
Abstract
Influenza virus infection is a public health problem, causing significant morbidity and mortality. Currently, zanamivir and oseltamivir are in common use, and there are already reports of antiviral resistance. Several studies have shown the antiviral potential of a wide variety of plant-based natural compounds, among them those of the quinone type. In this study, we evaluated the antiviral activity of naphthoquinones isolated from the stem bark of Diospyros anisandra, and we selected zeylanone epoxide (ZEP) to study its effects on influenza A and B viruses. Our results indicated that ZEP inhibits the replication of influenza A and B viruses, at early and middle stages of the replication cycle. Confined nuclear localization of the viral NP indicated that ZEP affects its intracellular distribution and reduces viral yield. This is the first report on the antiviral properties and possible mechanism of action of ZEP in vitro, showing its broad-spectrum activity against influenza A and B viruses.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Influenza viruses belong to the family Orthomyxoviridae and are characterized by a segmented negative-strand RNA genome. These enveloped viruses are divided into four types: A, B, C and D. Types A and B have eight genomic segments and three membrane proteins: neuraminidase (NA), hemagglutinin (HA) and matrix (M2), while influenza C and D viruses have seven genomic segments and two surface proteins: HEF and M2 [4, 6, 33].
Influenza virus treatment is currently limited to a small number of neuraminidase inhibitors whose effectiveness is threatened by the emergence of antiviral resistance [5, 21, 25]. The search for compounds with novel structures and antiviral properties has long included the screening of natural products [27]. Within this class of compounds, naphthoquinones represent an excellent group for study, exhibiting a diverse range of reported activities, including antibacterial, antifungal, antiparasitic, anticancer and antiviral activity [14, 26]. Previous studies focused on the antiviral effect of natural and synthetic quinones have shown fusion-inhibition properties towards the HA of influenza H3N2 viruses [2] or anti-NA activity [37]. Similarly, in silico studies of natural naphthoquinones, such as plumbagin and juglone, have identified these compounds as potential drugs capable of interacting with NA and HA proteins of influenza virus [10, 38].
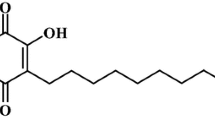
As part of a search for natural active compounds from the native flora of the Yucatan Peninsula, we selected Diospyros anisandra, a native species rich in quinone compounds. We purified a series of quinones, together with terpenoids, from the stem bark of this plant [3, 35]. Some of the quinone compounds showed anti-mycobacterial properties against resistant strains of Mycobacterium tuberculosis and displayed low cytotoxicity in peripheral human cells [34]. In this study, we investigated the antiviral activity of naphthoquinones isolated from D. anisandra, and identified zeylanone epoxide (ZEP) as a compound with promising activity (Fig. 1). It was determined that influenza A and B virus replication is inhibited by interfering with the early and middle stages of the virus life cycle by affecting the intracellular distribution of viral NP protein and reducing viral titres.

Chemical structure of ZEP isolated from D. anisandra
Materials and methods
Cells and viruses
Madin-Darby canine kidney (MDCK) cells (donated by Instituto de Diagnostico y Referencia Epidemiologico InDRE/IRR FR-58) were maintained in 1X Dulbecco´s modified Eagle’s medium (DMEM, GIBCO) supplemented with 10% fetal bovine serum (GIBCO) and 100 U of penicillin and 100 µg of streptomycin (GIBCO) per mL. Cells were grown at 37 °C with 5% CO2.
Influenza virus strains were provided by the Virology Laboratory of the Regional Research Center “Dr. Hideyo Noguchi” from the Universidad Autonoma de Yucatan. Viruses identified as A/Yucatan/2370/09 (H1N1) pdm, B/Yucatan/286/2010 (Victoria lineage), A/Mexico/InDRE797/10 (H1N1-H275Y) pdm and A/Sydney/5/97 (H3N2) were propagated in MDCK cells in the presence of 1 µg of TPCK-trypsin (SIGMA) per mL and stored at -70 °C until use. The viral titre was determined in MDCK cells by a standard plaque assay protocol.
