
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) is leading to huge losses in the swine industry worldwide. Its nonstructural protein 2 (Nsp2), with a cysteine protease domain (PL2), is crucial for virus replication and as a trigger to host innate immune regulation. In this study, three monoclonal antibodies (mAbs) to Nsp2, designated 4A12, 4G8, and 8H11, were generated. Subsequently, a sequence of recombinant peptides with partial overlap was utilized to determine the epitopes using these mAbs. We found three novel minimal linear Nsp2 B cell epitopes, 188ELSDDSNRPV197, 42HLKRYSPPAE51, and 54CGWHCISA61, which were identified by the antibodies 4A12, 4G8, and 8H11, respectively. Structure analysis indicates that 42HLKRYSPPAE51 and 188ELSDDSNRPV197 are located separately in hypervariable region 1 and hypervariable region 2 of Nsp2. Interestingly, 54CGWHCISA61 is located in the PL2 region, which is highly conserved in all arteriviruses, particularly at the expected conserved catalytic site at Cys54. Importantly, 54CGWHCISA61 is located in the inner region of the expected 3D structure of Nsp2, which reveals that the epitope is cryptic. These findings not only provide valuable insight for vaccine design and hold diagnostic potential for the identified epitopes, but also reveal a protective mechanism against variation under selective pressure in an important epitope.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Porcine reproductive and respiratory syndrome (PRRS) was first discovered almost simultaneously in North America and Europe in the late 1980s (Albina et al. 1992; Collins et al. 1992; Wensvoort et al. 1991; Morin and Robinson 1991). PRRS had been widely observed around the world and had caused severe economic losses for pig producers because it causes reproductive problems and growth retardation (Pejsak et al. 1997; Albina 1997). Studies have found that the causative pathogen is a single-stranded RNA virus, PRRSV, which was subsequently classified in the order Nidovirales, family Arteriviridae and genus Arterivirus, together with Equine Arterivirus (EAV) and two other arteriviruses, lactate dehydrogenase elevating virus (LDV) and simian hemorrhagic fever virus (SHFV) (Cavanagh 1997). Two genotypes have been identified via genome analysis: the European isolate and the North American isolate. These two isolates cause surprisingly similar overall disease phenotype, virion morphology, cellular tropism, and genomic organization although genetically divergent (Ramirez et al. 2008). PRRSV is characterized by a frequent mutation and recombination in vivo, which generates new strains, making it hard to control and eradicate (Wang et al. 2016; Chen et al. 2011). In 2006, a highly pathogenic strain of PRRSV (HP-PRRSV) led to devastating losses in China (Li et al. 2007; Tian et al. 2007; Zhou et al. 2008).
PRRSV genome is about 15 kb in size and includes 11 known open reading frames (ORFs) flanked by 5′ UTR and 3′ UTR. The first three-quarters of the viral genome generates four polyproteins (pp1a, pp1a-Nsp2N, pp1a-Nsp2TF, pp1ab), by two documented programmed ribosomal frame-shift (RFS), which are co-translated and post-translationally processed into more than 16 nonstructural proteins (Nsps) via four virus-encoded proteases involved in papain-like cysteine proteinases 1α (PL1α; Nsp1α), PL1β (Nsp1β), and PL2 (Nsp2) and the main serine proteinase (SP; Nsp4). Recognized polymerase motifs in pp1b are the Nsp9 (RNA-dependent RNA polymerase, RdRp), Nsp10 (RNA helicase), Nsp11 (endoribonuclease), and Nsp12 (functions unclear). The viral structural proteins are separately encoded via many sub-genomic RNAs (sgRNA) produced by an anti-sense intermediate (sgRNA2-7) (Lunney et al. 2016).
