
Abstract
Porcine circovirus type 2 (PCV2) is a major pathogen associated with swine diseases. It is the smallest single-stranded DNA virus, and its genome contains four major open reading frames (ORFs). ORF2 encodes the major structural protein Cap, which can self-assemble into virus-like particles (VLPs) in vitro and contains the primary antigenic determinants. In this study, we developed a high-efficiency method for obtaining VLPs and optimized the purification conditions. In this method, we expressed the protein Cap with a 6× His tag using baculovirus-infected silkworm larvae as well as the E. coli BL21(DE3) prokaryotic expression system. The PCV2 Cap proteins produced by the silkworm larvae and E. coli BL21(DE3) were purified. Cap proteins purified from silkworm larvae self-assembled into VLPs in vitro, while the Cap proteins purified from bacteria were unable to self-assemble. Transmission electron microscopy confirmed the self-assembly of VLPs. The immunogenicity of the VLPs produced using the baculovirus system was demonstrated using an enzyme-linked immunosorbent assay (ELISA). Furthermore, the purification process was optimized. The results demonstrated that the expression system using baculovirus-infected silkworm larvae is a good choice for obtaining VLPs of PCV2 and has potential for the development of a low-cost and efficient vaccine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Porcine circovirus type 2 (PCV2) is the smallest single-stranded DNA virus [7, 9] and it is a member of the family Circoviridae. The first porcine circovirus (PCV) was reported in Canada and the associated disease was described as a PCV disease [11, 21]. There are three types of PCVs. PCV1 and PCV2 were first found in the 1970s and the 1990s, respectively, while PCV3 was first found in 2016 [11, 26, 37]. The PCV2 genome consists of only 1.7 kb, and it contains four major open reading frames (ORFs). These ORFs encode two proteins associated with replication, a structural protein that forms the viral capsid, a protein that induces apoptosis of host cells, and a protein that inhibits caspase activity and regulates CD4 and CD8T lymphocytes [13, 31]. However, PCV2 lacks transcription factors, which means that the replication of PCV2 strongly relies on host factors [32]. Some factors, such as protein kinase C (PKC) and human 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), can affect the replication of PCV2 [27]. PCV2 is able to infect the porcine kidney cell line PK-15 [1, 17]. PCV2 infection in piglets is associated with postweaning multisystemic wasting syndrome [2] and has become a destructive factor in the swine industry.
The Cap protein is the only virion-associated structural protein, and it plays an important role in immunity and induction of antibodies [29]. Vaccination is a good strategy for controlling damage due to PCV2. There are some reports that have suggested that PCV2 virus-like particles (VLPs) are similar to the native virus in their structure and immunogenicity [4]. Due to the presence of multiple repeats of the surface epitopes, PCV2 VLPs provide high sensitivity in diagnostic tests and are suitable for the preparation of a vaccine [38]. There have been many studies reporting the production of the Cap protein using different expression systems, including E. coli, phage display, baculovirus, and pseudorabies virus expression systems [6, 8, 30, 39]. Methods such as CsCl and sucrose density gradient centrifugation are used to purify VLPs directly. Cap protein monomers can also be purified by nickel affinity chromatography and allowed to self-assemble into VLPs in vitro [8].
To improve cost efficiency, a baculovirus-silkworm larvae expression system that expresses multiple genes and produces VLPs was developed [20, 34]. In this system, recombinant bacmid DNA was introduced into insect cells without lipofection using invasive diaminopimelate (DAP) auxotrophic E. coli to infect insect cells [35]. In contrast to the traditional Bac-to-Bac method, this system contained 10 genes for expression [41]. A zero-background transposition system was used to construct recombinant bacmids rapidly and effectively [24]. In this study, we focused on the purification of PCV2 Cap proteins produced using two expression systems and tested whether the protein could self-assemble into VLPs.
Materials and methods
Bacterial strains, cells, plasmids, and reagents
E. coli BL21(DE3), E. coli TOP10, and the plasmid pET30a were purchased from Thermo Fisher, USA. The viral bacmid Ac/BmBacmid modified with a mini-attTn7 site was constructed previously in our laboratory. The PCV2 Cap gene containing restriction enzyme recognition sequences for NdeI and XhoI with a 6× His tag sequence was synthesized previously. Restriction enzymes were purchased from New England Biolabs (NEB), and the T4 ligase was purchased from Thermo Fisher. The rTaq and Primestar used for gene amplification were purchased from TaKaRa. Grace’s insect cell culture medium and fetal bovine serum were purchased from Gibco.
Construction of a prokaryotic expression vector and transfer vector
The recombinant plasmid pET30a-PCV2Cap was transferred into E. coli BL21(DE3) and verified by PCR and sequencing using specific primer pairs. The PCV2 Cap with the 6× His tag and eGFP gene was amplified by PCR using primers containing specific restriction enzyme recognition sequences. The PCV2 Cap gene was digested with XmaI and XhoI, and the eGFP gene was digested with XbaI and SacI. These fragments were then ligated into pFBDM. The recombinant plasmid was named pF-PCV2Cap-eGFP.
