN-pscFv5 or control, i.e., PEN-pscFv8 (background inhibition control), pig immune serum to PRRSV (positive inhibition control), poly(I:C) (positive interferon stimulating control), or medium (positive infection or negative inhibition control) for 3 days. Uninfected MARC-145 cells were also included as a negative infection control. The results of immunoperoxidase monolayer assay (IPMA) revealed that the infected cells treated with PEN-pscFv5 had the lowest number of infected cells, followed by pig immune serum, poly(I:C), control PEN-pscFv8, and medium alone (positive infection control) (Fig. 5A and B). Likewise, infected cells treated with PEN-pscFv5 had the lowest number of infectious PRRSV particles in their supernatant (Fig. 5C). The co-localization of PEN-pScFv and PRRSV particles is shown in Figure 5D.

Inhibition of PRRSV replication in MARC-145 cells by PEN-pscFv5. (A) IPMA results for PRRSV-infected MARC-145 cells after various treatments. (B) Comparison of the number of PRRSV-infected MARC-145 cells after various treatments. (C) Comparison of the number of infectious PRRSV particles in cell culture supernatants of PRRSV-infected MARC-145 cells after various treatments. (D) Co-localization of PEN-pscFv (green fluorescence) and PRRSV particles (red fluorescence) in infected MARC-145 cells. *, p < 0.05; **, p < 0.01
Computer simulation for determining residues and regions of NSP1 predicted to be bound by pscFv5
A computer docking simulation showed the pscFv5 was predicted to form a contact interface mainly with C-terminal papain-like cysteine protease (CLPro) and C-terminal extension motifs of NSP1. The residues involved in the putative interaction between NSP1 and pscFv5 are detailed in Figure 6A and B and in Table 2. In the model, K124 of CLPro interacts with D147 of VL-CDR1, and R128 and Q131 of CLPro interact with D149 of VL-CDR1. P185, L186, G188, K189, D190, R195, and S198 of the C-terminal extension motif interact with Y104 of VH-CDR3, Y59, Y60, and K65 of VH-FR3, Q212/D147 of VL-FR4/VL-CDR1, and S146 of VL-CDR1, respectively.

(A) Molecular docking of pscFv5 (green) to viral NSP1 (beige). The pscFv5 was predicted to have protruding CDRs that interact with the C-terminal papain-like cysteine protease (CLpro) and C-terminal extension motifs of NSP1 (red). (B) Residues predicted to form a contact interface between the two molecules (see also Table 2).
Discussion
The therapeutic and diagnostic potential of monoclonal antibodies has been evident since production of the first mouse monoclonal antibody [21], and the first therapeutic mouse monoclonal antibody was produced in 1986 [22]. Nowadays, various technologies allow in vitro production of different types of engineered antibodies, including chimeric, humanized, and fully human antibodies, as well as antibody fragments, such as Fab, single-chain, single domain, and multispecific antibody molecules and antibody-drug/prodrug/toxin/radioisotope conjugates for use in diagnostic and therapeutic applications. Single-chain variable fragment antibodies (scFvs), also called single-chain antibodies, are one of the most popular types of genetically engineered antibodies [23]. The advantages of scFvs are their small size (five times smaller than conventional IgG), which gives them relatively high tissue-penetrating ability, and their low immunogenicity. Fully human scFv molecules can be generated conveniently using phage display technology [24]. In humans, several different scFvs have been used to control both infectious and non-infectious maladies and as treatment for intoxication or envenomation [25,26,27,28]. Nevertheless, the use of antibodies for passive immunization of animal diseases is still in its infancy. Antibodies derived from mouse and human libraries are being developed for use in pigs, cows, sheep, and other livestock animals [29,30,31]. To prevent adverse events, therapeutic monoclonal antibodies must be biocompatible with the animal species to which they are administered. Therefore, in this study, a porcine-scFv phage display library was constructed for further use in generation of porcine isotypic antibodies for treatment of porcine diseases. The pscFv phage display library was successfully constructed using degenerate primers and Taq DNA polymerase, which has a relatively high error rate, allowing one immunoglobulin gene template to give rise to many different DNA amplicons, thereby increasing the diversity of the porcine antibody gene repertoire [14, 15, 32]. The deduced amino acid sequences of the genes encoded by the DNA amplicons matched those of immunoglobulin heavy and light variable chains of Sus scrofa (IMGT), indicating that they were porcine isotypes. A library constructed in this manner can therefore be a useful biological tool for in vitro production of porcine antibodies of any desired specificity.
NSP1β of PRRSV is a multifunctional protein whose amino acid sequence is highly conserved. It is an accessory papain-like cysteine protease (PCPβ) with endopeptidase activity. The PCPβ activity is important for processing of the viral polyprotein and subgenomic mRNA (sg mRNA) synthesis [33]. Correct processing of the NSP1α-NSP1β and NSP1β-NSP2 cleavage sites is essential for replication of the PRRSV genome [33]. NSP1β also plays a major role in evasion of the innate antiviral immunity of the host by antagonizing interferon functions [34]. It is therefore expected that targeting the PRRSV NSP1β would not only restore antiviral immunity but also suppress viral transcription and genome replication.