Identification and isolation of ZEP
ZEP and other naphthoquinones were isolated from a hexanic extract of stem bark (Online Resource 1) as described previously by our group [3, 35]. Briefly, powdered stem bark (951 g) from D. anisandra (voucher specimen deposited at the herbarium “Roger Orellana” under collection number 23) was extracted by static maceration at ambient temperature with n-hexane (5 L) for 24 h. Three extractions were performed to obtain 7.03 g (0.74%) of crude n-hexanic extract. This extract was subjected to vacuum liquid chromatography on silica gel and eluted with a gradient of increasing polarity consisting of n-hexane, acetone and methanol to produce 11 fractions. Fraction 4 (4.94 g) was suspended in an aqueous solution of 5% KOH and extracted seven times with CH2Cl2. The organic phase was evaporated, and the aqueous solution was acidified with 10% hydrochloric acid and extracted with CH2Cl2 to produce the quinone fraction (QF: 818.9 mg). The QF was subjected to normal-phase column chromatography (CC) on silica gel (eluted with an n-hexane-ethyl acetate gradient with increasing polarity) to produce 11 fractions. QF-7 was purified by normal-phase CC on silica gel (eluted with isocratic CH2Cl2) to yield ZEP (4.3 mg). ZEP and other naphthoquinones were dissolved in dimethyl sulfoxide (DMSO) and diluted with culture medium for the following assays.
Cytotoxicity assay
The cytotoxicity of each metabolite was evaluated in MDCK cells as described previously [24]. Briefly, MDCK cells were seeded in 96-well plates at a cell density of 5 × 104 cells/well and incubated for 24 h at 37 °C with 5% CO2. Cells were washed twice with phosphate-buffered saline (PBS) and incubated with 100 µL of six different dilutions of each compound (100-1.56 µM) in quadruplicate. Cells were incubated with the compounds at 37 °C for 72 h. Cell viability was determined by staining with 0.4% crystal violet in methanol and then reading the absorbance at 490 nm in a Multilabel Plate Reader (Victor 3x, Perkin Elmer 2030). Cell viability was determined by comparing optical density (OD) from treated cells with OD of control cells, which were arbitrarily considered 100% viable. The mean cytotoxic concentration (CC50) was determined by plotting percent cell viability against compound concentration (µM), followed by nonlinear regression analysis using GraphPad Prism 6.01 software (San Diego, CA). For each compound tested, a cell control with only DMEM and 1% DMSO was included.
Antiviral assay
MDCK cells were seeded at a density of 5 × 104 cells/well and incubated for 24 h at 37 °C with 5% CO2. To measure the antiviral activity, cells were infected with viruses at an MOI of 0.01 in the presence of various concentrations of each compound diluted in 1X DMEM supplemented with 1 µg of TPCK-trypsin per mL and incubated at 37 °C in 5% CO2 for 72 h. After this, CPE was observed microscopically, and the supernatant of each compound at each concentration was recovered and stored at -80 °C to be assessed in a plaque assay.
The 50% inhibitory concentration (IC50) was defined as the concentration of compound required to inhibit viral infection of 50% of the cells and was determined by nonlinear regression analysis using the GraphPad Prism Version 6.01 program (San Diego, CA).
Plaque assay
MDCK cells were seeded at a density of 5 × 105 cells/well in 12-well plates for 24 h at 37 °C with 5% CO2. Cells were infected with serial dilutions (1 × 10-1 to 1 × 10-6) of supernatants harvested from each concentration of ZEP and incubated for 1 h at room temperature. Then, the viral inoculum was removed and the cells were incubated with overlay medium in 3% agarose for 72 h at 37 °C in 5% CO2. Cells were stained with 0.4% crystal violet in methanol. The plaques were counted by visual examination.
Time-of-addition experiments
Time-of-addition experiments were performed to evaluate whether ZEP affects entry, replication, or release of the virus during one or multiple cycles of replication.
The first assay was performed for one cycle of replication [13]. Briefly, confluent monolayers of MDCK cells in 24-well tissue culture plates were inoculated with virus at an MOI of 1. After adsorption for 60 min, the monolayers were incubated in DMEM containing 1 µg of TPCK-trypsin per mL with 100% humidity and 5% CO2 at 37 °C (time zero). Test medium containing 12.5 µM ZEP was applied to the cells from 0 to 2, 2 to 4, 4 to 6, 6 to 8, 8 to 10 or 0 to 10 h. After incubation, the cells were incubated in fresh medium without ZEP. At 10 h postinfection (p.i.), culture supernatants were collected and virus production was measured using a plaque assay.