The recombinant plasmid pF-PCV2Cap-eGFP was introduced by Tn7 transposition into the asd-deleted E. coli sw106-inv containing AcMultiBac [23]. The recombinant strain was cultured on solid medium with gentamicin (Gm), kanamycin (Kan), tetracycline (Tet), spectinomycin (Spe), and diaminopimelic acid (DAP) and was subsequently verified by PCR.
Production of the recombinant baculovirus
The Sw106 strain with the recombinant bacmid was grown to an optical density at 600 nm (OD600) of 0.4–0.6 and collected by centrifugation at 5000 × g for 3 min. The pellet was resuspended in double-distilled water, and the procedure was repeated three times or until the culture medium had been removed. It was then and resuspended in 500 μL of Grace’s insect medium. The suspension was adjusted to three different densities (106–108 cells/mL) in Grace’s medium. Insect cells were incubated in a 24-well plate for 12 h (80% of the base area). After removing the medium, 500-μL bacterial suspensions were added to the Sf9 insect cells and 500 μL of Grace’s insect medium. After incubation for 4 h, the Sf9 cells were incubated with complete Grace’s insect medium with 10% fetal bovine serum (FBS) and 0.075% penicillin/streptomycin for 3–5 days. The recombinant baculovirus was named AcBV-PCV2Cap-eGFP and verified by fluorescence microscopy.
E. coli sw106 with the Tn7 site blocked contained BmNPV bacmids or AcNPV bacmids. The target genes, which were cloned into pFBDM plasmids, were introduced into the bacmids through Tn7 transposition. Using this technique, recombinant bacmids were obtained with zero background transposition. The recombinant bacmids from invasive diaminopimelate (DAP) auxotrophic E. coli were introduced into the insect cells after disrupting the cell walls of the E. coli to release the recombinant bacmids, which were then able to generate infectious recombinant baculovirus particles in insect cells.
Production of the PCV2 Cap protein in E. coli BL21(DE3)
The bacterial strains containing the recombinant plasmid pET30a-PCV2 Cap were cultured on OD600 of 0.4–0.6 (in 50 mL of LB medium with 10 μg of kanamycin per mL at 37 °C and 220 rpm). Then, isopropyl-β-D-1-thiogalactopyranoside (IPTG) was added to the bacteria at four concentrations (0.4 M, 0.6 M, 0.8 M, and 1.0 M). Samples of the bacterial suspensions were collected every hour from 0 h to 6 h (1 mL per sample). Samples were centrifuged at 12000 × g, 4 °C for 5 min, and the pellets were resuspended in 80 μL of phosphate-buffered saline (PBS) (pH = 7.4) containing 20 μL of SDS-PAGE loading buffer and then heated at 100 °C for 10 min and centrifuged at 4 °C, 12,000 × g for 10 min. Samples of the IPTG-treated recombinant bacteria were disrupted in a lysis buffer by 200 cycles of sonication at 400 W (operation for 5 s, pause for 10 s). The homogenate was centrifuged at 4 °C, 12,000 × g for 30 min, and the supernatants and pellets were collected. All of the samples were analyzed by SDS-PAGE followed by Coomassie blue staining or transfer to a 0.22-μM polyvinylidene fluoride (PVDF) membrane for western blotting. Protein binding the PVDF membrane was detected with anti-His mouse antibodies and HRP-labeled goat anti-mouse IgG (H+L; purchased from Beytime, China).
Expression of the PCV2 Cap protein in insect cells and silkworm larvae
A monolayer of Sf9 cells was cultured for three days in Grace’s insect medium with 10% FBS and 0.075% penicillin and streptomycin at 28 °C. The cultured cells were infected with a high titer of virus. The virus infection was confirmed by eGFP fluorescence after 72 h. The Sf9 cells were harvested by centrifugation at 4 °C, 1000 × g for 10 min. The supernatant and pellet were collected and resuspended in 0.1 M PBS (pH 7.4). The pellets were disrupted by sonication for 10 min (operation for 5 s, pause for 10 s, 30 W). The supernatant was frozen at -80 °C and concentrated for 40 h. The same process was applied to BmN cells.
The recombinant virus BmBV-PCV2Cap-eGFP was collected from the culture medium and centrifuged at 12,000 × g for 5 min. Fifth-instar silkworm larvae were divided into two groups. One group was injected with the virus BmBV-PCV2Cap-eGFP, and the other was injected with Grace’s insect medium. A total of 10 μL was used for injection. About 15 min later, the injected larvae were fed fresh mulberry leaves and allowed to grow at 28 °C with 60-70% humidity. After four days, the larvae were tested using a blue shield detector at 475 nm and bled to obtain skin and hemolymph, which were analyzed by 12% SDS-PAGE and western blotting.
SDS-PAGE and western blotting of PCV2 Cap in BL21(DE3), insect cells, and silkworm larvae
Protein samples were separated by 12% SDS-PAGE and stained either with Coomassie brilliant blue or transferred electrophoretically onto a PVDF membrane. To test the antigenicity of the PCV2 Cap protein, the membrane was incubated with anti-His mouse polyclonal antibodies (1:4000) for 12 h at 4 °C. Horseradish peroxidase (HRP)-conjugated goat anti-mouse antibodies were used as the second antibody.
