
Abstract
It is well-recognized that human immunodeficiency virus type-1 (HIV-1) mainly targets CD4+ T cells and macrophages. Nonetheless, during the past three decades, a huge number of studies have reported that HIV-1 can directly or indirectly target other cellular components of the immune system including CD8+ T cells, B cells, dendritic cells, natural killer cells, and polymorphonuclear neutrophils (PMNs), among others. PMNs are the most abundant leukocytes in the human circulation, and are known to play principal roles in the elimination of invading pathogens, regulating different immune responses, healing of injured tissues, and maintaining mucosal homeostasis. Until recently, little was known about the impact of HIV-1 infection on PMNs as well as the impact of PMNs on HIV-1 disease progression. This is because early studies focused on neutropenia and recurrent microbial infections, particularly, during advanced disease. However, recent studies have extended the investigation area to cover new aspects of the interactions between HIV-1 and PMNs. This review aims to summarize these advances and address the impact of HIV-1 infection on PMNs as well as the impact of PMNs on HIV-1 disease progression to better understand the pathophysiology of HIV-1 infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although more than three decades have passed since the discovery of human immunodeficiency virus (HIV)-1, the etiologic agent of acquired immunodeficiency syndrome (AIDS), HIV-1 infection is still an incurable disease. This is, at least in part, due to the fact that HIV-1 almost targets/negatively affects all the cell types of the immune system in infected individuals [1,2,3,4,5,6,7]. For instance, CD4+ T cells are the major target cells for HIV-1 infection and replication. Nonetheless, not all CD4+ T cells are preferred for HIV-1 replication, since it replicates very efficiently in activated but not resting CD4+ T cells [8]. The very rapid viral replication in activated CD4+ T cells ensures high viral load as well as the high mutation rate that enables to HIV-1 escape both immune responses and antiviral therapeutics. On the other hand, infection of resting CD4+ T cells enables HIV-1 to become latent(transcriptionally silent), and thus unrecognizable to the immune system and antiviral therapeutics, ensuring viral persistence in HIV-1 patients [9]. Macrophages are the second most favored cells for HIV-1 infection and replication [10, 11]. However, unlike CD4+ T cells, which are susceptible to HIV-1-related cytopathic effects that result in a massive depletion of CD4+ T cells during the course of HIV-1 infection, macrophages show much more resistance against these cytopathic effects [12, 13]. Interestingly, HIV-1 hijacks this property to ensure its persistence through establishment of a stable latent infection in these cells [13,14,15]. Moreover, HIV-1 can harness monocytes/macrophages as vehicles to spread throughout the body compartments (such as the central nervous system and gut) and between target cells, thereby supporting its persistence [16,17,18]. HIV-1 can also infect other immune cells such as dendritic cells albeit at a rate of 1 to 2 orders of magnitude lower than infection in CD4+ T cells [19, 20]. Despite the fact that dendritic cells are not considered major targets for direct HIV-1 infection, dendritic cells can enhance HIV-1 infectivity towards CD4+ T cells [16, 17]. In addition, during the course of HIV-1 infection, dendritic cell numbers, phenotypes, and functions are grossly altered resulting in various immunological alterations, (reviewed in [21]). These alterations subsequently inhibit potent anti-HIV-1 immune responses, all of which are required to support HIV-1 persistence. This is also the case with other immune cells such as natural killer cells and basophils/mast cells (reviewed in [21]). Hence, we can say that HIV-1 is able to hijack and exploit almost all cell types of the immune system for its advantage, taking into consideration the individual differences between these cells. Accordingly, it is not surprising to realise that polymorphonuclear neutrophils (PMNs) undergo similar peturbation during the course of HIV-1 infection.
This review aims to address critical issues related to PMNs and HIV-1 infection. However, to begin we will concisely introduce the reader to some of the major biological aspects of PMNs that include: (i) PMNs infiltration, pathogen recognition and elimination; (ii) PMNs cross-talking with other immune cells; and (iii) PMNs and mucosal homeostasis, especially in the gut compartment. This introduction will allow a better estimatation and understanding of the very critical impact of PMNs on health and disease (i.e. during HIV-1 infection). Next we will address the impact of HIV-1 infection on PMN number and function, and then we will address the consequences of PMN alterations on the pathogenesis of HIV-1 infection. Finally, this review will address the role of PMNs in the pathology of gut mucosa and microbial translocation during HIV-1 infection, to improve our understanding of the pathophysiology of HIV-1 infection.
PMNs: roles in pathogen elimination and immune mediation
PMNs are the most abundant leukocytes in the human circulation, constituting up to 60-70% of the total number of circulating leukocytes. These granulocytes are generated in the bone marrow during the process of hematopoiesis at a rate of ~ 100 billion cells per day under normal conditions; this number may reach ~ 1 trillion during serious infections. Relatively speaking, PMNs are short lived cells; however, recent studies have indicated that PMNs may have 10 times (5.4 days) longer lifespans than that previously reported under homeostatic conditions [22]. As professional innate immune cells equipped with several defense mechanisms, PMNs can mediate different effector functions against extracellular pathogens. Intriguingly, it has been recently revealed that PMNs have the capability to eliminate intracellular pathogens including viral pathogens such as HIV-1 [23].
PMNs are characterized by their ability to rapidly infiltrate to the site of infection/inflammation to mediate their effector functions [24, 25]. To accomplish this task, PMNs express a variety of receptors which includes those required for adhesion to endothelial cells during the infiltration process such as selectins, selectin-ligands and integrins, among others [25]. After leaving the vascular compartment, chemo-attractant receptors (such as chemokine and cytokine receptors) facilitate their migration to the site of pathogenic stimulation [26, 27]. In order to detect pathogens, PMNs express several classes of receptors such as toll-like receptors (TLRs), nod-like receptors (NLRs), dectin-1 [28,29,30], Fc-receptors (FcR) that recognize antibody-opsonized pathogens, and complement receptors that recognize complement-opsonized targets [31,32,33,34,35,36]. In addition, PMNs express receptors for granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) that participate in enhancing their responsiveness and a metabolic burst, while prolonging their survival once at the site of stimuli [31, 36]. In turn, this helps further recruitment of immune cells to the site of inflammation to effectively eliminate invading pathogens and terminate inflammation, indicating that PMNs can be seen as one of the first lines of defense against invading pathogens.