In the second assay, MDCK cells were seeded at a cell density of 5 × 104 cells/well and infected with influenza A virus at an MOI of 0.01 and treated with ZEP (12.5 µM) before, during and after viral infection [20, 22]. After a 72-h incubation, cells were stained with 50 µL of 0.4% of crystal violet in methanol. Absorbance was measured at a wavelength of 490 nm. The percentage of viral inhibition was calculated by the formula [(A-B) / (C-B)] × 100, where A is the OD of the infected cells treated with the compounds, B is the OD of the virus control, and C is OD of the control cells.
Hemagglutination inhibition assay (HAI)
An HAI assay was performed to assess whether ZEP inhibits hemagglutination activity. HAI assays were performed in 96-well U-bottom plates. ZEP was serially diluted in PBS (50-3.125 µM), mixed with 4 HA units of virus and incubated for 1 h at 4 °C. Then, 50 µL of a 1% suspension of turkey red blood cells (RBC) was added and incubated for 1 h at 4 °C. The HAI titre corresponded to the last dilution of ZEP that inhibited virus hemagglutination. Controls for the assay included virus + RBC, ZEP + RBC, and RBC alone.
Neuraminidase inhibition assay (NAI)
An NAI assay was performed to evaluate whether ZEP affects the neuraminidase activity of influenza virus. Viruses were titrated to a standard NA enzyme activity and mixed with tenfold serial dilutions of either ZEP or oseltamivir carboxylate (OC) according to WHO/CDC protocols [36]. The relative neuraminidase activity was calculated as follows: % relative activity of NA = (NAt / NAv) × 100, where NAt is the activity of virus in the presence of the compound and NAv is virus activity [31]. OC was kindly donated by Hoffmann, La Roche Ltd. (Basel, Switzerland).
Quantification of the influenza A virus NP genome segment by real-time RT-PCR
To assess whether ZEP had an effect on the early synthesis and replication of the virus, a standard curve for quantification of the NP segment was performed as follows: MDCK cells were seeded at 1.5 × 105 cells/well in 24-well plates until confluence. Cells were infected with virus at an MOI of 0.01 for 1 h at room temperature, the inoculum was removed, and the cells were incubated with 12.5 µM ZEP in DMEM supplemented with 1 µg of TPCK-trypsin per mL. At 8, 12 and 36 h p.i., cells were harvested, and total viral RNA was extracted using a QIAamp viral RNA Mini Kit (QIAGEN) according to manufacturer’s instructions. To quantify viral NP, total RNA was reverse transcribed with random hexamers using M-MLV reverse transcriptase (Promega). cDNA was amplified by real-time RT-PCR using a QuantiFast probe RT-PCR Kit (QIAGEN) according to the manufacturer’s instructions, using a StepOnePlus Real-Time PCR System (Applied Biosystems). The sequences of the primers used for detection and quantification of NP were as follows: forward, 5′-TTG TAG CAA CAG GTG GGC ATG A-3′; reverse; 5′-GAG CTG TGT TCT GGT TTG TTT CAT-3′; probe, 5’-TGA GGT ATG CCA C’’T’’A TCC GTG AGT CGA AC-3′. PCR conditions were as follows: one cycle at 95 °C for 5 min, followed by 40 cycles at 95 °C for 10 s and 60 °C for 30 s. For the standard curve, the plasmid PDZ-NP, containing NP from A/Yucatan/2370/09 (H1N1), was used: (R2 = 0.98 between 1 × 103 and 1 × 109 copies). Serial dilutions in duplicate from an initial concentration of 747.8 ng DNA/µL were performed.
Indirect immunofluorescence assay
MDCK cells were grown on glass coverslips, infected with virus (MOI = 1) and treated with 12.5 µM ZEP after infection. At 4, 6 or 8 h p.i., cells were fixed and permeabilized with methanol/acetone. Cells were blocked overnight with 3% bovine serum albumin (BSA)/PBS at 4 °C. After washing, cells were then incubated with anti-IAV NP antibody (NR-19868 BEI Resources for H1N1; MCA400 AbD Serotec for H3N2) and FITC-conjugated secondary antibody (3105 Millipore). Nuclei were stained with DAPI (Sigma) according to the manufacturer´s instructions. Cells were observed directly using a fluorescence microscope (DM4000 B LED; Leica, Germany) equipped with a CCD camera.