Purification of Cap protein in silkworm larvae
A total of 100 silkworm larvae injected with the recombinant virus Bm-PCV2Cap-eGFP were bled, and their skins were resuspended with PBS (pH 7.4) containing phenylmethylsulphonyl fluoride (PMSF), ground in liquid nitrogen, and centrifuged at 12,000 × g, 4 °C for 30 min. The supernatant was collected, and the recombinant protein was purified using an Ni-NTA affinity cartridge. The sample supernatant was loaded at a rate of 1.5 mL/min, and the cartridge was washed with 6 column volumes (CV) of buffer A (0.1 M PBS, 300 mM NaCl, 5 mM imidazole) at 5 mL/min. The cartridge was washed with 6 CV of buffer B (0.1 M PBS, 300 mM NaCl, 10 mM imidazole) at 5 mL/min, and the purified protein was eluted with 10 CV of 0.1 M PBS, 300 mM NaCl, and a concentration gradient of imidazole (from 0 to 500 mM) at 5 mL/min.
Purification of Cap proteins from BL21(DE3)
BL21 cells were cultured in 200 mL of LB medium and treated with 20 µL of 1.0 M IPTG for 6 h to induce Cap protein expression. The harvested cells were centrifuged at 4 °C, 5000 × g for 20 min, and the pellets were washed three times with washing buffer (0.1 M PBS, pH 7.5) and then resuspended in buffer A (300 mM NaCl, 0.1 M PBS, 6 M urea, 5 mM imidazole) and disrupted by 200 cycles of sonication (400 W) on ice. The homogenate was centrifuged at 4 °C and 12,000 × g for 30 min. The supernatant was then filtered through a 0.22-µm filter immediately before application to an Ni-NTA affinity cartridge (NGC, Bio-Rad). The cartridge was washed with ddH2O at a flow rate of 10 mL/min, for 5 min and equilibrated with buffer A. The sample supernatant was loaded at 2 mL/min and the cartridge was washed with 6 CV of buffer A at 5 mL/min. The cartridge was washed with 6 CV of 300 mM NaCl, 0.1 M PBS, 6 M urea, and 10 mM imidazole (buffer B) at 5 mL/min, and the purified protein was eluted with 10 CV of 300 mM NaCl, 0.1 M PBS, 6 M urea, and 350 mM imidazole (elution buffer) at 5 mL/min. The samples were collected and subjected to SDS-PAGE and western blot analysis.
Transmission electron microscopy (TEM) to detect VLPs of PCV2
The purified Cap protein was negatively stained with uranyl acetate for 45 s and fixed on a copper mesh. The samples were viewed using a 200-kV transmission electron microscope (FEI, USA).
Immunization of mice and enzyme-linked immunosorbent assay (ELISA)
The Cap protein purified from silkworm larvae was dialyzed in 0.1 M PBS, pH 7.5 to remove the imidazole. The Cap protein was then allowed to self-assemble into VLPs in this buffer. Six-week-old female Kunming mice were divided into two groups: treatment and control groups. For immunization, the treatment group mice were injected with 400 μL of VLPs (1 μg/mL), while the mice in the control group were injected with protein purified from larvae injected with viruses that expressed the His tag alone. The mice in both groups were injected once a week, four times in total. Before the first injection, the samples were mixed with complete Freund’s adjuvant. Subsequently, the samples used in the second, third, and fourth injections were mixed with incomplete Freund’s adjuvant. The mice were kept at 25 °C for a week.
All of the mice were bled to obtain serum. Five samples were obtained from each group (treatment and control). To measure the concentration of PCV2 Cap polyclonal antibodies, a standard curve was generated using a Porcine Circovirus Antibody (PCV-Ab) ELISA Kit (Ziker Biological Technology, China). The samples, standards, and HRP-labeled detection antibodies were sequentially added to microwells that had been pre-coated with PCV2 Cap, incubated, and thoroughly washed. The substrate 3,3′,5,5′-tetramethylbenzidine (TMB) was used for color development. TMB is converted to a blue product by peroxidase and becomes yellow when acidified. The absorbance was measured at 450 nm using a microplate reader (Bio-Rad, USA) for quantitation.
Results
Establishment of two expression systems
The recombinant construct pF-PCV2Cap-eGFP was made and confirmed by PCR scanning and DNA sequencing. PCV2 Cap and eGFP were placed under the control of the p10 and polyhedron promoter, respectively (Fig 1a). PCV2 Cap and eGFP were successfully introduced into bacmids (Fig. 1b) by mini-Tn7 transposition, resulting in the recombinant bacterial strains E. coli sw106MultiAcBac-pF-PCV2Cap-eGFP and E. coli sw106MultiBmBac-pF-PCV2Cap-eGFP (Fig. 1c). The recombinant bacmids (AcBac and BmBac) were introduced into insect cells successfully without lipofection. In the insect cells, the recombinant bacmids were assembled into the recombinant baculoviruses AcMNPV-PCV2Cap-eGFP and BmNPV-PCV2Cap-eGFP (Fig. 1d). The recombinant baculovirus BmNPV-PCV2Cap-eGFP was used in the baculovirus-silkworm larvae expression system (Fig. 1e and f). For the prokaryotic expression system, a recombinant bacterial strains E. coli BL21(DE3)-pET30a-PCV2Cap were constructed. The presence of the PCV2 Cap gene was confirmed by PCR scanning and DNA sequencing (Fig. 1g).