In PRRSV, Nsp2 is the largest and most genetically diverse nonstructural protein and is encoded by one quarter of the whole genome, including a PL2 domain around the N terminal. The literature reports that PL2 domain has a core size of about 100 aa and is highly conserved among all arteriviruses (Han et al. 2009). The PL2 domain is followed by the central hypervariable (HV) region characterized by strain-specific insertions or deletions, a putative transmembrane region (TM), and a relatively conserved carboxyl domain (Han et al. 2007; Han et al. 2009; Han et al. 2006). It is well known that Nsp2 has important and multifunctional roles in virus replication and host immune system disorder. This indicates an intracellular function for Nsp2. In addition, many B cell and T cell epitopes from Nsp2 have been identified using bioinformatics or experiments (de Lima et al. 2006; Fang et al. 2004; Fang et al. 2007; Oleksiewicz et al. 2001; Wang et al. 2017; Yan et al. 2007). This evidence suggests that Nsp2 might also have a crucial function in extracellular virus replication or immune regulation. Although Nsp2 is a nonstructural protein in virions, it may be closely associated with the virion structure (Kappes et al. 2013). This may be why antibodies that respond to Nsp2 are immunodominant in PRRSV-infected pigs.
In this study, we expressed and purified His-fused Nsp2 (1–233 aa) protein which encompassing all PL2 domain, then produced three mAbs against PRRSV Nsp2. Three novel linear Nsp2 B cell epitopes, 188ELSDDSNRPV197, 42HLKRYSPPAE51, and 54CGWHCISA61, were subsequently identified using these three mAbs. All of them could be recognized by anti-PRRSV positive sera. Moreover, 54CGWHCISA61 recognized by 8H11 is located in the PL2 region, which is highly conserved among all arteriviruses. These findings provide valuable information for vaccine design and virus diagnosis.
Materials and methods
Cells, viruses, and sera
Marc-145 and myeloma SP2/0 obtained from American Type Culture Collection were cultured using DMEM (Gibco-BRL) with 10% heat-inactivated FBS (Gibco-BRL) and 1% penicillin/streptomycin at 37 °C in a 5% CO2 incubator. PRRSV genotype 2 strain HuN4 (GenBank accession no. EF635006) and JXA1R (GenBank accession no. KM659203) were highly virulent strains. PRRSV genotype 2 strain CH1R (GenBank accession no. EU807840) and VR2332 (GenBank accession no. AY150564) were classical strains. The PRRSV HuN4 strain, JXA1R strain, and CH1R strain were acquired from the State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences. The VR2332 strain was kindly provided by Dr. Wenhai Feng (China Agricultural University). PRRSV-positive serum was collected from a PRRSV inoculation trial. PRRSV-free serum was collected from a specific pathogen-free piglet.
Expression and purification of recombinant PRRSV Nsp2 (1–233 aa) protein
A truncated DNA sequence covering 1–233 aa of Nsp2 was amplified from cDNAs of PRRSV and inserted into the pET-28a vector. The primers Nsp2 (1–233 aa)-F/R are listed in Table 1. The constructed His-fused Nsp2 (1–233 aa) protein expressing plasmid pET-28a-Nsp2 (1–233 aa) was validated by DNA sequencing, followed by transformation into Escherichia coli (E. coli) BL21 (Novagen) and induction with 1 mM IPTG for 4 h in LB medium with kanamycin. The pellets were harvested by centrifugation and then washed twice with 1× PBS. Bacterial pellets were then resuspended and lysed by sonication. After centrifugation at 12,000 rpm, the insoluble fractions were mixed with 5× loading buffer. The mixture was boiled for 10 min and analyzed using 12% SDS-PAGE gel. The expressed His-fused Nsp2 (1–233 aa) protein was then isolated and purified by cutting the gel slices that contained the right bands which were stained by 0.25 mol/L KCl and followed by repeated freezing and thawing. The purity and concentration of the target protein were further evaluated and calculated by Western blot (WB) analysis by anti-His mAb and SDS-PAGE.
SDS-PAGE and Western blot
The IPTG-induced His-fused Nsp2 (1–233 aa) or purified His-fused Nsp2 (1–233 aa) were mixed with 5× loading buffer, boiled for 10 min and then applied to SDS-PAGE and WB analysis. For WB, the protein was transferred to PVDF membrane (Millipore) and blocked using 5% skim milk. After washing three times with TBST, the membrane was incubated with anti-His mAb (diluted 1:5000 in blocking buffer) for 2 h at RT. After washing 3 times using TBST, the membrane was incubated with HRP-conjugated secondary antibody (Sigma-Aldrich) 1 h at RT. The membrane was developed using electrochemiluminescence (ECL) reagents (Pierce).
