
Abstract
A new member of the genus Alphacarmovirus was detected in Stellaria aquatica using high-throughput RNA sequencing analysis. The complete genome sequence of this new virus isolate, tentatively named "Stellaria aquatica virus A" (StAV-A), comprises 4,017 nucleotides with five predicted open reading frames (ORFs) and has a typical alphacarmovirus genome organization. Pairwise comparison of StAV-A with selected members of family Tombusviridae showed 44–58%, 32–64%, and 19–49% sequence identity for the overall nucleotide sequence, polymerase, and coat protein, respectively. Phylogenetic analysis of polymerase sequences places StAV-A alongside other members of the genus Alphacarmovirus in the family Tombusviridae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Stellaria aquatica is a perennial angiosperm in the carnation family Caryophyllaceae and is commonly known as water or giant chickweed [1]. S. aquatica is cultivated in Korea for the use of its roots and leaves as a medicinal herb called “Soe-byeol-kkot”, which has been used for treatment of various diseases, including gastrointestinal disorders, asthma, diarrhea, measles, jaundice, and inflammation of the renal, digestive, reproductive, and respiratory tracts [2, 3]. S. aquatica is affected by various pathogens, including viruses such as cucumber mosaic virus (CMV), impatiens necrotic spot virus (INSV), and tomato yellow leaf curl virus (TYLCV) [4,5,6].
According to the International Committee on Taxonomy of Viruses (ICTV) 2022 report, members of the family of Tombusviridae are taxonomically classified into three subfamilies and 18 genera: Alphacarmovirus, Alphanecrovirus, Aureusvirus, Avenavirus, Betacarmovirus, Betanecrovirus, Dianthovirus, Gallantivirus, Gammacarmovirus, Luteovirus, Macanavirus, Machlomovirus, Panicovirus, Pelarspovirus, Tombusvirus, Tralespevirus, Umbravirus, and Zeavirus [7, 8]. Viruses in the family Tombusviridae have a monopartite genome, with the exception of the dianthoviruses, which have a bipartite genome, in a virion exhibiting icosahedral symmetry [8]. Members of the genus Alphacarmovirus, subfamily Procedovirinae, have a genome that is about 4.0 kb in size and contains five open reading frames (ORFs). All members are readily transmitted by mechanical inoculation and through plant material used for propagation. Transmission is soil-dependent and does not require a biological vector [8].
Here, we report the biological characterization and molecular properties, including the complete nucleotide sequence, of a novel virus, which we have tentatively named "Stellaria aquatic virus A" (StAV-A).
On July 14, 2021, S. aquatica with mottle symptoms and other plants exhibiting virus-like symptoms were collected from a farm in Jeongseon-gun, Gangwon-do, South Korea (Fig. 1A). In total, 37 samples were ground individually in liquid nitrogen and stored at -80°C until used. The samples were pooled for high-throughput sequencing (HTS) to identify a possible viral agent infecting the plants. The total RNA was isolated from the pooled sample using a Plant RNA Mini Kit (Wizbiosolution, Seongnam, South Korea) and subsequently treated using a Ribo-Zero rRNA Removal Kit (Plant Leaf) (Epicentre, Madison, WI, USA). A library was constructed from the ribosome-depleted RNA sample using an Illumina TruSeq RNA Sample Prep Kit (San Diego, CA, USA) and sequenced on an Illumina NovaSeq 6000 platform by Macrogen (Seoul, South Korea). A total of 66,802,871,570 paired-end reads were obtained and filtered to remove low-quality reads and adaptor sequences. After trimming, the total number of reads was 65,066,524,331. Using the Trinity program, high-quality reads were assembled de novo into transcript contig sequences. The resulting contig sequences were functionally annotated using the Basic Local Alignment Search Tool (BLAST)x of the GenBank database. BLASTx (https://blast.ncbi.nlm.nih.gov/Blast.cgi) analysis revealed a long contig, consisting of 4,076 nucleotides (nt), showing the highest similarity to genomic RNA sequences previously reported for carnation mottle virus (CarMV, GenBank no. MT682299.1) and adonis mosaic virus (AdMV, GenBank no.LC171345), which are members of the genus Alphacarmovirus, family Tombusviridae, sharing 61.19% and 64.3% nt sequence identity, respectively. These findings suggested that the contigs could be partial sequences of a novel alphacarmovirus-like virus that is distantly related to previously reported members of the family Tombusviridae. To confirm the HTS sequencing results, RNA was isolated individually from samples stored at -80°C, which was then subjected to reverse transcription PCR (RT-PCR) using SuPrimeScript RT-PCR Premix (GeNet Bio, Daejeon, South Korea). To detect StAV-A-infected plant samples, a set of contig-specific primers were designed (Supplementary Table S1). The expected 430-bp amplicon was detected by RT-PCR only in S. aquatica among the tested samples (n = 37). To confirm and determine the complete genome sequence of StAV-A, an additional four contig-specific primer pairs were designed. (Supplementary Table S1). Each of the contig-specific primer sets yielded an amplification product of the expected size when S. aquatica samples were tested. All amplicons were purified using an AccuPrep PCR Purification Kit (Bioneer, Daejeon, South Korea) and subsequently cloned into the RBC T&A Cloning Vector (RBC Bioscience, Taipei, Taiwan). At least five positive clones of each RT-PCR amplicon were sequenced independently by the Sanger method at GenoTech (Daejeon, Korea). The overlapping amplicon sequences were analyzed and successfully assembled using DNAMAN software (Lynnon Biosoft, Quebec, Canada).