Establishment of baculovirus-silkworm and E. coli expression systems for expression of PCV2 Cap proteins. a. Construction of recombinant vector pF-p10-PCV2Cap-polyh-eGFP. PCV2Cap and eGFP were placed under the control of the p10 and polyhedron promoter, respectively, and a recombinant vector was used for Tn7 transposition. b. and c. Construction of a recombinant bacmid containing the PCV2 Cap and eGFP genes through Tn7 transposition. The recombinant bacteria were screened with Tet, Spe, Gm, and Kan. d. Production of the recombinant virus Ac/BmNPV-PCV2Cap-eGFP. e. Production of Cap proteins using the baculovirus-silkworm larvae system. The silkworm larvae using injected with the recombinant virus BmNPV-PCV2Cap-eGFP. f. Three-dimensional structure of the Cap proteins. g. E. coli BL21(DE3) expression system with the Cap gene under the control of the T7 promoter
Expression of Cap proteins in insect cells and silkworm larvae
The recombinant bacterial strains sw106-Ac-pF-PCV2Cap-eGFP and sw106-Bm- pF-PCV2Cap-eGFP, which were incubated with Sf9 and BmN insect cells, successfully produced the recombinant viruses AcNPV-pF-PCV2Cap-eGFP and BmNPV-pF-PCV2Cap-eGFP. At 72 h postinfection, eGFP fluorescence was observed under a 450-nm-wavelength light (Fig. 2a and b). Using the multiBac expression system, the recombinant virus was constructed successfully, which was demonstrated by the fluorescence of eGFP.

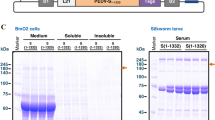
Observation of infected insect cells and silkworm larvae and confirmation of Cap protein expression. a. Sf9 cells infected with the recombinant virus AcNPV-PCV2Cap-eGFP after 72 h. b. BmN cells infected with the recombinant virus BmNPV-PCV2Cap-eGFP after 72 h. The images were taken with bright and blue light (450 nm). Bar = 2.5 μm. c. Western blot analysis of the Cap proteins expressed in Sf9 cells. d. Western blot analysis of the Cap proteins expressed in BmN cells. e. Silkworm larvae infected with the recombinant virus BmNPV-PCV2Cap-eGFP observed using a blue shield instrument at 450 nm. f. Western blot analysis of Cap proteins expressed in silkworm larvae
The insect cells (Sf9 and BmN) were infected with the recombinant viruses AcNPV-pF-PCV2Cap-eGFP and BmNPV-pF-PCV2Cap-eGFP. After the cells were disrupted and centrifuged, the supernatant and medium were subjected to western blotting. As shown in Fig. 2c and d, almost all of the Cap protein was located inside the cells.
The recombinant virus BmNPV-pF-PCV2Cap-eGFP was injected into silkworm larvae, which were observed continuously for four days, after which most of the larvae exhibited behavioral changes and their skin became transparent. The larvae were observed under a 450-nm wavelength light (Fig. 2e). Silkworm larvae were dissected, and their skin and hemolymph were collected for SDS-PAGE and western blotting, which showed that the Cap protein was expressed successfully in silkworm larvae (Fig. 2f).
Cap protein expression in E. coli BL21(DE3)
Treatment with 0.8 mM IPTG for 5 h at 37 °C resulted in maximal expression of Cap proteins in bacteria (Fig. 3a–d). SDS-PAGE analysis revealed that the Cap proteins induced by both 0.4 and 0.8 mM IPTG were present in the form of inclusion bodies (Fig. 3e). Western blotting confirmed the expression of Cap protein induced by 0.8 mM IPTG collected after 5 h (Fig. 3f)

Optimization of conditions for Cap protein expression in E. coli and solubility analysis. a. Expression of Cap proteins in E. coli BL21(DE3) induced using 0.4 mM IPTG. Lane 1, bacteria harvested at 0 h. Lanes 2 to 7, bacteria harvested 1 to 6 h after induction. b. Expression of Cap proteins in E. coli BL21(DE3) induced using 0.6 mM IPTG. Lanes 2 to 7, bacteria harvested 1 to 5 h after induction. c. Expression of Cap proteins in E. coli BL21(DE3) induced using 0.8 mM IPTG. Lane 1, bacteria harvested at 0 h. Lanes 2 to 7, bacteria harvested 1 to 6 h after induction. d. Expression of Cap protein in E. coli BL21(DE3) induced using 1.0 mM IPTG. Lane 1, bacteria harvested at 0 h. Lanes 2 to 7, bacteria 1 to 6 h after induction. e. Solubility of Cap proteins induced using 0.4 and 0.8 mM IPTG for 5 h. Lanes 1 and 4, total protein from the bacteria. Lanes 2 and 5, the supernatant of suspensions disrupted by sonication. Lanes 3 and 6, the pellet after suspension. f. Western blot analysis of Cap protein expressed in E. coli BL21(DE3) induced using IPTG for 5 h. Lane 1, bacteria at 0 h. Lanes 2 to 5, 1 induction using 0.4, 0.6, 0.8, and 1.0 mM IPTG for 5 h
Purification of Cap proteins
Samples from silkworm larvae were purified using an NGC medium-pressure chromatography system (Bio-Rad, USA). Imidazole gradients from 0 mM to 500 mM were used for elution (Fig. 4a). SDS-PAGE and western blotting showed that Cap proteins had been purified successfully and completely eluted with 350 mM imidazole (Fig. 4b and c). About 55 mg of the Cap protein was purified from 60 silkworm larvae injected with the recombinant virus. Cap protein produced in bacteria purified in a similar manner (data not shown).