Generation and features of monoclonal antibodies
The Nsp2-specific mAbs were produced by immunizing 6-week-old female BALB/c mice using 100 μg recombinant His-fused Nsp2 (1–233 aa) protein emulsified with incomplete Freund’s adjuvant (Sigma-Aldrich). Two booster immunizations containing purified recombinant His-fused Nsp2 (1–233 aa) protein at the same volume of the adjuvant were performed every 2 weeks. Three days after the final booster, spleen cells were used to fuse with SP2/0 cells using polyethylene glycol (PEG 4000; Sigma-Aldrich). These animal experiments were performed under protocol IACUC No. HSY-IACUC-2015-006. All The fused cells were then seeded in 96-well plates with DMEM containing hypoxanthine-aminopterin-thymidine (HAT; Sigma-Aldrich) and 20% FBS. Five days later, the medium was replaced with DMEM with hypoxanthine-thymidine (HT; Sigma-Aldrich) and 20% FBS; Then, the supernatant was collected to identify Nsp2-specific antibodies with ELISA assay. Positive clones were sub-cultured three times using limited dilution assay and injected intraperitoneally into mice treated with an equal volume of incomplete Freund’s adjuvant for the collection of ascites fluid. The subtype of the mAb was determined by the SBA Clonotyping™ System/HRP (Southern Biotechnology Associates Inc).
Enzyme-linked immunosorbent assay
Ninety-six-well microtiter plates were coated with 100 μL, well purified, His-fused Nsp2 (1–233 aa) protein or synthesized peptides at 4 °C overnight, blocked using 5% skim milk in PBS for 2 h at 37 °C and washed four times using PBST. One hundred-microliter diluted supernatants of hybridoma or ascite fluid were set in the plates for 2 h at 37 °C, followed by PBST rinsing four times, and clean up. Samples were then plated with HRP-conjugated goat anti-mouse IgG (1:5000 dilution in PBST) (Sigma-Aldrich) for incubation of 1 h, followed by washing four times. Tetramethylbenzidine (TMB) was added and the signal was collected at 450 nm.
Indirect immunofluorescence assay
Marc-145 cells were infected using HP-PRRSV HuN4 (MOI = 0.1). At 24 h post-inoculation, Marc-145 cells were washed using PBS and fixed with 4% PFA for 20 min and permeabilized with 0.3% Triton X-100 for 5 min, followed by blocking using 10% FBS in PBS for 2 h. The primary antibody was added and incubated for 2 h at RT. Cells were then rinsed 3 times with cold PBS and incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (Zsbio, China). Data was collected with a fluorescence microscope (Nikon TS100, Japan).
Epitope mapping
Ten overlapping GST-tagged peptides spanning the Nsp2 (1–233 aa) (P1-P10) were expressed and analyzed by WB to identify the epitopes recognized by 4A12, 4G8, and 8H11. Then, another eight overlapping GST-tagged peptides (P11-P18) were expressed based on the first mapping results and analysis of hydrophilicity using PROTEAN software. A series of peptides (F1-F7) were synthesized as coating antigens for ELISA assays to identify the minimal epitopes recognized by 4A12 and 4G8. The minimal linear epitope recognized by 8H11 was further mapped by expressing 18 overlapping GST-fused fragments (P19–P36). All primers used in this study are listed in Table 1 and Table 2.
Bioinformatics
To explore Nsp2 epitope conservation among different PRRSV reference strains, the peptide sequence of epitopes from different reference strains were matched using DNASTAR Megalign software (DNASTAR Inc., USA). The spatial position of Nsp2 epitopes was determined by mapping epitope locations on the structure of PRRSV Nsp2 with PyMOL software based on data from the SWISS-MODEL online server. Nsp2 structural features were determined with PROTEAN software (DNASTAR Inc., USA).
Results
His-fused Nsp2 (1–233 aa) protein expression and purification
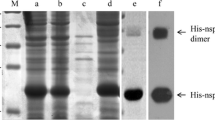
Recombinant His-fused Nsp2 (1–233 aa) protein was successfully expressed in E. coli BL21 cells (Fig. 1a, b). Analysis showed that the protein was expressed in the inclusion bodies, but not in the supernatants (Fig. 1c, d), indicating that His-fused Nsp2 (1–233 aa) protein was mainly present in the inclusion bodies. His-fused Nsp2 (1–233 aa) protein was then isolated and purified by cutting the gel slices that contained the right bands (Fig. 1e). WB analysis showed the purified His-fused Nsp2 (1–233 aa) protein and was identified by anti-His mAb (Fig. 1f).