After producing fully porcine scFvs against a recombinant NSP1β protein using a pscFv-phage display library, the pscFvs were modified by linking the antibody to a cell-penetrating peptide, penetratin (PEN), which would allow the antibody to penetrate the target cell to gain access to the intracellular NSP1β and interfere with the bioactivities of this viral protein. A PEN-pscFv5 transbody that showed relatively strong binding to recombinant NSP1β in an indirect ELISA was found to be more effective for inhibiting replication of intracellular virus than an anti-PRRSV immune serum, poly(I:C), and a nonspecific control transbody. The immune serum, which contained intact non-cell-penetrable four-chain antibodies, is presumed to exert its antiviral activity outside the cells, i.e., by prevention of entry of the virus into the cultured cells. Poly(I:C), which stimulates expression of interferons [35], was found to inhibit virus replication significantly less efficiently than the NSP1β-specific PEN-pscFv.
A computer simulation showed that the antibody was predicted to use several residues in multiple CDRs to interact with the residues of the C-terminal papain-like cysteine protease and C-terminal extension motifs of the NSP1 that are involved in replicase polyprotein processing and subgenomic mRNA production [33]. Thus, interference with these functions might be the mechanism by which viral replication is inhibited by the PEN-pscFv. These predictions need to be tested experimentally. To the best of our knowledge, this is the first study to examine the antiviral activity of a porcine transbody against an intracellular viral protein during PRRSV infection. Although the results presented here were obtained using PRRSV-2, similar transbody-based drugs are expected to be applicable for treatment and prevention of infections by other PRRSV types as well.
Data Availability
Data is available.
References
Polson DD, Marsh WE, Dial GD (1992) Financial evaluation and decision making in the swine breeding herd. Vet Clin N Am Food Anim Pract 8(3):725–747
Christianson WT, Choi CS, Collins JE, Molitor TW, Morrison RB, Joo HS (1993) Pathogenesis of porcine reproductive and respiratory syndrome virus infection in mid-gestation sows and fetuses. Can J Vet Res 57(4):262–268
Rossow KD, Collins JE, Goyal SM, Nelson EA, Christopher-Hennings J, Benfield DA (1995) Pathogenesis of porcine reproductive and respiratory syndrome virus infection in gnotobiotic pigs. Vet Pathol 32(4):361–373
Rowland RR, Lunney J, Dekkers J (2012) Control of porcine reproductive and respiratory syndrome (PRRS) through genetic improvements in disease resistance and tolerance. Front Genet 3:260. https://doi.org/10.3389/fgene.2012.00260
Brockmeier SL, Loving CL, Vorwald AC, Kehrli ME Jr, Baker RB, Nicholson TL, Lager KM, Miller LC, Faaberg KS (2012) Genomic sequence and virulence comparison of four Type 2 porcine reproductive and respiratory syndrome virus strains. Virus Res 169(1):212–221
Meulenberg JJ, van Nieuwstadt AP, van Essen-Zandbergen A, Bos-de Ruijter JN, Langeveld JP, Meloen RH (1998) Localization and fine mapping of antigenic sites on the nucleocapsid protein N of porcine reproductive and respiratory syndrome virus with monoclonal antibodies. Virology 252(1):106–114
Snijder EJ, Kikkert M, Fang Y (2013) Arterivirus molecular biology and pathogenesis. J Gen Virol 94(Pt 10):2141–2163
Meng XJ, Paul PS, Halbur PG, Lum MA (1995) Phylogenetic analyses of the putative M (ORF 6) and N (ORF 7) genes of porcine reproductive and respiratory syndrome virus (PRRSV): implication for the existence of two genotypes of PRRSV in the U.S.A. and Europe. Arch Virol. 140(4):745–755
Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK, Osorio FA (2010) Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J Virol 84(3):1574–1584
den Boon JA, Faaberg KS, Meulenberg JJ, Wassenaar AL, Plagemann PG, Gorbalenya AE, Snijder EJ (1995) Processing and evolution of the N-terminal region of the arterivirus replicase ORF1a protein: identification of two papain-like cysteine proteases. J Virol 69(7):4500–4505
Fang Y, Snijder EJ (2010) The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res 154(1–2):61–76
Song C, Krell P, Yoo D (2010) Nonstructural protein 1α subunit-based inhibition of NF-κB activation and suppression of interferon-β production by porcine reproductive and respiratory syndrome virus. Virology 407(2):268–280
Jantafong T, Boonsoongnern A, Poolperm P, Urairong K, Lekcharoensuk C, Lekcharoensuk P (2011) Genetic characterization of porcine circovirus type 2 in piglets from PMWS-affected and -negative farms in Thailand. Virol J. 8:88