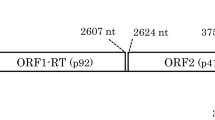
Stellaria aquatica exhibiting mottle virus-like symptoms and genome organization of StAV-A. (A) Mottle symptoms of a S. aquatica plant in which Stellaria aquatica virus A (StAV-A) was detected. (B) Genome organization of Stellaria aquatica virus A (StAV-A). Different colored boxes represent the ORFs, and the readthrough product is indicated.
In addition, contig-specific primer sets were designed for 5′ rapid amplification of cDNA ends (5′-RACE) and 3′-RACE (Supplementary Table S1). Both ends of the genome sequence were determined using the 5′/3′ RACE System (Invitrogen, Carlsbad, CA, USA) [9]. All of the amplicons were cloned into the RBC T&A Cloning Vector (RBC Bioscience, Taipei, Taiwan). To obtain consensus sequences, at least five positive clones of each PCR amplicon were sequenced at GenoTech (Daejeon, Korea). DNAMAN software (Lynnon Biosoft, Quebec, Canada) was used to assemble and analyze overlapping amplicon sequences. The complete genome sequence of StAV-A has been deposited in the NCBI GenBank database under the accession number ON816018.
The StAV-A genomic RNA consists of 4,017 nucleotides (nt), a size consistent with those of other alphacarmoviruses. The StAV-A genome sequence contains five ORFs and 5′ and 3′ untranslated regions (UTRs) of 44 and 321 nucleotides, respectively (Fig. 1B). The 5′-proximal ORF1 encodes a 27-kDa protein (260 aa, nt 45–785), which terminates with an amber codon at nt 783–786. The suppression of the p27 amber codon would result in translation of an ORF1-RT 86-kDa polymerase (497 aa, nt 45–2339), which terminates at an amber codon at nt 2337–2339. The 5′AAA-UAG-GGA3′ amber codon and the surrounding sequence is consistent with the consensus sequence AA(A/G)-UAG-G(G/U)(G/A) required for efficient readthrough [10,11,12]. The partially overlapping ORFs 2 and 3 encode a putative movement protein of 7 kDa (61 aa, nt 2321–2506) and a 10-kDa protein (87 aa, nt 2412–2675), respectively. ORF4 encodes the 37-kDa coat protein (343 aa, nt 2665–3696).
Pairwise comparison of the nucleotide sequence with members of the genus Alphacarmovirus and other genera in the family Tombusviridae, using DNAMAN, showed overall nucleotide sequence identity of 44–58%. Additionally, alignment of the amino acid sequences of the StAV-A ORFs with those of the other members of the genus Alphacarmovirus and selected members of the family Tombusviridae yielded 32–64% sequence identity for the polymerase and 19–49% sequence identity for the coat protein (Supplementary Table S2).
To examine the relationship of StAV-A to members of genus Alphacarmovirus, family Tombusviridae, the amino acid sequences of the polymerase (Fig. 2A) and coat protein (Fig. 2B) were used to construct a phylogenetic tree by the maximum-likelihood method, based on the LG+G model, using MEGA X software with 1,000 bootstrap replicates [13]. CLUSTALW 2.1 was used for the pairwise sequence alignment of the other Alphacarmovirus members and selected members from family Tombusviridae [14].