His-fused Nsp2 (1–233 aa) protein expression and purification(a, b), expression of His-fused Nsp2 (1–233 aa) protein without IPTG induction (a), and induced using of 1 mM/mL IPTG (b). c, d Expression of His-fused Nsp2 (1–233 aa) protein from the supernatant (c) and from the inclusion bodies (d) of IPTG-induced E. coli. (e) His-fused Nsp2 (1–233 aa) protein was purified. (f) WB analysis of purified His-fused Nsp2 (1–233 aa) protein with anti-His antibody
Screening and features of the anti-Nsp2 monoclonal antibodies
Five female BALB/c mice were separately immunized with purified His-fused Nsp2 (1–233 aa) protein to generate hybridomas producing specific antibodies against the PRRSV Nsp2. We identified three hybridoma clones, 4A12, 4G8, and 8H11, by screening supernatants using PRRSV Nsp2-specific indirect ELISA. Antibody isotype analysis indicated that 4A12 and 4G8 belong to the IgG2a/κ-type, whereas 8H11 belongs to the IgG1/κ-type. To verify the specificity of these three mAbs, Marc-145 cells were infected by HP-PRRSV HuN4 for determination of mAbs specificity by WB (Fig. 2a) and IFA (Fig. 2b) separately. The results showed that 4A12, 4G8, and 8H11 can all react well with Nsp2 of HP-PRRSV HuN4.

Features and identification of the anti-Nsp2 monoclonal antibodies, Marc-145 were infected with control or HP-PRRSV HuN4 at a multiplicity of infection (MOI) of 0.1 for 36 h. Then, cell lysates were applied to WB for the test of anti-Nsp2 mAbs, anti-N mAb, and anti-GAPDH mAb (a), or PFA- fixed Marc-145 was for immunostaining analysis with the anti-Nsp2 mAbs (a, c, e for control; b, d, f for PRRSV infection) (b)
Epitope mapping
To identify the epitopes recognized by these three mAbs, ten overlapping GST-tagged peptides spanning the Nsp2 (1–233 aa ) were expressed and analyzed by WB. The results showed that the epitopes recognized by 4A12, 4G8, and 8H11 were located between 181–210 aa, 41–51 aa, and 41–71 aa, respectively (Fig. 3a). Subsequently, another eight overlapping GST-tagged peptides spanning the 21–90 aa and 181–210 aa of Nsp2 were expressed based on analysis of hydrophilicity using PROTEAN software. The epitopes recognized by 4A12, 4G8, and 8H11 were further mapped separately between 188–199 aa, 42–51 aa, and 45–71 aa (Fig. 3b). To further confirm the minimal linear epitopes recognized by 4A12 and 4G8, a series of peptides spanning 188–199 aa and 42–51 aa of Nsp2 were synthesized as coating antigens for ELISA assays (Table 3). In the results shown in Fig. 3c, d, Nsp2-F2 and Nsp2-F3 were recognized by 4A12 (P < 0.001) and Nsp2-F6 and Nsp2-F7 were not recognized by 4G8. Based on the results of WB and ELISA assays, we concluded that the minimal linear epitopes recognized by 4A12 and 4G8 were 188ELSDDSNRPV197 and 42HLKRYSPPAE51, separately. The minimal linear epitope recognized by 8H11 was further mapped by expressing 18 overlapping GST-fused fragments spanning the Nsp2 (45–71 aa). The results shown in Fig. 3e indicate that 54CGWHCISA61 is the minimal linear epitope recognized by 8H11.