Purification and identification of Cap protein and VLPs. a. Elution of Cap proteins with 500 mM imidazole, as indicated by the absorption peak. b. and c. Analysis of purified Cap proteins using 12% SDS-PAGE and western blotting. Lane 1, an unpurified E. coli BL21(DE3) sample. Lanes 2 to 6, proteins eluted with 500 mM imidazole. d. Cap proteins that were purified from silkworm larvae skin and allowed to assemble into VLPs. e. Higher magnification with the PCV2 VLPs indicated by arrows. Bar = 50 nm
Examination of PCV2 VLPs by TEM
Cap protein purified from silkworm larvae was dialyzed against 0.1 M PBS, pH 7.4. TEM images (Fig. 4d) revealed that the purified Cap protein self-assembled into VLPs in vitro. The PCV2 VLPs were about 25 nm in diameter, and were nearly round in shape (Fig. 4e). The Cap protein purified from E. coli BL21(DE3) did not self-assemble into VLPs (data not shown).
Immunization of mice and ELISA
The mice from the treatment and control groups were bled to obtain serum. A standard curve was constructed to determine the concentration of polyclonal antibodies (Fig. 5a), which was found to be about 1500 ng/mL (Fig. 5b). Mice injected with VLPs made from PCV2 Cap proteins were successfully immunized (Fig. 5c).

Detection and quantitation of PCV2 antibodies. a. The standard curve for measuring the concentration of PCV2 antibodies based on the OD450 and the standard samples. b. Five antibody samples obtained from mice. c. Western blot analysis of the five samples. The antibodies were detected using an HRP-conjugated goat anti-mouse antibody
Discussion
The Cap protein of the PCV2 is the sole structural protein of the viral capsid and plays an important role in immune responses during the PCV2 infection [19]. Three expression systems for PCV2 Cap protein expression were established: prokaryotic expression, insect cell expression, and silkworm larvae expression systems. For the prokaryotic expression system, the Cap gene was introduced into the vector pET30a, which was used for gene expression. The prokaryotic expression system is a low-cost and convenient way to produce the recombinant protein, but the formation of inclusion bodies is a problem [10]. It is easier to introduce a recombinant virus into insect cells using the insect cell expression system. However, this is not a good approach for expressing large amounts of recombinant proteins. It is usually used for multigene expression and antigen display systems [40]. Our results showed that the baculovirus-silkworm larvae expression system yielded a higher level of expression than insect cells, suggesting that using baculovirus-silkworm larvae expression system is a good strategy for obtaining Cap proteins.
The fatal disease called postweaning multisystemic wasting syndrome (PMWS), which occurs in piglets, is associated with PCV2 [2]. This disease is prevented by vaccination or by treatment of infected piglets with specific antibodies [3]. Due to their structural similarity to native virions, VLPs can be used for immunization. The 60-monomer VLPs of the Cap protein are strongly immunoreactive and bind to specific antibodies [4, 18]. In this study, we optimized the purification conditions to produce and purify large amounts of Cap protein. VLPs containing PCV2 produced in silkworm larvae were used for immunization and detection of antibodies. Examination by TEM showed the Cap proteins of PCV2 from silkworm larvae self-assembled into VLPs. The fact that a large amount of VLPs can be efficiently produced with the baculovirus-silkworm larvae expression system suggests that this system can be used for antibody detection and vaccine preparation.
Wu and Yin obtained PCV2 VLPs from E. coli. Other reports have described Cap protein expression in insect cells. Cap protein produced in silkworm pupae can also self-assemble into VLPs [12, 15, 33, 36]. In our research and other studies [14], Cap protein expressed in E. coli failed to self-assemble into VLPs. We suggest that this could be due to differences in post-translational modifications in different expression systems. For example, N-glycosylation and phosphorylation occur in eukaryotic but not prokaryotic expression systems. Such post-translational modifications might be necessary for the protein to attain its proper structure [25]. In addition, in this study, the Cap proteins expressed in the bacterial system were present in inclusion bodies, which, during the process of purification, were denatured by treatment with urea, which also might have prevented the formation of VLPs.