There are several different mechanisms by which PMNs can eliminate pathogens. These include: phagocytosis, neutrophil extracellular traps (NETs) formation, antibody-dependent cellular cytotoxicity, degranulation and the release of antimicrobial peptides [24, 25, 31, 37, 38]. For instance, once a pathogen is encountered by a PMN, particularly in the circulation, phagocytosis takes place. The presence of serum favors triggering of phagocytosis while inhibiting the induction of NETs [39]. This information indicates that the extracellular milieu may significantly affect the mechanisms of killing employed by PMNs. It is also of considerable importance to realise that PMNs are extremely potent and very efficient phagocytes that can internalize IgG-opsonized particles within less than 60 seconds when compared to other professional phagocytes such as macrophages, which require several minutes to digest similar amounts and types of ingested particles [40,41,42]. The degradation process takes place once a pathogen or a microorganism is inside the phagosome of a PMN. This is accomplished by mediating the fusion of the PMNs’ granules with the phagosome. These granules contain several digesting and hydrolyzing enzymes that act as weapons to destroy the phagocytized pathogens [31]. PMNs further recruit nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase (NOX) to the phagosome to optimally destruct their contents [43, 44].
In another example, PMNs have suicidal capabilities, i.e. to capture and kill invading microorganisms in order to limit their spread. PMNs release highly sticky net-like structures upon infiltration to the site of invasion [45, 46]. These structures have been designated as NETs and are composed of genomic DNA, histones, and various antimicrobials such as calprotectin, α-defensin, and myeloperooidase (MPO) among others, which combine to efficiently eliminate invading pathogens and prohibit their dissemination [45, 46]. Intriguingly, intact PMNs can also release NETs, indicating that NET formation is not only associated with PMNs cell death [47]. Furthermore, PMNs can mediate the killing of infected cells in an antibody-dependent manner via engagement of FcγR with the Fc portion on IgG-opsonized infected cells [48]. Taken together, these data briefly clarify how PMNs can infiltrate, recognize and eliminate pathogens and also highlight their role as key effector cells in the innate immune system.
Indeed, PMNs function is not only restricted to their ability to eliminate pathogens; they can also engage with several types of immune cells to orchestrate immune responses. For instance, in vitro and in vivo studies have shown that the direct interaction of lipopolysaccharides (LPS)-stimulated PMNs with dendritic cells induces their activation (maturation) and production of tumor necrosis factor alpha (TNF-α) and interleukin-12 (IL-12) [48]. Other studies have also revealed that PMNs are involved in the induction of dendritic cell activation upon the direct interaction of PMNs’ surface molecules, such as macrophage antigen-1 (MAC1) and carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM1), with corresponding molecules on dendritic cells, namely dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) [49, 50]. Moreover, PMNs have been demonstrated to promote dendritic cell survival through a manner dependent on cell-to-cell contact [51]. Alternatively, activated PMNs release different molecules that can mediate dendritic cell activation in a manner independent on direct contact; these include α-defensins, cathelicidins, lactoferrin, and high-mobility group proteins [52]. Accordingly, these activated dendritic cells can then mediate T cell proliferation and shape their polarization towards distinct helper T cell phenotypes [50, 53], thereby shaping the adaptive immune response. However, under certain circumstances, PMN interaction with dendritic cells may not result in their activation. For example, PMNs-released elastases and ectosomes interfere with dendritic cell activation, in part, through increasing the production of transforming growth factor-β1 (TGF-β1), an immunosuppressive cytokine [54, 55]. These data indicate that PMNs are involved in both immune activation and suppression, which seems to be dependent on the particular microenvironment where interactions take place.
In another example, Silva has shown that PMNs and monocytes/macrophages work together in harmony to mediate effective downstream innate immune responses against both extracellular and intracellular pathogens [56]. Activated PMNs recruit monocytes/macrophages to the site of inflammation through secretion of different attractant molecules such as macrophage inflammatory protein-1α (MIP-1α) and MIP-1β, among others [57,58,59]. PMNs can then activate recruited monocytes/macrophages and mediate their polarization toward anti-inflammatory or pro-inflammatory subsets according to the microenvironment of the interaction [60]. In turn, these activated macrophages release G-CSF and GM-CSF that prolong the survival of PMNs [61, 62], maximising the PMNs’ effector functions. Furthermore, these activated macrophages can then mediate and shape the adaptive immune responses, since macrophages are well-recognized to act as professional antigen presenting cells. Interestingly, recent studies have also shown that NET formation by PMNs is regulated by macrophages in a time- and phenotype-dependent manner (for more details see ref. [63]), which reflects the vital relationship between these types of cells, and also highlights the functional complementarily between these cells [56].
In terms of PMN cross-talking with other types of innate immune cells, such as natural killer cells, Spӧrri and colleagues have shown that mice PMNs are critical activators of natural killer cells [64]. They have shown that IL-18-derived from PMNs in combination with IL-12, a dendritic cell-derived cytokine, are critical for triggering the secretion of interferon-γ (IFN-γ) from natural killer cells in Legionella pneumophila-infected mice. Interestingly, the lack of IFN-γ, as a result of neutropenia in infected mice, has been implicated in their inability to clear the bacterial infection [64]. In line with these data, a later study revealed that natural killer cells from neutropinic mice exhibit hyperproliferation, poor survival, and hyporesponsiveness due to a block in their maturation process at an immature stage [65]. The critical impact of PMNs on natural killer cell functions has also been confirmed in neutropinic-related disorders such as autoimmune neutropenia and severe congenital neutropenia [65]. However, once the natural killer cells are activated by PMNs, they can activate dendritic cells by releasing IFN-γ and TNF-α, or through a contact-dependent activation manner [66]. In turn, these activated dendritic cells can then activate adaptive immune responses, as previously discussed. Furthermore, activated natural killer cells have been observed to promote the activation and survival of PMNs in culture studies, both of which rely on direct cell-to-cell contact and cytokine-dependent mechanisms [67]. These data briefly reflect the very critical relationship between these two types of cells. For more details, Costantini and Cassatella have comprehensively reviewed the defensive alliance between PMNs and natural killer cells [68].