Identification of epitopes detected by 4A12, 4G8, 8H11 (a, b). Ten GST-tagged peptides spanning the Nsp2 (1–233 aa) (a) and eight GST-fused peptides spanning the 21–90 aa and 181–210 aa of Nsp2 (b) were expressed and applied to WB analysis. c, d Seven peptides by reducing single amino acid gradually from the N- or C-terminus of the 21–90 aa and 181–210 aa peptide were synthesized and analyzed with indirect ELISA with 4A12 (c) and 4G8 (d) (*P < 0.05; **P < 0.01; ***P < 0.001; ns, no significance). (e) Eighteen GST-tagged peptides covering the Nsp2 (45–71 aa) were expressed and applied to WB with 8H11
Serological tests for PRRSV Nsp2 and the identified epitopes
To further confirm the immunogenic reactivity of Nsp2 and novel epitopes after PRRSV infection, we detected the reactivity between His-fused Nsp2 (1–233 aa) protein and anti-PRRSV sera using WB. Results showed that His-fused Nsp2 (1–233 aa) protein can react with anti-PRRSV positive sera, but not PRRSV negative sera (Fig. 4a). Furthermore, we used either His-fused Nsp2 (1–233 aa) protein or synthetic peptides 188ELSDDSNRPV197, 42HLKRYSPPAE51, and 54CGWHCISA61 as coating antigens for the ELISA assay, and inoculation-induced anti-PRRSV sera as the primary antibody with mAbs 4A12, 4G8, and 8H11 as control, separately. As shown in Fig. 4b–e, the signal of PRRSV-positive sera significantly increased in both the His-fused Nsp2 (1-233 aa) protein- and the peptide-coated ELISA assay compared with the signal of PRRSV negative sera (P < 0.001). These results suggest that PRRSV Nsp2 can induce humoral immune response in the PRRSV-infected pig and that anti-PRRSV sera can recognize epitopes 188ELSDDSNRPV197, 42HLKRYSPPAE51, and 54CGWHCISA61.

Serological detection and identification of epitopes of PRRSV Nsp2. (a) His-fused Nsp2 (1–233 aa) protein was applied to WB for analyzing the response to anti-PRRSV sera. b–e ELISAs was performed using either His-fused Nsp2 (1–233 aa) protein (b) or synthetic peptides 188ELSDDSNRPV197 (c), 42HLKRYSPPAE51 (d), and 54CGWHCISA61 (e) to analyzing the anti-PRRSV sera, with the mAbs 4A12, 4G8, and 8H11 as control, separately (*P < 0.05; **P < 0.01; ***P < 0.001)
The conservation of novel epitopes among different PRRSV isolates
Sequence alignment analysis of novel epitopes from the different PRRSV isolates was performed to determine if the novel epitopes identified by 4A12, 4G8, and 8H11 were conserved among different PRRSV reference strains. As shown in Fig. 5a–c, the linear epitope 188ELSDDSNRPV197 identified by 4A12 is conserved among HP-PRRSV isolates of type 2 PRRSV, though there is one amino acid mutation in JXA1R and SY0608, separately. The epitopes 42HLKRYSPPAE51 and 54CGWHCISA61, recognized separately by 4G8 and 8H11, are conserved among type 2 PRRSV. Interestingly, there are one or two amino acid mutations in epitope recognized by 4G8 of classical isolates of type 2 PRRSV. We performed WB analysis to further identify whether these mutations influence the conservation of the three novel epitopes among PRRSV reference isolates. Results showed that 4A12 can recognize non-classical HP-PRRSV isolates of type 2 PRRSV; 4G8 and 8H11 can recognize all four tested type 2 PRRSV.

The conservation of identified epitopes. a–c Sequence analysis of the epitope recognized by 4A12 (a), 4G8 (b), and 8H11 (c) of different PRRSV isolates. (d) WB assay for the immune response to different PRRSV isolates of 4A12, 4G8, and 8H11
Spatial structures and position of the identified epitopes
To understand the structural mechanism of the epitopes identified by these three mAbs, the 3D structure was predicted using an online computer software program. Analysis revealed that the 4A12 and 4G8-recognized epitope is fully exposed on the surface of the predicted PRRSV Nsp2 (1–233 aa) structure (Fig. 6a) and has no secondary structure (Fig. 6b). Interestingly, the epitope recognized by 8H11 was located in the inner region of the predicted structure and formed a beta-sheet structure, which revealed that the epitope 54CGWHCISA61 recognized by 8H11 is a cryptic epitope. This was the first reported cryptic epitope in a PRRSV-encoded protein. Moreover, 54CGWHCISA61 is located in the PL2 region, which is highly conserved in all arteriviruses, particularly at the predicted catalytic site at Cys54.