Statistical analysis
Data are presented as the mean ± standard deviation of three independent experiments. Statistical significance was calculated in GraphPad Prism 6.01 software using one-way ANOVA analysis with Dunnett´s test, with P-values <0.05 considered significant.
Results
Cytotoxicity of quinones
Six natural naphthoquinones, plumbagin, zeylanone epoxide, 3,3-biplumbagin, droserone, 2,3-epoxiplumbagin, and cis-isoshinanolone, were all isolated from a hexanic extract of Diospyros anisandra with a purity of >95% [35]. Prior to antiviral analysis, the effect of quinones (in the range of 100-1.56 µM) on MDCK cell viability was evaluated. 3,3’-Biplumbagin was the most cytotoxic quinone, followed by plumbagin, zeylanone epoxide and 2,3-epoxiplumbagin, with CC50 values in the range of 2.80 to 30 µM. Droserone (>100 µM) and cis-isoshinanolone (>100 µM) displayed higher CC50 values, indicating 100% cell viability at all concentrations tested (Online Resource 1). No significant cytotoxicity was observed when ZEP was used at ≤ 12.5 µM. These results were used to determine the range of concentrations of quinones to be used in the subsequent experiment.
Inhibition of influenza A and B virus multiplication by ZEP
The antiviral activity of all of the compounds against influenza A virus was evaluated in vitro. Briefly, MDCK cells were infected with influenza A (H1N1) pdm09 virus (MOI: 0.01) and treated with compounds at the indicated concentrations. After 72 h, virus titres were determined by plaque assay. ZEP was the most active, with an IC50 value of 0.65 µM and a selective index of 33.3, suggesting that ZEP possesses anti-IAV activity, as shown in Fig. 2a. The other naphthoquinones did not show activity against influenza A viruses (Online Resource 1).

Inhibitory effect of ZEP on influenza virus titres. MDCK cells were infected with a) A/Yucatán/2370/09 (H1N1) pdm, b) A/México/InDRE797/10 (H1N1) pdm, c) A/Sydney/5/97 (H3N2), or d) B/Yucatán/286/10 (MOI: 0.01) and treated with serial twofold dilutions of ZEP. Culture supernatants were harvested at 72 h, and virus titers were determined by plaque assay. Data represent mean values from three independent replicates. A significant difference was observed between ZEP-treated cells and the untreated virus control. ****, p < 0.0001
To evaluate whether ZEP is effective against a broad spectrum of viral strains, the same experiment described above was performed using other influenza viruses: an H1N1 strain resistant to oseltamivir (A/Mexico/InDRE797/10), an H3N2 strain (A/Sydney/5/97) and an influenza B virus strain (B/Yucatan/286/2010). As shown in Fig. 2b-d, ZEP significantly inhibited plaque formation by all three viruses at concentrations of 3.125-12.5 µM. The level of plaque reduction was almost 100% at concentrations of 12.5 and 6.25 µM. These results show that ZEP has activity against a broad spectrum of influenza A and B viruses (Table 1).
Effect of ZEP on influenza virus infection under different treatment conditions
In order to investigate the stage(s) at which ZEP inhibits influenza virus replication in vitro, time-of-addition experiments were performed.
First, to determine at which step of the replication cycle after adsorption ZEP exerts its effect, we performed a single-cycle replication assay. Briefly, MDCK cells were infected (MOI: 1) and treated with 12.5 µM ZEP for different lengths of time, and at 10 h p.i., the virus yield was measured by plaque assay. The results showed that ZEP treatment significantly reduced the virus yield, with its greatest effect between 0 and 6 h p.i., irrespective of the virus strain (Fig. 3a-d). No infectious progeny were detected when the treatment was constantly maintained for 10 h, probably due to inhibition of different stages of the influenza virus infectious cycle. The maximum inhibitory activity was also observed during the first 4 h of infection, suggesting that ZEP acts at the early (internalization, fusion or uncoating) and/or middle (nuclear transport/replication) stages, with a consequent reduction in the number of released infectious particles.