PMNs depletion (neutropenia)
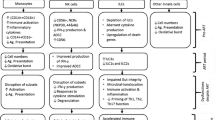
In terms of numerical-alteration, some studies have reported that about 17% of HIV-1 patients exhibit neutropenia [73, 74], while others have reported that up to 50% of HIV-1 patients exhibit neutropenia [75], indicating that neutropenia is relatively common in HIV-1 patients. Recent studies have also indicated that the incidence of severe neutropenia is high in HIV-1 patients living in West Africa, even in those treated with antiretroviral therapy (ART) [76]. Of note ethnic neutropenia is prevalent in individuals of African ancestry [77, 78], which is, at least in part, related to genetic factors [78]. Of particular importance, longitudinal analysis has found that HIV-1 disease progression is directly associated with the severity of neutropenia [75]. Of note, neutropenia has not been only implicated in the disease progression of HIV-1 infection but is also considered as a possible risk factor for HIV-1 transmission during the perinatal period, since a higher PMN count in HIV-1 positive women has been demonstrated to be inversely associated with perinatal HIV-1 transmission risk [79]. PMNs were also shown to play a role in protection against sexual HIV-1 acquisition in adults, as demonstrated by a Taiwanese cohort (which studied HIV-1-exposed but uninfected individuals) [80]. These data reflect the possible impact of neutropenia on disease progression in HIV-1 infected individuals and also the transmission risk to uninfected individuals. Therefore additional investigations are required to further establish the role of neutropenia on HIV-1 disease progression and transmission. However, in this paper we will only address the role of PMNs during HIV-1 infection. Several possible mechanisms, by which HIV-1 infection can contribute to the neutropenia, are mentioned as follows:
-
Direct cytopathic effects related to direct HIV-1 infection. Some early studies indicated that HIV-1 could directly infect PMNs, due to the detection of HIV-1 DNA in these cells [81]. This was further supported by the findings of Biswas et al. who showed that 7.8% of HIV-1 patients and 12% of healthy individuals express the CD4 molecule (the primary receptor for HIV-1 entry into target cells) on 39-97% of their PMN populations [82]. In addition, PMNs constitutively express C-X-C chemokine receptor type 4 (CXCR4 or X4), a major co-receptor involved in HIV-1 entry [82]. Even though this suggests that PMN depletion during the course of HIV-1 infection could, in part, be due to direct HIV-1-related cytopathic effects on PMNs in some patients, there is still no clear indication that HIV-1 can directly infect PMNs. Nonetheless, as professional phagocytes, PMNs can internalize HIV-1 by phagocytosis. HIV-1 might also be able to escape destruction by endosomal compartments withini PMNs, as it does in macrophages [83]. Furthermore, HIV-1 can also its Nef protein to inhibit the formation of phagosomes in macrophages by altering endosomal compartment membrane recycling [84]; thus, it could be assumed that HIV-1 could use the same strategy in PMNs to establish a non-canonical (indirect) mechanisms of infection. These hypotheses remain assumptions and cannot fully explain PMN depletion during HIV-1 infection. Therefore, it is of particular importance to highlight that studying the capability of HIV-1 to infect and mediate cytopathic effects in PMNs remain important questions that need to be answered in the near future. However, other explanations do exist to explain neutropenia during the course of HIV-1 infection.
-
PMNs apoptosis (bystander apoptosis). Early ex vivo studies have demonstrated that PMNs from AIDS patients exhibit remarkable increased rates of apoptosis; however, in vitro incubation of PMNs from AIDS patients with G-CSF significantly decreased the rate of apoptosis [85], suggesting a potential benefit of G-CSF in this situation. Other studies have assessed programmed PMN cell-death at different HIV-1 disease stages using TUNEL assays and propidium iodide, and have shown that accelerated PMN apoptosis occurs at different clinical stages, with a remarkable increase in advanced disease stages [86]. Importantly, Fas-mediated apoptosis in PMNs from HIV-1 patients was proposed to be a mechanism that contributes to neutropenia during HIV-1 infection [87]. It is noteworthy that apoptosis in PMNs from HIV-1 patients was shown to be closely associated with the levels of Fas-FasL surface molecules expressed, which are directly associated with viral load [87], indicating that HIV-1 indirectly mediates PMN apoptosis. Other studies have demonstrated that oxidative stress secondary to HIV-1 infection is associated with increased spontaneous PMN apoptosis during the course of HIV-1 infection, since the inhibition of reactive oxygen species (ROS) resulted in decreased PMN apoptosis [88]. Furthermore, the inhibition of ROS decreased caspase-3 hydrolysis, connecting oxidative stress with the intrinsic (caspase-3), but not the extrinsic (caspase-8), apoptotic pathway in mediating PMN apoptosis during HIV-1 infection [88]. Studies in non-human primates (Rhesus macaques) infected with a pathogenic simian immunodeficiency virus (SIV) strain have also demonstrated that PMNs undergo apoptosis [89]. Intriguingly, SIV-infected Rhesus macaques with increased PMN apoptosis rates were shown to be associated with faster disease progression [89]. PMN apoptosis in SIV-infected Rhesus macaques was also demonstrated to be associated with PMN activation state and ROS production [89]. These data emphasize that the remarkably increased rate of PMN apoptosis during the course of HIV-1 infection is likely, at least in part, to be a possible explanation for their observed depletion during HIV-1 infection [88, 90, 91]; however, other factors may also contribute to neutropenia in HIV-1 patients.
-
Affecting the hematopoiesis process. HIV-1 may directly decrease PMNs counts through affecting the hematopoiesis process in the bone marrow. This is thought possible because several studies have demonstrated that HIV-1 can infect certain CD34+ hematopoietic stem cells that express CD4, CCR5 and CXCR4 on their surfaces [92,93,94,95,96,97,98,99]. Moreover, different viral proteins can also directly affect the hematopoiesis process. For example, in vitro studies have revealed that the HIV-1 envelope glycoprotein 120 (gp120) can suppress the growth of CD34+ hematopoietic stem cells by inducing endogenous TGF-β, which is a growth inhibitory cytokine [100]. Other studies demonstrated that HIV-1 gp120 can also induce apoptosis in CD34+ hematopoietic stem cells in a Fas-manner dependent [101]. In another example, Nef and Tat viral proteins can suppress the growth of granulomonocytic and myeloid progenitor cells, thereby contributing to the neutropenia observed in HIV-1 infection [102,103,104]. Deductively, these direct suppressive effects of HIV-1 and HIV-1 proteins on the hematopoiesis process are consistent with the pan-leukocytopenia and other cytopenias such as anemia and thrombocytopenia observed in HIV-1 patients [105]. Interestingly, HIV-1 can also indirectly affect the hematopoiesis process by altering the bone marrow microenvironment (through modulation of cytokines and growth factors) [106,107,108]. This, in part, can be attributed to the ability of HIV-1 to infect different types of bone marrow stromal cells (e.g. monocytes/macrophages and megakaryocytes) [109,110,111], which are the source of cytokines, growth factors, and other regulators involved in hematopoiesis.