Spatial structure and position of the identified epitopes. Localization and spatial distribution of the identified epitopes. a, b The relative spatial position of the identified epitopes is presented in spheres (a) and cartoon (b) from a partially predicted 3D structure of PRRSV Nsp2, epitopes recognized by 4A12, 4G8, and 8H11 are shown in blue, yellow, purple, separately. (c) An enlargement of Nsp2 (1–233 aa) is presented below the full length Nsp2. Nsp2 protein includes five different parts: HV-1, the putative PL2 domain, the predicted transmembrane domains HV2 region, and an uncertain C terminal. At the bottom is a 1–233 aa sequence comparison of PRRSV reference strain VR2332 and PRRSV HuN4 strain used in our study. Identified epitopes are marked with shaded boxes. Potential catalytic residues are presented in red
Discussion
PRRSV infection in pigs causes significant economic loss worldwide. To date, there has been progress in the understanding of its epidemiology and transmission. Recent research indicates that PRRSV nonstructural proteins act in various important roles in the regulation of self-replication, pathogenesis, and immune reactivity (Lunney et al. 2016; Rascon-Castelo et al. 2015). Among the proteins, PRRSV Nsp2 is multifunctional in its viral replication and immune reactivity, composed of four regions: PL2, a 500- to 700-amino-acid middle HV domain, a TM domain, and a C-terminal region (Han et al. 2007; Han et al. 2009; Han et al. 2006). In our study, the purified truncated His-fused Nsp2 (1–233 aa) protein which encompasses the PL2 domain is a part of the hypervariable region used for producing mAbs by immunizing BALB/c mice. These three specific mAbs that recognize Nsp2 were then successfully produced. After a series of screenings, the minimal epitopes recognized by these three mAbs were also identified.
Previous study had identified many epitopes in two types of PRRSV Nsp2. Most of these epitope regions showed natural deletions/insertions and hypervariability in their epitope sequences. Further research indicated that these epitopes are not necessary for replication, but may have a crucial function for the regulation of the host immune system (Chen et al. 2010). For the current study, Nsp2 epitope recognized by 4A12 is also located in the hypervariable region and can only recognize the HP-PRRSV isolates of type 2 PRRSV. Nsp2 epitope recognized by 4G8 contains a part of PL2 region which is highly conserved among type 2 PRRSV. WB analysis indicated that 4G8 can recognize all tested type 2 reference PRRSV strains though its ability to recognize classical isolates that are lower than the HP-PRRSV due to one or two non-critical residual mutations in the classical isolates. The epitope 54CGWHCISA61 recognized by 8H11 is located in the PL2 region. This epitope is highly conserved among type 2 PRRSV, which indicates diagnostic potential. Moreover, 54CGWHCISA61 is composed mostly of hydrophobic amino acid, which indicates that this epitope is cryptic. This is the first reported cryptic epitope in PRRSV-encoded proteins, providing valuable insight for vaccine design.
Among different strains, the homologous protein functional domain is highly conserved, leading to the homology of structure and sequence (Schmitt et al. 2002). In contrast, buried motifs in the structure with a less similar sequence may not influence the whole protein structure (Mortimer and Minchin 2016). However, highly homologous buried sequences may suggest physiological function. Typically, the regions of the HIV-1 envelope glycoprotein are evolutionarily highly conserved, with hidden structural motifs playing an important role in structural formation. These motifs can mediate cellular interactions by providing the attachment position for infection of target cells and by making adjustments for changes of external conditions (Wyatt et al. 1998). Therefore, this is an important protective mechanism in the evolutionary formation of the hidden epitopes, characterized by the conformational flexibility of the structures, which protect the cryptic region from non-specific binding, but interact exclusively with a specific motif or sequence (Rizzuto et al. 1998; Sullivan et al. 1998; Wu et al. 1996). For the 8H11-recognized hidden epitope that is highly conserved among different PRRSV isolates and locates in the Nsp2 PL2 core domain, functional importance would be expected.