Comparing the baculovirus-silkworm larvae expression system to the E. coli BL21(DE3) expression system, we concluded that the baculovirus silkworm expression system is preferable for production of PCV2 VLPs. An advantage of the E. coli expression system is that Cap proteins can be easily obtained in large quantities. However, in this research, we focused on the production of VLPs for immunization. Our study showed that the Cap protein from silkworm larvae self-assembled into VLPs, whereas that from E. coli did not. It is possible that the Cap protein from silkworm larvae underwent post-translational modifications that are necessary for protein structure and self-assembly into VLPs. Expression of the Cap protein in fifth-instar baculovirus silkworm larvae saves time, and large amounts of recombinant protein can be produced. In this study, about 55 mg of Cap protein was purified from 60 injected silkworm larvae. After injection, expression was observed by monitoring eGFP fluorescence.
Using the E. coli BL21(DE3) system, we also expressed Cap proteins using a T7 promoter and pelB signal peptide to localize the Cap protein to the periplasmic space [5, 16, 22, 28]. This approach successfully solved the inclusion body problem, but the Cap proteins were expressed at a low level.
To summarize, two systems were applied to produce a large quantity of Cap proteins. After purification, only the Cap protein monomers from the silkworm larvae system assembled into VLPs. To determine the best conditions for expressing Cap protein in E. coli, the concentration of IPTG, induction time, and temperature were optimized. For large-scale production, it was best is to perform induction using 0.8 mM IPTG at 37 °C for 5 h. For expression of the Cap protein in the baculovirus-silkworm larvae system, it was advantageous to delete the asd gene, which saved time and increased the output of recombinant protein. Using the baculovirus-silkworm larvae expression system is an efficient method for producing Cap proteins and VLPs for vaccines and antibody detection.
References
Allan G, Meehan B, Todd D, Kennedy S, McNeilly F, Ellis J, Clark EG, Harding J, Espuna E, Botner A, Charreyre C (1998) Novel porcine circoviruses from pigs with wasting disease syndromes. Vet Rec 142(17):467–468
Allan GM, Ellis JA (2000) Porcine circoviruses: a review. J Vet Diagn Investig 12(1):3–14. https://doi.org/10.1177/104063870001200102
Blanchard P, Mahé D, Cariolet R, Keranflec HA, Baudouard MA, Cordioli P, Albina E, Jestin A (2003) Protection of swine against post-weaning multisystemic wasting syndrome (PMWS) by porcine circovirus type 2 (PCV2) proteins. Vaccine 21(31):4565–4575. https://doi.org/10.1016/S0264-410X(03)00503-6
Bucarey SA, Noriega J, Reyes P, Tapia C, Sáenz L, Zuñiga A, Tobar JA (2009) The optimized capsid gene of porcine circovirus type 2 expressed in yeast forms virus-like particles and elicits antibody responses in mice fed with recombinant yeast extracts. Vaccine 27(42):5781–5790. https://doi.org/10.1016/j.vaccine.2009.07.061
Cao W, Li H, Zhang J, Li D, Acheampong DO, Chen Z, Wang M (2013) Periplasmic expression optimization of VEGFR2 D3 adopting response surface methodology: antiangiogenic activity study. Protein Expr Purif 90(2):55–66. https://doi.org/10.1016/j.pep.2013.04.010
Chi J, Wu C, Chien M, Wu P, Wu C, Huang C (2014) The preparation of porcine circovirus type 2 (PCV2) virus-like particles using a recombinant pseudorabies virus and its application to vaccine development. J Biotechnol 181:12–19. https://doi.org/10.1016/j.jbiotec.2014.04.006
Crowther RA, Berriman JA, Curran WL, Allan GM, Todd D (2003) Comparison of the structures of three circoviruses: chicken anemia virus, porcine circovirus type 2, and beak and feather disease virus. J Virol 24(77):13036–13041. https://doi.org/10.1128/JVI.77.24.13036
Fan H, Ju C, Tong T, Huang H, Lv J, Chen H (2007) Immunogenicity of empty capsids of porcine circovius type 2 produced in insect cells. Vet Res Commun 31(4):487–496. https://doi.org/10.1007/s11259-007-3469-7
Hamel AL, Lin LL, Gopi PSN (1998) Nucleotide sequence of porcine circovirus associated with postweaning multisystemic wasting syndrome in pigs. J Virol 72(6):5262–5267
Kumar S, Jain KK, Singh A, Panda AK, Kuhad RC (2015) Characterization of recombinant pectate lyase refolded from inclusion bodies generated in E. coli BL21(DE3). Protein Expr Purif 110:43–51. https://doi.org/10.1016/j.pep.2014.12.003