On the other hand, secondary and opportunistic infections (bacterial, fungal, protozoal, or viral infections) are frequently reported in HIV-1 patients, particularly in advanced disease stages [70, 112,113,114,115]. Certain infections may directly contribute to the neutropenia in HIV-1-infected individuals by targeting hematopoietic stem cells, myeloid progenitors, and bone marrow stromal cells, all of which would impair the normal hematopoiesis process. Alternatively, these secondary infections may directly target mature PMNs in the blood circulation. For secondary bacterial infections, HIV-1 patients exhibit an increased risk of Salmonella infection [114,115,116], which is known to cause neutropenia by infecting and suppressing the development of bone marrow hematopoietic stem cells [117]. Similarly, mycobacterial infections such as infection with Mycobacterium tuberculosis were also shown to negatively affect the bone marrow and hematopoiesis [118, 119], taking into consideration that Mycobacterium tuberculosis infection is relatively prevalent in HIV-1 patients [119, 120]. Cytomegalovirus (CMV) infection is another example of a frequently reported secondary viral infection in HIV-1 patients [121]. Of considerable importance, CMV infection is known to target both bone marrow stromal cells and hematopoietic stem cells, which, in turn, suppress the normal hematopoiesis process resulting in cytopenia, including neutropenia [122]. In addition, studies have shown that HIV-1 patients infected with opportunistic fungal pathogens such as Pneumocystis carinii, Candida albicans, or Cryptococcus neoformans show suppressed myelopoiesis and injured bone marrow [123, 124], thereby affecting the generation of new PMNs at the level of hematopoiesis in the bone marrow, also resulting in neutropenia.
-
Increased PMN infiltration rate. The continuous infiltration of PMNs into inflamed lymphatic tissues (e.g. mucosa-associated lymphatic tissues and lymph nodes) and other non-lymphatic tissues that harbor HIV-1 may, in part, provide another explanation for the depletion of PMNs from the circulation of HIV-1 patients (discussed later) [125], especially because PMNs are among the first cells to infiltrate to the site of an immune stimulus, as previously discussed. This assumption, in part, arose from the observation that increased infiltration of macrophages into the gut mucosa was shown to be associated with depleted circulating monocytes in HIV-1 infected individuals [126]. In addition, increased dendritic cell homing to lymphatic tissues was also suggested as an explanation, at least in part, for the decreased dendritic cell numbers in the circulation of HIV-1 patients (reviewed in ref [21]).
-
Therapeutic drugs. Some, but not all, antiretroviral drug classes and other drugs that are used to treat co-infections, opportunistic infections, and/or HIV-1-related or unrelated malignancies can also cause neutropenia [72, 76, 127,128,129,130,131,132,133,134,135]. For antiretroviral drugs, it is well-known that certain antiretroviral drugs such as Zidovudine (AZT), which is a nucleoside analog reverse-transcriptase inhibitor, have bone marrow toxicity and myelosuppression properties [132, 133]. Importantly, studies have shown that HIV-1 patients receiving AZT-containing highly active antiretroviral therapy (HAART) are more likely to experience neutropenia [134]. Other antiretroviral drugs, particularly protease inhibitors, are also known to cause neutropenia [135]. Therefore, during the treatment of neutropenia in ART-treated HIV-1 patients, health care providers should consider the bone marrow associated myelosuppression as side effects of these drugs.
-
Miscellaneous factors. Other factors such as age, ethnicity, genetics, and advanced disease stage may also contribute to neutropenia during the course of HIV-1 infection [78, 136, 137].
Taken together, these data indicate that neutropenia among HIV-1 patients is multifactorial (Fig. 1). Several clinical and experimental investigations have indicated that using cytokines that act as hematopoietic growth factors such as G-CSF and/or GM-CSF may significantly increase PMN counts by overcoming the myelosuppression observed in HIV-1 infection [70, 138]. For G-CSF, several studies have demonstrated that the application of filgrastim, a recombinant human methionyl G-CSF, to HIV-1 patients can significantly alleviate neutropenia [137, 139,140,141]. For GM-CSF, a clinical study has shown that treatment of leukopenic HIV-1 patients with recombinant human GM-CSF significantly increased the total leukocyte count, including PMNs [142]. Another clinical study (a phase III trial) has also demonstrated that administration of GM-CSF to HIV-1 patients at an advanced disease stage significantly increased PMN and CD4+ T cell counts [143]. These hematopoietic growth factors can increase PMN generation at the level of bone marrow hematopoiesis and alleviate apoptosis in the peripheral circulation. Further investigations have shown that using other cytokines such as IL-15 can also significantly reduce the rate of PMN apoptosis [144]. Of note, clinical application of IL-15 should not be considered, because increased plasma levels of IL-15 is associated with HIV-1 disease progression [145].

Factors that drive neutropenia in HIV-1 patients
PMNs: functional defects in HIV-1 infection
It is well-established that PMNs from HIV-1 patients exhibit multiple functional defects [23, 71, 146,147,148,149,150]. For example, several studies have reported an impaired anti-microbial killing activity for PMNs from HIV-1 patients, especially from patients in advanced stages of disease. For instance, the capacity of PMNs from HIV-1 patients to phagocytize bacteria (e.g. Escherichia coli and Staphylococcus aureus) was significantly reduced in patients with low CD4+ T cells count when compared to patients with higher CD4+ T cells count and healthy individuals [151,152,153]. Similarly, investigators have also reported defects in the anti-fungal (e.g. Aspergillus fumigatus and Cryptococcus neoformans) activity of PMNs obtained from HIV-1 patients [154, 155]. HIV-1 and its accessory proteins such as Tat have been implicated directly in the impairment of phagocytosis and respiratory burst in different phagocytes, including PMNs [149, 150]. Of note, the level of impairment of the phagocytic activity and respiratory burst of PMNs during the course of HIV-1 infection has shown to be directly associated with the viral load and indirectly with CD4+ T cell counts. PMNs from patients successfully treated with highly active antiretroviral therapy (HAART) were shown to have better functions than patients suffering from HAART failure [150], indicating that such defects are associated with faster disease progression. Other studies have reported defects in PMN development, cell structure, adhesion, chemotaxis and recruitment during HIV-1 infection [147, 148, 156, 157]. Still others reported dysregulated cytokine production and defects in degranulation in PMNs from HIV-1 patients [158,159,160]. Furthermore, PMNs from HIV-1 patients exhibit dysregulated responses to endotoxin stimulation and a reduced inhibitory response to S100A8 and S100A9, calcium-binding proteins that are abundant in the cytosolic compartment of human PMNs which can inhibit oxidative metabolism. This is likely to be associated with an increased risk of oxidative stress-related illnesses such as cardiovascular diseases [161]. HIV-1 can also indirectly impair PMN functions, for example, HIV-1 down-regulates NET-mediated effector functions by inhibiting their formation, through suppressing the production of ROS via IL-10 produced by dendritic cells following HIV-1 binding to DC-SIGN [23]. One should also realise that NET formation by PMNs is initiated after viral stimulation of PMNs’ TLR-7 and TLR-8 receptors [23]. Furthermore, HIV-1 can impair antibody-dependent cellular cytotoxicity, mediated by phagocytes including PMNs, partially, by enhancing the shedding of and/or down-regulating the expression level of CD16 on PMNs [162, 163]. Indeed these functional defects can negatively affect a wide range of immune responses against HIV-1, thereby contributing to the pathogenesis of HIV-1 infections.