Effect of different treatment conditions on the inhibitory effect of ZEP on influenza virus infection. Single-cycle assay with a) A/Yucatán/2370/09 (H1N1) pdm, b) A/México/InDRE797/10 (H1N1) pdm, c) A/Sydney/5/97 (H3N2) and d) B/Yucatán/286/10) at an MOI of 1. Cells were treated with 12.5 µM ZEP for the specified time period after adsorption: 0-2, 2-4, 4-6, 6-8, 8-10, 0-10 h. Viral yields were determined at 10 h p.i. by plaque assay. Multiple-cycle assay for e) A/Yucatán/2370/09 (H1N1) pdm at an MOI of 0.01 under different treatment conditions with 12.5 µM ZEP. After a 72-h incubation, cells were stained and the absorbance was read. Data are expressed as the mean ± standard deviation of three independent replicates. Asterisks represent significant differences between treatment and the control. ***, P < 0.001; ****, P < 0.0001
The second experiment was performed to evaluate the effect on multiple cycles of replication. Briefly, MDCK cells were treated with 12.5 µM of ZEP before, during, and after infection with the virus at an MOI of 0.01. This allowed us to assess, first, whether ZEP interacts with sialic acid receptors on the host cell or inhibits the virus-cell binding step (pre-treatment of cells before virus infection), second, whether it affects virus entry into cells (co-treatment during virus infection) or has a virucidal effect (pre-treatment of virus before infection), and third, whether the compound inhibits viral replication or virus release (post-treatment of cells) [1, 11]. As shown in Fig. 3e, pre-treatment of cells with ZEP did not prevent infection, suggesting that there is no direct interaction with the cell surface. However, when cells were infected with viruses in the presence of ZEP, an inhibitory effect of 50% was observed. Interestingly, this effect increased proportionally with the length of time that the virus was exposed to the compound, increasing 100% of inhibition. This suggests that ZEP may act either by direct binding to the virus particle (virucidal activity by pre-treatment of virus) or by blocking at a post-adsorption stage, as we showed during the single-cycle assay (Fig. 3a-d).
A direct interaction of ZEP with the surface proteins HA and NA can be ruled out because neither inhibition of hemagglutination (Table 2) nor of neuraminidase activity (Fig. 4a) was observed. However, as shown in Fig. 4b, in the presence of 12.5 µM ZEP, there is a significant decrease (p < 0.0001) in the number copies of the genome segment NP compared to the viral control, with an inhibitory effect greater than 90% even after four replication cycles (36 h), suggesting that ZEP inhibits viral RNA synthesis or possibly an even earlier step.

Measurement of antiviral activity of ZEP by NA assay and qRT-PCR. a) Effect of ZEP on the NA activity of influenza A and B viruses. b) Effect of ZEP on the synthesis of viral RNA. The NP segment of the virus was amplified by real-time qRT-PCR using total RNA extracted from MDCK cells at 8, 12 and 36 h after the inoculation. The results are shown as the mean of two replicates ± standard deviation. Significant differences between the virus control and ZEP-treated cells were observed. ****, P < 0.0001
Next, we used indirect immunofluorescence to investigate the effect of ZEP on the intracellular trafficking of the viral NP protein at 4, 6 and 8 h p.i. As shown in Fig. 5, in the virus control, at 4 h p.i., NP protein was mainly localized in the nucleus, showing a strong green fluorescence in infected cells, and then, by 8 h p.i., NP was translocated to the cytoplasm, as indicated by green fluorescence both in the cell body and the nucleus (Fig. 5, panels a1-c3). In contrast, in ZEP-treated cells, NP was hardly detectable at 4 h p.i. (Fig. 5 panels d1-d3), and at 6 and 8 h p.i. it was still retained in the nucleus, as indicated by a strong fluorescence signal (Fig. 5, panels d1-d3). The same effect was observed with H3N2 viruses, although the number of infected cells was higher than was observed for H1N1 (Fig. 6). These results suggest that ZEP might affect transport from the nucleus during the middle and late stages of the replication cycle, which would not only affect the number of cells infected but would also reduce the number of infectious particles released.