Li G, He W, Zhu H, Bi Y, Wang R, Xing G, Zhang C, Zhou J, Yuen K, Gao GF, Su S (2018) Origin, genetic diversity, and evolutionary dynamics of novel porcine circovirus 3. Adv Sci 5(9):1800275. https://doi.org/10.1002/advs.201800275
López-Vidal J, Gómez-Sebastián S, Bárcena J, Nuñez MDC, Martínez-Alonso D, Dudognon B, Guijarro E, Escribano JM (2015) Improved production efficiency of virus-like particles by the baculovirus expression vector system. PLoS One 10(10):e140039. https://doi.org/10.1371/journal.pone.0140039
Mankertz A, Mankertz J, Wolf K, Buhk HJ (1998) Identification of a protein essential for replication of porcine circovirus. J Gen Virol 79(Pt 2):381–384. https://doi.org/10.1099/0022-1317-79-2-381
Marcekova Z, Psikal I, Kosinova E, Benada O, Sebo P, Bumba L (2009) Heterologous expression of full-length capsid protein of porcine circovirus 2 in Escherichia coli and its potential use for detection of antibodies. J Virol Methods 162(1–2):133–141. https://doi.org/10.1016/j.jviromet.2009.07.028
Masuda A, Lee JM, Miyata T, Sato T, Hayashi S, Hino M, Morokuma D, Karasaki N, Mon H, Kusakabe T (2018) Purification and characterization of immunogenic recombinant virus-like particles of porcine circovirus type 2 expressed in silkworm pupae. J Gen Virol 99(7):917–926. https://doi.org/10.1099/jgv.0.001087
Menezes ACSC, Suzuki MF, Oliveira JE, Ribela MTCP, Furigo IC, Donato J, Bartolini P, Soares CRJ (2017) Expression, purification and characterization of the authentic form of human growth hormone receptor antagonist G120R-hGH obtained in Escherichia coli periplasmic space. Protein Expr Purif 131:91–100. https://doi.org/10.1016/j.pep.2016.12.001
Morozov I, Sirinarumitr T, Sorden SD, Halbur PG, Morgan MK, Yoon KJ, Paul PS (1998) Detection of a novel strain of porcine circovirus in pigs with postweaning multisystemic wasting syndrome. J Clin Microbiol 36(9):2535–2541
Nainys J, Lasickiene R, Petraityte-Burneikiene R, Dabrisius J, Lelesius R, Sereika V, Zvirbliene A, Sasnauskas K, Gedvilaite A (2014) Generation in yeast of recombinant virus-like particles of porcine circovirus type 2 capsid protein and their use for a serologic assay and development of monoclonal antibodies. BMC Biotechnol 14(1):100. https://doi.org/10.1186/s12896-014-0100-1
Nawagitgul P, Morozov I, Bolin SR, Harms PA, Sorden SD, Paul PS (2000) Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J Gen Virol 81(Pt 9):2281
Qi Q, Yao L, Liang Z, Yan D, Li Z, Huang Y, Sun J (2016) Production of human type II collagen using an efficient baculovirus-silkworm multigene expression system. Mol Genet Genomics 291(6):2189–2198. https://doi.org/10.1007/s00438-016-1251-7
Segalés J (2012) Porcine circovirus type 2 (PCV2) infections: clinical signs. Pathol Lab Diagn Virus Res 164(1–2):10–19. https://doi.org/10.1016/j.virusres.2011.10.007
Selas Castiñeiras T, Williams SG, Hitchcock A, Cole JA, Smith DC, Overton TW (2018) Development of a generic Β-lactamase screening system for improved signal peptides for periplasmic targeting of recombinant proteins in Escherichia coli. Sci Rep. https://doi.org/10.1038/s41598-018-25192-3
Sun J, Yao L, Yao N, Xu H, Jin P, Kan Y (2010) Production of recombinant Bombyx mori nucleopolyhedrovirus in silkworm by intrahaemocoelic injection with invasive diaminopimelate auxotrophic Escherichia coli containing BmNPV-Bacmid. Biotechnol Appl Biochem 57(3):117–125. https://doi.org/10.1042/BA20100148
Sun JC, Zhang EH, Yao LG, Zhang HL, Jin PF (2009) A high efficient method of constructing recombinant Bombyx mori (silkworm) multiple nucleopolyhedrovirus based on zero-background Tn7-mediated transposition in Escherichia coli. Biotechnol Prog 25(2):524–529. https://doi.org/10.1002/btpr.125
Swartz JR (2001) Advances in Escherichia coli production of therapeutic proteins. Curr Opin Biotechnol 12(2):195–201. https://doi.org/10.1016/s0958-1669(00)00199-3
Tischer I, Bode L, Apodaca J, Timm H, Peters D, Rasch R, Pociuli S, Gerike E (1995) Presence of antibodies reacting with porcine circovirus in sera of humans, mice, and cattle. Arch Virol 140(8):1427–1439. https://doi.org/10.1007/BF01322669
Tischer I, Rasch R, Tochtermann G (1974) Characterization of papovavirus-and picornavirus-like particles in permanent pig kidney cell lines. Zentralbl Bakteriol Orig A 226:153–167
Tong X, Oh EK, Lee B, Lee JK (2019) Production of long-chain free fatty acids from metabolically engineered Rhodobacter sphaeroides heterologously producing periplasmic phospholipase A2 in dodecane-overlaid two-phase culture. Microb Cell Fact. https://doi.org/10.1186/s12934-019-1070-8