Functional defects in PMNs from HIV-1 patients may, in part, result from the direct binding of HIV-1 or HIV-1 proteins to the cell membrane of PMN as declared by Muñoz and coworkers [164], or indirectly by altering the plasma cytokine network and cellular components of the immune system, or even due to the presence of intrinsic defects in the PMNs themselves during development [165]. Importantly, application of G-CSF and GM-CSF has been shown to abrogate PMN functional defects in HIV-1 patients [138, 139].
PMNs and the pathogenesis of HIV-1 infection
Resting PMNs, even those negative for CD4, were shown to bind to HIV-1 and efficiently enhance viral transfer to CD4+ T cells in a manner dependent on cell-to-cell contact [166]. Interestingly, studies have revealed that activation of PMNs can increase the binding of HIV-1 at least twofold. These events were shown to be associated with increased transfer of HIV-1 to CD4+ T cells, when compared to HIV-1 bound to resting PMNs and to free HIV-1 particles [167]. In addition, PMNs were demonstrated to significantly increase C-C chemokine receptor type 5 (CCR5 or R5)/macrophage tropic HIV-1 replication when cultured with monocyte-derived macrophages through the production of IL-10 and CCL2 in a manner independent on direct cell-to-cell contact [168, 169]. At mucosal tissues of the genital tract and in draining lymph nodes, the attachment of HIV-1 to PMNs may support the establishment of new infection by facilitating trans-infection of CD4+ T cells. This, in part, could be achieved by the aid of peripheral blood mononeuclear cells, since they produce GM-CSF that prolongs PMN survival, which in turn may facilitate the interaction of HIV-1-bound PMNs with CD4+ T cells [170]. Consistently, increased infiltration of PMNs to the penile foreskin in the presence of CD4+ T cells has been shown to be correlated with an increased risk of HIV-1 infection [171].
PMNs can also contribute to the chronic immune activation observed during the course of HIV-1 infection, which is a hallmark of pathogenesis in HIV-1 disease progression [172], through the increased production of α-defensins in the circulation of HIV-1 patients [173]. Although high levels α-defensins could play a beneficial role in protection against HIV-1 acquisition in highly exposed but uninfected individuals [80], they may have detrimental effects in the course of natural HIV-1 infection [173]. Moreover, PMNs have been shown to express high levels of programmed death-1 ligand (PD-L1) on their surfaces [146]. Importantly, the elevated expression level of PD-L1 on PMNs has been shown to be associated with: (i) increased PD-1 expression on both CD4+ and CD8+ T lymphocytes; (ii) increased levels of PMN degranulation markers; and (iii) an increased frequency of PMNs expressing the granulocytic myeloid-derived suppressor cell phenotype [146]. Of note, PD-L1 interaction with PD-1 on T cells has been implicated in immune exhaustion, lower CD4+ T cell counts and faster disease progression, all of which are critical in the pathogenesis of both HIV-1 and SIV infections [146, 174,175,176,177]. This may be because the interaction of PD-1 on T cells with its ligand (PD-L1) expressed on several types of immune cells, such as monocytes/macrophages and dendritic cells, negatively affects T cell function through down-regulating cytokine production and proliferation. Similarly, increased PD-L1 expression on PMNs in HIV-1 infection has been shown to suppress T cell immune responses, through the PD-L1/PD-1 pathway and ROS production [146]. As such, PD-1 blockade is suggested as a strategy to abrogate PD-1/PD-L1 mediated immune activation, exhaustion, and impairment [174,175,176,177].
Chronic immune activation is strongly believed to be associated with the development of non-HIV/AIDS-related inflammatory conditions in HIV-1 patients, even in those with well-controlled viremia (HIV-1 elite controllers or treatment responders). Furthermore, immune activation may be the underlying cause of continuous loss of CD4+ T cells, especially in those with undetectable viremia [178]. To assess the role of PMNs in this context, Campillo-Gimenez and coworkers enrolled two groups of ART-treated HIV-1 patients, with and without inflammatory disorders, as well as a group of healthy individuals as a control group. Importantly, they showed that hyperactivation of PMNs was greater in those patients with inflammatory conditions [179]. Hyperactivation of PMNs was also shown to be associated with imbalanced PMNs apoptosis/necrosis [179], which, in part, could be related to impaired macrophages failing to phagocytize apoptotic PMNs in HIV-1 patients [180], thereby contributing to the chronic inflammation in HIV-1 infection. One useful marker for PMN activation and systemic inflammation during HIV-1 infection is the increased expression of CD64 (FcγRI) on PMNs, as has been recently revealed [181, 182]. Activation of PMNs during HIV-1 infection can be mediated by direct HIV-1 contact with TLRs expressed on PMNs [183]. The interaction of PMNs’ TLRs with HIV-1 or HIV-1-derived single-stranded RNA has been shown to induce the production of inflammatory cytokines (such as TNF-α and IL-6) and ROS [183, 184]. Furthermore, HIV-1 Nef can also activate PMNs and induce the production of ROS [185]. Importantly, it has been revealed that there is a relationship between ROS production and TLRs [186, 187]. Consistently, increased PMN ROS production following HIV-1 interaction with TLR7/8 has also been revealed [188], indicating that HIV-1 enhances the production of ROS, in part, through TLRs. This increased inflammatory cytokine production and ROS generation could have detrimental impacts on the site of stimulution leading to tissue damage such as epithelial barrier damage resulting in microbial translocation (discussed below), which is a critical contributor to the chronic immune activation during HIV-1 infection. These data indicate that the activation of PMNs is a consequence of HIV-1 infection and that activated PMNs, in turn, contribute to the chronic immune activation during the course of HIV-1 infection. Hence, using antioxidants and/or anti-inflammatory agents could provide a potential therapeutic strategy for HIV-1 infection, at least in part, through containing chronic immune activation.