This is the first time three novel mAbs, 4A12, 4G8, and 8H11, were produced against Nsp2 of HP-PRRSV HuN4 and identified three linear B cell epitopes recognized specifically via these mAbs. Among them, 54CGWHCISA61, recognized by 8H11 located in the Nsp2 PL2 core domain, is the first identified cryptic epitope in PRRSV-encoded proteins. Our findings may be valuable for vaccine design and may suggest diagnostic potential for these identified epitopes. Moreover, these findings also reveal a protective mechanism for an important epitope against variation under selective pressure.
References
Albina E (1997) Epidemiology of porcine reproductive and respiratory syndrome (PRRS): an overview. Vet Microbiol 55:309–316
Albina E, Leforban Y, Baron T, Plana Duran J (1992) Blue-eared pig disease in Brittany: a new test. Vet Rec 130:83–84
Cavanagh D (1997) Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol 142:629–633
Chen Z, Zhou X, Lunney JK, Lawson S, Sun Z, Brown E, Christopher-Hennings J, Knudsen D, Nelson E, Fang Y (2010) Immunodominant epitopes in nsp2 of porcine reproductive and respiratory syndrome virus are dispensable for replication, but play an important role in modulation of the host immune response. J Gen Virol 91:1047–1057
Chen N, Cao Z, Yu X, Deng X, Zhao T, Wang L, Liu Q, Li X, Tian K (2011) Emergence of novel European genotype porcine reproductive and respiratory syndrome virus in mainland China. J Gen Virol 92:880–892
Collins JE, Benfield DA, Christianson WT, Harris L, Hennings JC, Shaw DP, Goyal SM, McCullough S, Morrison RB, Joo HS, Gorcyca D, Chladek D (1992) Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J Vet Diagn Investig 4:117–126
de Lima M, Pattnaik AK, Flores EF, Osorio FA (2006) Serologic marker candidates identified among B-cell linear epitopes of Nsp2 and structural proteins of a north American strain of porcine reproductive and respiratory syndrome virus. Virology 353:410–421
Fang Y, Kim DY, Ropp S, Steen P, Christopher-Hennings J, Nelson EA, Rowland RR (2004) Heterogeneity in Nsp2 of European-like porcine reproductive and respiratory syndrome viruses isolated in the United States. Virus Res 100:229–235
Fang Y, Schneider P, Zhang WP, Faaberg KS, Nelson EA, Rowland RR (2007) Diversity and evolution of a newly emerged north American type 1 porcine arterivirus: analysis of isolates collected between 1999 and 2004. Arch Virol 152:1009–1017
Han J, Wang Y, Faaberg KS (2006) Complete genome analysis of RFLP 184 isolates of porcine reproductive and respiratory syndrome virus. Virus Res 122:175–182
Han J, Liu G, Wang Y, Faaberg KS (2007) Identification of nonessential regions of the nsp2 replicase protein of porcine reproductive and respiratory syndrome virus strain VR-2332 for replication in cell culture. J Virol 81:9878–9890
Han J, Rutherford MS, Faaberg KS (2009) The porcine reproductive and respiratory syndrome virus nsp2 cysteine protease domain possesses both trans- and cis-cleavage activities. J Virol 83:9449–9463
Kappes MA, Miller CL, Faaberg KS (2013) Highly divergent strains of porcine reproductive and respiratory syndrome virus incorporate multiple isoforms of nonstructural protein 2 into virions. J Virol 87:13456–13465
Li Y, Wang X, Bo K, Wang X, Tang B, Yang B, Jiang W, Jiang P (2007) Emergence of a highly pathogenic porcine reproductive and respiratory syndrome virus in the mid-eastern region of China. Vet J 174:577–584
Lunney JK, Fang Y, Ladinig A, Chen N, Li Y, Rowland B, Renukaradhya GJ (2016) Porcine reproductive and respiratory syndrome virus (PRRSV): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci 4:129–154
Morin M, Robinson Y (1991) Porcine reproductive and respiratory syndrome in Quebec. Vet Rec 129:367–368
Mortimer GM, Minchin RF (2016) Cryptic epitopes and functional diversity in extracellular proteins. Int J Biochem Cell Biol 81:112–120