Trible BR, Kerrigan M, Crossland N, Potter M, Faaberg K, Hesse R, Rowland RRR (2011) Antibody recognition of porcine circovirus type 2 capsid protein epitopes after vaccination, infection, and disease. Clin Vaccine Immunol 18(5):749–757. https://doi.org/10.1128/CVI.00418-10
Trundova M, Celer V (2007) Expression of porcine circovirus 2 ORF2 gene requires codon optimized E. coli cells. Virus Genes 34(2):199–204. https://doi.org/10.1007/s11262-006-0043-2
Truong C, Mahe D, Blanchard P, Le Dimna M, Madec F, Jestin A, Albina E (2001) Identification of an immunorelevant ORF2 epitope from porcine circovirus type 2 as a serological marker for experimental and natural infection. Arch Virol 146(6):1197–1211. https://doi.org/10.1007/s007050170115
Wei L, Kwang J, Wang J, Shi L, Yang B, Li Y, Liu J (2008) Porcine circovirus type 2 induces the activation of nuclear factor kappa B by IκBα degradation. Virology 378(1):177–184. https://doi.org/10.1016/j.virol.2008.05.013
Wu P, Chen T, Chi J, Chien M, Huang C (2016) Efficient expression and purification of porcine circovirus type 2 virus-like particles in Escherichia coli. J Biotechnol 220:78–85. https://doi.org/10.1016/j.jbiotec.2016.01.017
Yao L, Wang S, Su S, Yao N, He J, Peng L, Sun J (2012) Construction of a baculovirus-silkworm multigene expression system and its application on producing virus-like particles. PLoS One 7(3):e32510. https://doi.org/10.1371/journal.pone.0032510
Yao LG, Sun JC, Xu H, Kan YC, Zhang XM, Yan HC (2010) A novel economic method for high throughput production of recombinant baculovirus by infecting insect cells with bacmid-containing diminopimelate-auxotrophic Escherichia coli. J Biotechnol 145(1):23–29. https://doi.org/10.1016/j.jbiotec.2009.10.003
Yin S, Sun S, Yang S, Shang Y, Cai X, Liu X (2010) Self-assembly of virus-like particles of porcine circovirus type 2 capsid protein expressed from Escherichia coli. Virol J 7:166. https://doi.org/10.1186/1743-422X-7-166
Zhai S, Lu S, Wei W, Lv D, Wen X, Zhai Q, Chen Q, Sun Y, Xi Y (2019) Reservoirs of porcine circoviruses: a mini review. Front Vet Sci 6:319. https://doi.org/10.3389/fvets.2019.00319
Zhang Y, Wang Z, Zhan Y, Gong Q, Yu W, Deng Z, Wang A, Yang Y, Wang N (2016) Generation of E. coli-derived virus-like particles of porcine circovirus type 2 and their use in an indirect IgG enzyme-linked immunosorbent assay. Arch Virol 161(6):1485–1491. https://doi.org/10.1007/s00705-016-2816-9
Zheng G, Lu Q, Wang F, Jin Q, Teng M, Zhang N, Ren T, Ding P, Zhang G (2017) Selection of affinity peptides for the purification potential of porcine circovirus type 2 (PCV2) cap virus-like particles (VLPs). RSC Adv 7(62):38911–38914. https://doi.org/10.1039/C7RA05790C
Zheng H, Ren F, Lu Q, Cao Z, Song J, Feng M, Liu J, Sun J (2019) An efficient method for multigene co-interference by recombinant Bombyx mori nucleopolyhedrovirus. Mol Genet Genomics 294(1):111–120. https://doi.org/10.1007/s00438-018-1491-9
Zheng H, Wang X, Ren F, Zou S, Feng M, Xu L, Yao L, Sun J (2018) Construction of a highly efficient display system for baculovirus and its application on multigene co-display. Mol Genet Genomics 293(5):1265–1277. https://doi.org/10.1007/s00438-018-1459-9
Acknowledgements
The authors acknowledge the assistance of Bo Ning in the maintenance and collection of field samples for this work. The assistance of Dr. Feifei Ren in experimental technology is also greatly appreciated.
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 31872426 and, 31372373), the Natural Science Foundation of Guangdong Province, China (grant number 2016A030311018).
Author information
Authors and Affiliations
Contributions
JS coordinated the project. QH and ZC performed the research. QH HZ and JS wrote the manuscript. QL and PW contributed to new methods. QH, ZC and JS performed data analysis. QH, ZC, and JS interpreted the context of results. All of the authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Roman Pogranichniy.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
He, Q., Cao, Z., Wang, P. et al. Efficient application of a baculovirus-silkworm larvae expression system for obtaining porcine circovirus type 2 virus-like particles for a vaccine. Arch Virol 165, 2301–2309 (2020). https://doi.org/10.1007/s00705-020-04754-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04754-9