The role of PMNs in maintaining gut epithelial barrier integrity in homeostasis and HIV-1 infection
PMNs were shown to critically impact mucosal homeostasis, especially within the gut compartment [189, “reviewed in 190, 191”, 192]. This is because they play a major role in controlling microbes translocated across impaired gut epithelial barriers. This is based on their exceptional capacity to eliminate extracellular pathogens, as aforementioned, as well as their ability to participate in the healing of damaged tissues (discussed below). However; PMNs may have detrimental effects on inflamed parts of the gut mucosa, especially during certain chronic inflammatory conditions. Hence, one can conclude that PMNs may act as a double-edged sword (reviewed in [190]).
The importance of maintaining gut epithelial barrier integrity in human health
In normal adult humans, approximately, a 400 m2 monolayer of columnar epithelial cells covers the gastrointestinal tract, also known as the gut epithelial barrier, representing the largest environmentally-exposed part of the body. Maintaining an intact gut epithelial barrier is essential for maintaining gut homeostasis (reviewed in [193,194,195,196]). This is because the gut epithelial barrier acts as a physical barrier to prevent the non-physiological passage or translocation of the gut’s lumenal content (i.e. commensal microorganisms and their byproducts, and other noxious substances) to the lamina propria, deeper tissues, and/or into the blood circulation. Indeed, microbial translocation is a consequence and/or a cause of different pathological conditions such as: inflammatory bowel diseases, celiac disease, obesity, diabetes, and certain cancers (e.g. colorectal cancer) [197,198,199,200,201,202,203,204]. Furthermore, the intact gut epithelial barrier is essential for orchestrating the immune responses within the gut compartment [193,194,195,196]. These data underscore the extreme importance of maintaining both the functional and physical integrity of this barrier.
The continuous exposure of this barrier to noxious substances present in the gut lumen can compromise its integrity as time passes. To avoid this, this barrier is entirely regenerated, every three to five days on average in humans, through a strictly balanced process of senescent epithelial cells shedding at the intestinal villi and differentiation of new epithelial cells from the intestinal stem cells that reside within the intestinal crypt [205,206,207,208]. Of note, gut epithelial cells are held together by tight junctions. These junctions are composed of multi-protein complexes that form a selective permeable barrier between adherent cells [209]. To further support the integrity of this layer, a massive number of immune cells are localized within gut mucosal tissues; ready to ‘accommodate’ any invasion that could impair the integrity of the gut epithelial barrier [210].
The role of PMNs in controlling microbial translocation under normal conditions
During gut epithelial barrier regeneration, some of the gut lumen contents translocate across this layer into the lamina propria. Once this occurs, phagocytes (especially PMNs and macrophages) and other immune cells are rapidly recruited. These phagocytes, particularly PMNs, will contribute to the clearance of translocated microbes and prevent their dissemination into the lamina propria or deeper to the draining lymph nodes. Most importantly they prevent them from reaching the blood circulation. In fact, evidence for the very critical role of PMNs in controlling intestinal microbial translocation arose from early investigations which indicated that 50% of the infections in neutropenic cancer patients result from intestinal microbiota [211]. More recent studies have also indicated that chemotherapy-induced neutropenia in mice models is also associated with increased intestinal microbial translocation [212,213,214,215]. However, under normal conditions, recruited PMNs participate in the healing of injured intestinal epithelial barriers to prevent further microbial translocation. Nonetheless, prolonged immune activation and continuous immune cell recruitment to the site of infection/inflammation can negatively affect these tissues, which in turn can lead to certain pathological conditions (discussed below), depending on the site of inflammation. Hence, upon clearance of translocated microbes, PMNs release several agents such as lipid mediators including resolvins, protectins, and lipoxins to counteract the recruitment of other phagocytes, including PMNs. Moreover, these lipid mediators participate in the healing of damaged epithelial barriers (reviewed in [216]). At the same time, PMNs release some proteases that degrade cytokines and chemokines at the site of cleared microbes to limit the recruitment of additional phagocytes, including PMNs, to in effect terminate inflammation. Of note, these data could provide an explanation for why the gut of healthy individuals contains a small number of PMNs, when compared to other innate effector cells [42].
Indeed, other types of immune cells are now well-appreciated to play a central role in maintaining mucosal tissue homeostasis, particularly within the gut compartments, such as the helper T cells type 17 (TH17). These cells secrete different cytokines (such as IL-17 and IL-22) and chemokines that are involved in maintaining the integrity of gut compartments [217, 218]. These cytokines can act as chemo-attractants to PMNs, and play a crucial role in antimicrobial production such as β–defensin and S100, a calcium-binding protein that participates in immune defense against bacterial pathogens [218]. In addition, they participate in the healing of injured intestinal epithelial barriers by inducing the proliferation, differentiation, and tight-junction formation of epithelial cells [218, 219]. Interestingly, under normal conditions, PMNs could be involved in maintaining normal TH17 cells counts, since studies on mice have demonstrated that PMNs can instruct the polarization of naive helper T cells to differentiate into TH17 cells [220]. Taken together, these data highlight the indispensable role of PMNs in maintaining gut homeostasis under physiological conditions.