Effect of ZEP on the intracellular distribution of the viral NP protein of influenza A (H1N1) pdm virus. MDCK cells were infected with A/Yucatán/2370/09 (H1N1) pdm (MOI: 1) and treated with 12.5 µM ZEP. At 4, 6 and 8 h p.i., cells were labeled with anti-IAV NP. Scale of images, 200 µm

Effect of ZEP on the intracellular distribution of viral NP protein of influenza A (H3N2) virus. MDCK cells were infected with A/Sydney/5/97 (H3N2) (MOI: 1) and treated with 12.5 µM ZEP. At 4, 6 and 8 h p.i., cells were labeled with anti-IAV NP. Scale of images, 200 µm
Discussion
In this study, we report the antiviral activity of natural quinones from Diospyros anisandra, including zeylanone epoxide (ZEP), which was previously described for the first time by our group [35]. Studies have shown that quinones have two main mechanisms of toxicity: either by forming reactive oxygen species (ROS) or by behaving as electrophiles capable of forming covalent bonds with nucleophilic groups in biological molecules such as glutathione (GSH) [18, 19]. All compounds evaluated in our study differed in their cytotoxicity depending on their chemical structure. Therefore, and based on the chemical structure of the naphthoquinones evaluated (Online Resource 1), we suggest that the cytotoxicity we observed was probably attributable to a redox effect [18, 30].
An initial screening to detect antiviral activity indicated that, of the six naphthoquinones, only ZEP inhibited the replication of influenza A (H1N1) pdm virus. Importantly, it was found that ZEP was also effective against H1N1 pdm-H275Y, H3N2 and influenza B viruses, suggesting broad-spectrum anti-influenza activity, resulting in a significant reduction in virus yield in MDCK cells, as determined by plaque assays. Reduced release of viral particles of some strains of influenza virus was confirmed by 1) reduction in the amount of viral genomic RNA, as determined by quantitative RT-PCR, and 2) inhibition of nuclear NP export, as determined by indirect immunofluorescence. In an attempt to determine its possible mode of action, ZEP activity was assessed using a series of assays that demonstrated that in assays involving either one cycle of replication or multiple cycles of replication, influenza A and B viruses were significantly inhibited. Some of our results indicate a possible virucidal effect by direct interaction of ZEP with the virus that does not affect HA or NA activity. In this regard, it has been reported that quinone compounds such as the anthraquinone aloe-emodin are capable of affecting the integrity of the viral envelope, preventing absorption [32].
Immunofluorescence ZEP-treated infected cells suggested that this compound inhibits the nuclear export of the NP protein, thereby reducing the number of viral particles. The NP protein of influenza virus, encoded on segment five of the viral genome, is one of the most abundant viral proteins in infected cells. Among its main functions is to bind stoichiometrically to the viral RNA, stabilizing the ribonucleoprotein (RNP) complex, but there is also evidence of its involvement in nuclear trafficking and vRNA transcription and replication [7, 15, 23]. The traffic of the RNP is reflected in the distribution of its major protein component, NP. At early times postinfection, NP is found predominantly in the nucleus of infected cells, but at later times postinfection, substantial amounts accumulate in the cytoplasm [29]. It has been reported recently that the nuclear export of the RNPs that are incorporated into the progeny virion is a process that is mediated via the interaction of a protein assembly comprising the cellular chromosome region maintenance 1 receptor (CRM1) and the viral M1, NEP, and vRNP [12, 15]. Based on the results of this study, it is likely that ZEP inhibits the nuclear export of RNP, interfering with the daisy chain complex (MI/NEP/vRNP) before it is released to the cytoplasm [9]. However, the direct interaction of ZEP with this complex needs to be examined further.
Numerous compounds have been reported to be NP inhibitors, such as verdinexor, a selective inhibitor of nuclear protein [28], TGBG, a natural product that suppress the nuclear export of the vRNP complex via the PI3K/Akt signaling pathway [8], dapivirine, which affects influenza A and B viruses by inhibiting the nuclear entry of NP in the early stage of viral replication [16], and ZBMD-1, which blocks the nuclear export of NP by impeding the binding of NP to CRM1 [17].