Oleksiewicz MB, Botner A, Toft P, Normann P, Storgaard T (2001) Epitope mapping porcine reproductive and respiratory syndrome virus by phage display: the nsp2 fragment of the replicase polyprotein contains a cluster of B-cell epitopes. J Virol 75:3277–3290
Pejsak Z, Stadejek T, Markowska-Daniel I (1997) Clinical signs and economic losses caused by porcine reproductive and respiratory syndrome virus in a large breeding farm. Vet Microbiol 55:317–322
Ramirez E, Moreno V, Diaz N, Osorio F, Ruiz A, Neira V, Quezada M (2008) Evaluation of the pathogenicity and transmissibility of a chilean isolate of porcine reproductive and respiratory syndrome virus. Transbound Emerg Dis 55:115–124
Rascon-Castelo E, Burgara-Estrella A, Mateu E, Hernandez J (2015) Immunological features of the non-structural proteins of porcine reproductive and respiratory syndrome virus. Viruses 7:873–886
Rizzuto CD, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong PD, Hendrickson WA, Sodroski J (1998) A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280:1949–1953
Schmitt S, Kuhn D, Klebe G (2002) A new method to detect related function among proteins independent of sequence and fold homology. J Mol Biol 323:387–406
Sullivan N, Sun Y, Sattentau Q, Thali M, Wu D, Denisova G, Gershoni J, Robinson J, Moore J, Sodroski J (1998) CD4-induced conformational changes in the human immunodeficiency virus type 1 gp120 glycoprotein: consequences for virus entry and neutralization. J Virol 72:4694–4703
Tian K, Yu X, Zhao T, Feng Y, Cao Z, Wang C, Hu Y, Chen X, Hu D, Tian X, Liu D, Zhang S, Deng X, Ding Y, Yang L, Zhang Y, Xiao H, Qiao M, Wang B, Hou L, Wang X, Yang X, Kang L, Sun M, Jin P, Wang S, Kitamura Y, Yan J, Gao GF (2007) Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS One 2:e526
Wang X, Yang X, Zhou R, Zhou L, Ge X, Guo X, Yang H (2016) Genomic characterization and pathogenicity of a strain of type 1 porcine reproductive and respiratory syndrome virus. Virus Res 225:40–49
Wang F, Yang Y, Liu X, He M, Liu Y, Sun N, Zhu H, Ren J, Wu H, Wen Y (2017) Development of monoclonal antibody for differentiating porcine reproductive and respiratory syndrome virus and identification of a novel non-structural protein 2 epitope peptide. Virusdisease 28:408–415
Wensvoort G, Terpstra C, Pol JM, ter Laak EA, Bloemraad M, de Kluyver EP, Kragten C, van Buiten L, den Besten A, Wagenaar F, Broekhuijsen JM, Moonen PL, Zetstra T, de Boer EA, Tibben HJ, de Jong MF, van Veld P, Greenland GJ, van Gennep JA, Voets MT, Verheijden JH, Braamskamp J (1991) Mystery swine disease in the Netherlands: the isolation of Lelystad virus. Vet Q 13:121–130
Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J (1996) CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179–183
Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG (1998) The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705–711
Yan Y, Guo X, Ge X, Chen Y, Cha Z, Yang H (2007) Monoclonal antibody and porcine antisera recognized B-cell epitopes of Nsp2 protein of a Chinese strain of porcine reproductive and respiratory syndrome virus. Virus Res 126:207–215
Zhou YJ, Hao XF, Tian ZJ, Tong GZ, Yoo D, An TQ, Zhou T, Li GX, Qiu HJ, Wei TC, Yuan XF (2008) Highly virulent porcine reproductive and respiratory syndrome virus emerged in China. Transbound Emerg Dis 55:152–164
Funding
This study was supported by National Key R&D Program of China (2017YFD0501600), the Chinese Academy of Agricultural Sciences Foundation (1610302017010 and Y2016JC23), the National Natural Science Foundation of China (31640083 and 31872448), and the State Key Laboratory of Veterinary Biotechnological Foundation (SKLVBP2018002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics statement
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bi, C., Shao, Z., Li, J. et al. Identification of novel epitopes targeting non-structural protein 2 of PRRSV using monoclonal antibodies. Appl Microbiol Biotechnol 103, 2689–2699 (2019). https://doi.org/10.1007/s00253-019-09665-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-09665-7