PMNs and the pathology of certain intestinal inflammatory conditions
Under abnormal conditions, PMNs can participate in the pathogenesis of certain diseases. For instance, inflammatory bowel diseases are characterized by chronic inflammation of the intestinal tract (reviewed in [221, 222]), microbial translocation [223, 224], abnormal function of PMNs [225, 226], as well as increased infiltration and activation of PMNs and other phagocytes to the gut compartments [190, 227], all of which contribute to the pathogenesis of inflammatory bowel diseases. This is because PMN infiltration to the site of infection/inflammation is associated with increased pro-inflammatory cytokine secretion (such as IL-1β, IL-6, and TNF-α), ROS generation, and MPO releasing, which is the major constituent of the PMNs’ primary granules [156, 228,229,230,231,232]. These products, when increased at the gut mucosa, are associated with the severity of inflammatory bowel diseases [230, 233, 234], because they not only impact translocated microbes/invaders, if present, but also negatively impact the tissues at the sites of stimulation. For instance, the interaction of MPO with mannose receptors on residual macrophages leads to pro-inflammatory cytokine and ROS release by macrophages (reviewed in [53]). In turn, pro-inflammatory cytokines then increase the permeability of the gut epithelial barrier by manipulating the expression of genes responsible for tight junction formation, or through biochemical or morphological reorganization of tight junction proteins, as illustrated in (Fig. 2) [230,231,232, 235,236,237]. Equally, excessive ROS production within these compartments participates in the induction of a high rate of epithelial cells shedding/apoptosis, thereby dysregulating the balance of apoptotic intestinal epithelial cells shedding at the intestinal villi and the differentiation of new epithelial cells from the intestinal stem cells at the intestinal crypt. In other words, the increased apoptosis/shedding rate of intestinal epithelial cells exceeds the capacity of intestinal epithelial stem cell differentiation to compensate for this high rate of cell death, manifested by crypt hypertrophy [238,239,240]. Indeed, not only do the PMNs’ secreted molecules impact on gut epithelial barrier function, but also the PMNs themselves can significantly affect epithelial barrier function and homeostasis, since PMN transepithelial infiltration can modulate the expression, conformation, and distribution of adhesion molecules; for more details see [241]. Accordingly, increased PMN infiltration rates into the gut compartment, as a result of persistent gut inflammation and/or microbial translocation, has been demonstrated to play a central role in the impairment of gut epithelial barrier.

HIV-1 infection, gut epithelial barrier integrity, microbial translocation, and a possible role for PMNs
In the case of HIV-1 infection, there is more to say about microbial translocation, immune activation and disease progression. This is because it is well-recognized that HIV-1 mainly resides and replicates in the gut compartment (gut associate lymphatic tissues, GALT), where a huge number of immune target cells are present [242,243,244]. After HIV-1 infection and before microbial translocation (pre-microbial translocation stage), HIV-1 induces local inflammation in GALT, which, in part, could be mediated by pyroptosis, a form of inflammatory programmed cell death pathway induced by caspase-1, caspase-4, or caspase-5 in humans in response to different stimuli, which is associated with increased IL-1β and IL-18 production. This assumption arose from recent investigations that have shown that the intestinal paneth cells of Rhesus macaques, which are located at the intestinal epithelial crypt, produce IL-1β in response to SIV [232], inducing local inflammation. As a consequence of both viral replication and inflammation of GALT, the integrity of the gut epithelial barrier becomes partially impaired resulting in microbial translocation. From this point, HIV-1 infection actually shifts into another stage, specifically the post-microbial translocation stage. As a result, activation and death pathways are changed, as demonstrated by Steele and coworkers, who revealed that microbial exposure alters HIV-1-induced pyroptosis (i.e. through the caspase-1 pathway) in bystander CD4+ T cells in the gut mucosa towards apoptosis (i.e. a caspase-3 pathway) in infected CD4+ T cells, as a result of increasingly productive infected T cells [245]. Similarly, it has also been revealed that the exposure to Lactobacillus plantarum can reverse the damage mediated by IL-1β [232]. These are consistent with the recent observations that microbial translocation increases HIV-1 replication in CD4+ T cells [246]. Indeed, the continuous HIV-1 replication in GALT results in (i) a massive depletion of immune cells mainly CD4+ T cells, including TH17 and TH22 cells, and (ii) a great alteration in the immune components, as demonstrated both in HIV-1-infected humans and SIV-infected non-human primates [219, 238, 243, 244, 247,248,249,250]. This, in turn, alters the anatomical structure and functional activities of these compartments leading to impaired GALT integrity [238, 250,251,252,253,254,255,256,257], supporting additional microbial translocation [238, 249].
It is important to realise that although the alteration and depletion of immune cells in GALT contributes to the impairment of gut epithelial barrier integrity, HIV-1 and its proteins (such as Gp120) can also do this, leading to microbial translocation [254]. Two decades ago, Delézay and coworkers demonstrated that exposure of HIV-1 to HT-29-D4, a human colonic epithelial cell line, can impair their differentiation, in part, by affecting epithelial barrier function [255]. Of note, some in vitro studies have also indicated that HIV-1 could directly infect epithelial cells [256,257,258,259]. Interestingly, according to these studies, HIV-1 infection of epithelial cells could be associated with inflammatory cytokine secretion [258, 259]. Other studies have shown that epithelial cells naturally resist HIV-1 infection but instead have demonstrated that HIV-1 can bind to and interact with epithelial cells [254, 260, 261]. For instance, Nazli and coworkers have demonstrated that the exposure of T84, an intestinal cell line, to HIV-1 or its gp120, but not Tat protein, increased their permeability as a result of disruption to the tight junctions [254]. These events were also shown to be associated with increased inflammatory cytokine secretion, including TNF-α [254]. Furthermore, HIV-1 replication in GALT has been implicated in Wnt and TGF-β signaling pathway dysregulation, pathways which are involved in intestinal epithelial cell migration and differentiation [238]. This could explain why ‘well-controlled’ HIV-1 replication in GALT was shown to associate with better gut epithelial barrier function [238], and thus less microbial translocation. This process is actually observed in HIV-1 elite controllers (who do not progress to AIDS naturally and have considerably lower levels of microbial translocation and immune activation), reflecting the critical impact of HIV-1 replication and gut epithelial barrier integrity in the pathogenesis of HIV-1 infection in humans [262, 263]. Similarly, the absence of microbial translocation and immune activation in chronically SIV-infected Soot mangabeys (monkeys that do not progress to AIDS naturally) provides evidence for the critical correlation between viral replication and gut epithelial barrier integrity and disease progression [263, 264].
On the other hand, a study was conducted to assess the function of the intestinal epithelial barrier using HT-29/B6, a colonic epithelial cell line, upon exposure to HIV-1-infected immune cells [265]. In this study, Stockmann and colleagues have shown that HIV-1 infected immune cells can impair the function of the intestinal epithelial barrier, at least in part, by secreting pro-inflammatory cytokines [265]. Similarly, the continuous infiltration of immune cells, particularly phagocytes (such as PMNs and macrophages), to the site of inflammation can worsen the inflammatory status of these tissues, at least in part, by increasing pro-inflammatory cytokine secretion (Fig. 3). This adds another mechanism that can explain the pathogenesis of GALT damage and increased microbial translocation in HIV-1 infection.