In summary, in this study, we demonstrated that ZEP possesses antiviral activity against influenza A and B viruses in vitro. It was found to reduce viral titers and block the extranuclear transport of NP, indicating that it might be considered a promising antiviral agent against influenza viruses. However, it is necessary to investigate in more detail what other proteins involved in the transport of vRNP are affected by ZEP.
References
Aoki-Utsubo C, Chen M, Hotta H (2018) Time-of-addition and temperature-shift assays to determine particular step(s) in the viral life cycle that is blocked by antiviral substance(s). Bio-protocol 8(9):e2830
Bodian D, Yamasaki B, Buswell R, Stearns J, White J, Kuntz D (1993) Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochem 32:2976–2978
Borges R, Uc A, Quintal C, Canché G, Cáceres M (2013) A selective chemical method for the separation of quinones from the stem bark of Diospyros anisandra. Int J Curr Pharm Res 5:13–17
Bouvier N, Palese P (2008) The biology of influenza virus. Vaccine 12:49–53
Burnham A, Baranovich T, Govorkova E (2013) Neuraminidase inhibitors for influenza B virus infection: Efficacy and resistance. Antiviral Res 100:520–534
CDC (2017) Types of influenza viruses. https://www.cdc.gov/flu/about/viruses/types.htm
Cianci C, Gerritz S, Deminie C, Krystal M (2013) Influenza nucleoprotein: promising target for antiviral chemotherapy. Antivir Chem Chemother 23:77–91
Chang S, Park J, Kim Y, Kang J, Min J (2016) A natural component from Euphorbia humifusa Willd displays novel, broad-spectrum anti-influenza activity by blocking nuclear export of viral ribonucleoprotein. Biochem Biophys Res Commun 471:282–289
Chaimayo C, Hayashi T, Underwood A, Hodges E, Takimoto T (2017) Selective incorporation of vRNP into influenza A virions determined by its specific interaction with M1 protein. Virology 505:23–32
Chavan R, Shinde B, Girkar K, Mandage R, Chowdhary A (2014) Identification of potent natural inhibitors against H1N1/A/2009 virus using in silico subtractive genomics approach and docking technology. Int J Pharm Res 6:105–113
Dayem AA, Choi HY, Kim YB, Cho S-G (2015) Antiviral effect of methylated flavonol isorhamnetin against influenza. PLoS One 10(3):e0121610
Dou D, Revol R, Östbye H, Wang H, Daniels R (2018) Influenza A virus cell entry, replication, virion assembly and movement. Front Immunol 9:1581–1597
Furuta Y, Takahashi K, Sangawa H, Kuno-Maekawa M, Uehara S, Nomura N, Kozaki K, Egawa H, Shiraki K (2005) Mechanism of Action of T-705 against influenza virus. Antimicrob Agents Chemother 49:981–986
Hook I, Mills C, Sheridan H (2014) Bioactive naphthoquinones from higher plants. In: Rahman A (ed) Studies in natural products chemistry. Elsevier, New York, pp 119–160
Hu Y, Sneyd H, Dekant R, Wang J (2017) Influenza A virus nucleoprotein: a highly conserved multifunctional viral protein as a hot antiviral drug target. Curr Top Med Chem 17:2271–2285
Hu Y, Zhang J, Musharrafieh R, Ma C, Hau R, Wang J (2017) Discovery of dapivirine, a nonnucleoside HIV-1 reverse transcriptase inhibitor, as a broad-spectrum antiviral against both influenza A and B viruses. Antiviral Res 145:103–113
Huang F, Chen J, Zhang J, Tan L, Lu G, Luo Y, Pan T, Liang J, Li Q, Luo B, Zhang H, Lu G (2018) Identification of a novel compound targeting the nuclear export of influenza A virus nucleoprotein. J Cell Mol Med 22:1826–1839
Klots L, Hou X, Jacob C (2014) 1,4-Naphthoquinones: from oxidative damage to cellular and inter-cellular signaling. Molecules 19:14902–14918
Kumagai Y, Shinkai Y, Miura T, Cho A (2012) The chemical biology of naphthoquinones and its environmental implications. Annu Rev Pharmacol Toxicol 52:221–247