Phylogenetic analysis of Stellaria aquatica virus A (StAV-A), viruses form the genus Alphacarmovirus, and selected members of family Tombusviridae. (A) Polymerase amino acid sequence. (B) Coat protein amino acid sequence
In conclusion, we regard StAV-A as a novel member of the genus Alphacarmovirus, family Tombusviridae. The results fully comply with the ICTV species demarcation criteria, which require less than 52% coat protein amino acid sequence identity and less than 57% polymerase amino acid sequence identity. To our knowledge, this is the first report of the complete genome sequence of StAV-A infecting S. aquatica in the Republic of Korea.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Gray A, 1810-1888, Robinson BL, et al (1908) Gray’s new manual of botany
Oladeji OS, Oyebamiji AK (2020) Stellaria media (L.) Vill.- A plant with immense therapeutic potentials: phytochemistry and pharmacology. Heliyon 6:e04150. https://doi.org/10.1016/j.heliyon.2020.e04150
Cho H-D, Kang W-S, Kim D-H et al (2019) Comparison of biological activity between Stellaria aquatica seed extracts. Korean J Food Preserv 26:228–237. https://doi.org/10.11002/kjfp.2019.26.2.228
Sastry KS, Mandal B, Hammond J, et al (2019) Stellaria spp. (Stellaria aquatica, S. media) (Chickweed) BT—encyclopedia of plant viruses and viroids. In: Sastry KS, Mandal B, Hammond J, et al (eds). Springer India, New Delhi, pp 2501–2503
Mertelik J, Mokra V, Gotzova B, Gabrielova S (2000) First report of impatiens necrotic spot virus in the Czech Republic. Plant Dis 84:1045. https://doi.org/10.1094/PDIS.2000.84.9.1045B
Kil E-J, Byun H-S, Kim S et al (2015) Tomato yellow leaf curl virus can overwinter in Stellaria aquatica, a Winter-Hardy TYLCV-Reservoir Weed. Plant Dis 99:588–592. https://doi.org/10.1094/PDIS-04-14-0352-RE
Adams MJ, Lefkowitz EJ, King AMQ et al (2016) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch Virol 161:2921–2949
Walker PJ, Siddell SG, Lefkowitz EJ et al (2021) Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch Virol 166:2633–2648. https://doi.org/10.1007/s00705-021-05156-1
Gudeta WF, Igori D, Belete MT et al (2022) Complete genome sequence of pueraria virus A, a new member of the genus Caulimovirus. Arch Virol 167:1481–1485. https://doi.org/10.1007/s00705-022-05431-9
Guilley H, Carrington JC, Balàzs E et al (1985) Nucleotide sequence and genome organization of carnation mottle virus RNA. Nucleic Acids Res 13:6663–6677. https://doi.org/10.1093/nar/13.18.6663
Gulati-Sakhuja A, Rains L, Tian T, Liu H-Y (2011) The complete nucleotide sequence and genome organization of a novel carmovirus—honeysuckle ringspot virus isolated from honeysuckle. Arch Virol 156:1635. https://doi.org/10.1007/s00705-011-1009-9
Harrell L, Melcher U, Atkins JF (2002) Predominance of six different hexanucleotide recoding signals 3′ of read-through stop codons. Nucleic Acids Res 30:2011–2017. https://doi.org/10.1093/nar/30.9.2011
Gudeta WF, Belete MT, Igori D et al (2022) Complete genome sequence of cnidium closterovirus 1, a novel member of the genus Closterovirus infecting Cnidium officinale. Arch Virol. https://doi.org/10.1007/s00705-022-05448-0
Belete MT, Gudeta WF, Kim SE et al (2021) Complete genome sequence of Codonopsis torradovirus A, a novel torradovirus infecting Codonopsis lanceolata in South Korea. Arch Virol 166:3473–3476. https://doi.org/10.1007/s00705-021-05244-2
Acknowledgements
This work was supported by IPET (Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry (IPET) through Crop Viruses and Pests Response Industry Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA), Republic of Korea (321104-3). We thank Barbara Spathelf, PhD, from Edanz (www.edanz.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with animals or human participants performed by any of the authors.
Additional information
Handling Editor: Massimo Turina.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gudeta, W.F., Kim, S.E., Seo, H.K. et al. The complete genome sequence of Stellaria aquatica virus A, a new member of the genus Alphacarmovirus, family Tombusviridae. Arch Virol 168, 90 (2023). https://doi.org/10.1007/s00705-022-05651-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-022-05651-z