Thanongsaksrikul J, Srimanote P, Tongtawe P, Glab-Ampai K, Malik AA, Supasorn O, Chiawwit P, Poovorawan Y, Chaicumpa W (2018) Identification and production of mouse scFv to specific epitope of enterovirus-71 virion protein-2 (VP2). Arch Virol 163(5):1141–1152
Kulkeaw K, Sakolvaree Y, Srimanote P, Tongtawe P, Maneewatch S, Sookrung N, Tungtrongchitr A, Tapchaisri P, Kurazono H, Chaicumpa W (2009) Human monoclonal ScFv neutralize lethal Thai cobra, Naja kaouthia, neurotoxin. J Proteomics 72(2):270–282
Derossi D, Joliot AH, Chassaing G, Prochiantz A (1994) The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem 269(14):10444–10450
Thueng-in K, Thanongsaksrikul J, Srimanote P, Bangphoomi K, Poungpair O, Maneewatch S, Choowongkomon K, Chaicumpa W (2012) Cell penetrable humanized-VH/V(H)H that inhibit RNA dependent RNA polymerase (NS5B) of HCV. PLoS ONE 7(11):e49254
Elmar Krieger, Molecular graphics, modeling and simulation program for Linux, Windows and MacOS. http://www.yasara.org
Xue F, Sun Y, Yan L, Zhao C, Chen J, Bartlam M, Li X, Lou Z, Rao ZJ (2010) The crystal structure of porcine reproductive and respiratory syndrome virus nonstructural protein Nsp1beta reveals a novel metal-dependent nuclease. Virol 84(13):6461–6471
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12(1):7–8
Köhler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256(5517):495–497
Norman DJ, Shield CF 3rd, Barry JM, Henell K, Funnell MB, Lemon J (1987) Therapeutic use of OKT3 monoclonal antibody for acute renal allograft rejection. Nephron 46(Suppl 1):41–47
Nelson AL, Dhimolea E, Reichert JM (2010) Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 9(10):767–774
Osbourn J, Groves M, Vaughan T (2005) From rodent reagents to human therapeutics using antibody guided selection. Methods 36(1):61–68
Agrawal AG, Petersen LR (2003) Human immunoglobulin as a treatment for West Nile virus infection. J Infect Dis 188(1):1–4
Rasetti-Escargueil C, Avril A, Chahboun S, Tierney R, Bak N et al (2015) Development of human-like scFv-Fc antibodies neutralizing Botulinum toxin serotype B. MAbs 7(6):1161–1177
Wang H, Yu R, Fang T, Yu T, Chi X et al (2016) Tetanus neurotoxin neutralizing antibodies screened from a human immune scFv antibody phage display library. Toxins 8(9):266
Lee CH, Lee YC, Lee YL et al (2017) Single chain antibody fragment against venom from the snake Daboia russelii formosensis. Toxins (Basel). 9(11):347
Sapats SI, Trinidad L, Gould G, Heine HG, Van Den Berg TP, Eterradossi N, Jackwood D, Parede L, Toquin D, Ignjatovic J (2006) Chicken recombinant antibodies specific for very virulent infectious bursal disease virus. Arch Virol 151:1551–1566
Braganza A, Wallace K, Pell L, Parrish CR, Siegel DL, Mason NJ (2011) Generation and validation of canine single chain variable fragment phage display libraries. Vet Immunol Immunopathol 139:27–40
Zhang F, Chen Y, Yang L, Zhu J (2019) Construction and characterization of porcine single-chain fragment variable antibodies that neutralize transmissible gastroenteritis virus in vitro. Arch Virol 164:983–994
Li F, Aitken R (2004) Cloning of porcine scFv antibodies by phage display and expression in Escherichia coli. Vet Immunol Immunopathol 97(1–2):39–51
Kroese MV, Zevenhoven-Dobbe JC, Bos-de Ruijter JNA, Peeters BPH, Meulenberg JJM, Cornelissen LAHM, Snijder EJ (2008) The nsp1alpha and nsp1 papain-like autoproteinases are essential for porcine reproductive and respiratory syndrome virus RNA synthesis. J Gen Virol 89(Pt 2):494–499
Sun Z, Chen Z, Lawson SR, Fang Y (2010) The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J Virol 84(15):7832–7846
Farina GA, York MR, Di Marzio M et al (2010) Poly(I:C) drives type I IFN- and TGFβ-mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Invest Dermatol 130(11):2583–2593
Acknowledgements
This work was supported by the Thailand Research Fund, Office of the Higher Education Commission and Kasetsart University, Thailand (Grant number MRG5680053).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Additional information
Handling Editor: Tim Skern.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.

Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
Cite this article
Thueng-in, K., Theerawatanasirikul, S., Meechan, P. et al. Cell-penetrating porcine single-chain antibodies (transbodies) against nonstructural protein 1β (NSP1β) of porcine reproductive and respiratory syndrome virus inhibit virus replication. Arch Virol 168, 133 (2023). https://doi.org/10.1007/s00705-023-05760-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-023-05760-3