Kwon H, Kim H, Yoon S, Ryu Y, Chang J, Cho K, Rho M, Park S, Lee W (2010) In vitro inhibitory activity of Alpinia katsumadai extracts against influenza virus infection and hemagglutination. Virol J 7:307–316
Li T, Chan M, Lee N (2015) Clinical implications of antiviral resistance in influenza. Viruses 7:4929–4944
Liu G, Xion S, Xiang Y, Gou C, Ge F, Yang C, Zhang Y, Wang Y, Kitazato K (2011) Antiviral activity and possible mechanisms of action of pentagalloylglucose (PGG) against influenza A virus. Arch Virol 156:1359–1369
Loregian A, Mercorelli B, Nannetti G, Compagnin C, Palù G (2014) Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell Mol Life Sci 71:3659–3683
Makau J, Watanabe K, Kobayashi N (2013) Anti-influenza activity of Alchemilla mollis extract: possible virucidal activity against influenza virus particles. Drug Discov Ther 7:189–195
McKimm-Breschkin J (2013) Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance. Influenza Other Respir Viruses 7:25–36
Nematollahi A, Aminimoghadamfarouj N, Wiart C (2012) Reviews on 1,4-naphthoquinones from Diospyros L. J Asian Nat Prod Res 14:80–88
Newman D, Cragg G (2012) Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75:311–335
Perwitasari O, Johnson S, Yan X, Howerth E, Shacham S, Landesman Y, Baloglu E, McCauley D, Tamir S, Tompkins M, Tripp R (2014) Verdinexor, a novel selective inhibitor of nuclear export, reduces influenza A virus replication in vitro and in vivo. J Virol 88:10228–10243
Portela A, Digard P (2002) The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol 83:723–734
Rodriguez C, Shinyashiki M, Froines J, Yu R, Fukuto J, Cho A (2004) An examination of quinone toxicity using the yeast Saccharomyces cerevisiae model system. Toxicology 201:185–196
Song J, Lee K, Seong B (2005) Antiviral effect of catechins in green tea on influenza virus. Antiviral Res 68:66–74
Sydiskis R, Owen D, Lohr J, Rosler K, Blomster R (1991) Inactivation of enveloped viruses by anthraquinones extracted from plants. Antimicrob Agents Chemother 35:2463–2466
Szewczyk B, Bienkowska-Szewczyk K, Król E (2014) Introduction to molecular biology of influenza A viruses. Acta Biochim Pol 61:397–401
Uc A, Borges R, Said S, Vargas J, González F, Méndez M, Cáceres M, Molina G (2014) Naphthoquinones isolated from Diospyros anisandra exhibit potent activity against pan-resistant first-line drugs Mycobacterium tuberculosis strains. Pulm Pharmacol Ther 27:114–120
Uc A, Molina GM, Said S, Méndez M, Cáceres M, Borges R (2013) A new dimeric naphthoquinone from Diospyros anisandra. Nat Prod Res 27:1174–1178
WHO (2011) Manual for the laboratory diagnosis and virological surveillance of influenza. http://apps.who.int/iris/bitstream/10665/44518/1/9789241548090_eng.pdf
Yang Y, Zhao D, Yuan K, Zhou G, Wang Y, Xiao Y, Wang C, Xu J, Yang W (2014) Two new dimeric naphthoquinones with neuraminidase inhibitory activity from Lithospermum erythrorhizon. Nat Prod Res 29:908–913
Yang Z, Yang Y, Wu F, Feng X (2013) Computational investigation of interaction mechanisms between juglone and influenza virus surface glycoproteins. Mol Simul 39:788–795
Acknowledgments
This work was supported by CONACYT (project number 126763). Cetina-Montejo also acknowledges CONACYT for grant number 394861. We would like to thank technician M. Cáceres-Farfán for providing technical support in the GC-MS.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have not conflict of interest.
Additional information
Handling Editor: Diego G. Diel.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cetina-Montejo, L., Ayora-Talavera, G. & Borges-Argáez, R. Zeylanone epoxide isolated from Diospyros anisandra stem bark inhibits influenza virus in vitro. Arch Virol 164, 1543–1552 (2019). https://doi.org/10.1007/s00705-019-04223-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04223-y