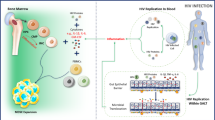
A proposed model describing the role of PMNs in the impairment of gut epithelial barrier integrity and microbial translocation during HIV-1 infection. After HIV-1 infection (sexual or non-sexual in nature) HIV-1 disseminates to the gut associated lymphatic tissues where a pool of immune cells is present, as seen in case number 1. Exposure of the gut epithelial barrier to HIV-1 particles can impair barrier integrity. Furthermore, HIV-1 replication, depletion of immune cells (particularly CD4+ T cells) and increased inflammatory cytokine production can also contribute to epithelial barrier integrity damage (such as decreased tight-junctions expression), resulting in microbial translocation as seen in case number 2. Microbial translocation increases immune activation, inflammation and HIV-1 replication supporting additional microbial translocation and triggering the infiltration of phagocytes such as PMNs and macrophages, case number 3. Unfortunately, as in case number 4 the increased infiltration of PMNs and macrophages within the gut mucosa, can only worsen the inflammatory condition of these tissues leading to permanent damage to the gut epithelial barrier
To further support the role of increased PMN infiltration into an organ (tissues) in the pathology of that organ (tissues), Puerta-Arias et al. [266] showed that PMNs themselves can contribute to the pathogenesis of fibrosis and pulmonary inflammation in mice, in which increased PMN infiltration was observed, through secreting pro-inflammatory cytokines. As such, depletion of PMNs was proposed as a strategy to promote the resolution of fibrosis and pulmonary inflammation in mice, in part, by down-regulating the production of pro-inflammatory cytokines [265]. Consistently, the continuous infiltration of PMNs has also been implicated in lung-pathology in HIV-1-infected humanized mice co- or not infected with Mycobacterium tuberculosis [266]. Interestingly, in both cases, there was a remarkable increase in pro-inflammatory cytokine (IL-1β, IL-6, TNF-α, and IL-8) secretion [267]. On the other hand, the increased infiltration rate of phagocytes (i.e., macrophages) to the gut mucosa has been reported in HIV-1 patients, which was also shown to be associated with increased pro-inflammatory molecules related to macrophages within these compartments [126]. As such, the accumulation of macrophages in the gut mucosa has been suggested as a contributor in the pathogenesis of HIV-1 infection through promotion of inflammation [126]. Interestingly, macrophages that accumulate in the colon of AIDS patients were demonstrated to be responsive to LPS and express inflammatory cytokines such as IL-1β and TNF-α, supporting the role of macrophages in the pathogenesis of the gut mucosa in HIV-1 infected individuals [268]. Similarly to macrophages, increased PMN infiltration to the gut compartments of chronically SIV-infected Rhesus macaques has also been demonstrated [264]. Importantly, this study showed that the lamina propria of SIV-infected Rhesus macaques contains an increased level of MPO+ PMNs; this observation was shown to be associated with epithelial barrier damage and increased microbial translocation [264]. These infiltrated PMNs could participate in the pathology of gut mucosa by increasing the secretion of inflammatory cytokines (Fig. 2 and Fig. 3), MPO, and by generating ROS. Moreover, Somsouk and coworkers have demonstrated that HIV-1 infected individuals, even those treated with ART, have significantly high rates of PMN infiltration into gut mucosal tissues, and this event was shown to be associated with increased mucosal apoptosis [269], both of which can also contribute to microbial translocation (Fig. 3). Microbial translocation contributes to the pathogenesis of HIV-1 infection by driving chronic immune activation, which is now recognized as a critical predictive marker for faster disease progression in HIV-1 and SIV infections. Indeed, certain translocated microbes, namely Gram negative bacteria, can enhance viral replication by increasing the expression of the CCR5 receptor on CD4+ T cells present in the lamina propria [246], amplifying this vicious cycle. It should be noted that HIV-1 is among those pathogens that thrive under highly inflammatory conditions (high pro-inflammatory cytokines and ROS) [270].
Taken together, these data indicate that impairment of gut mucosal integrity and increased microbial translocation in HIV-1 infection is, at least in part, a result of increased inflammatory conditions (inflammatory cytokines and ROS) mediated by an increased infiltration rate of phagocytes, including PMNs, to the gut mucosa of HIV-1-infected individuals. However, additional investigation is needed to further establish the role of PMNs in gut epithelial barrier integrity and microbial translocation during the course of HIV-1 infection at different clinical stages of disease (acute, chronic, and AIDS).
Conclusion
PMNs are critical innate immune cells involved in the clearance of pathogens. They are considered the most powerful immune cells in eliminating pathogens, especially extracellular ones. Additionally, they play a vital role in regulating innate immune responses, since they cross-talk with different innate immune cells. Furthermore, PMNs can directly instruct polarization and activation of specific adaptive immune responses. These data underscore the critical role that PMNs play in pathogen elimination and immune response mediation and regulation.
In the case of HIV-1 infection, neutropenia is relatively common in HIV-1 patients. Neutropenia is known to be associated with recurrent microbial infections, particularly, during the advanced stages of HIV-1 disease. Of note, several factors can lead to neutropenia in HIV-1 patients, including increased peripheral apoptosis rates, decreased production rates at the level of hematopoiesis, increased rates of infiltration, as well as certain drug treatments. Of central importance, the neutropenia observed during HIV-1 infection is not only known associated with increased microbial infections but also contributes to defects in immune function. On the other hand, PMNs become defective as HIV-1 disease progresses, and these defects are associated with immune response impairment and increased microbial infection. In addition, PMNs from HIV-1 patients exhibit hyperactivation that can contribute to chronic immune activation and immune exhaustion, both of which are known to contribute to disease progression in HIV-1 patients. Therefore restoring normal PMN count and function is essential for preventing microbial infection and immune impairment. To this end the therapeutic application of G-CSF and GM-CSF to HIV-1 patients is suggested.
Finally, HIV-1 mainly resides and replicates in lymphatic tissues, especially, in the GALT. These tissues become chronically inflamed during the early events of HIV-1 infection. This, in turn, leads to gut integrity damage, and as a consequence, microbial translocation occurs. Both events lead to an increased phagocytic infiltration rates, particularly of PMNs. Unfortunately, once at the GALT, PMNs become fully trapped in the viral ‘illusion’. These cells worsen the inflammation status of the GALT, by increasing the production of inflammatory cytokines and ROS that results in further damage to the integrity of the gut mucosa. Hence, therapeutic application of antioxidants and/or anti-inflammatory agents could provide a potential strategy for inhibiting HIV-1 infection through containment of chronic immune activation, particularly within the GALT, which could limit phagocyte infiltration, including that of PMNs.
References
Moir S, Ho J, Malaspina A (2008) Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med 205:1797–1805
Moir S, Malaspina A, Ogwaro KM, Donoghue ET, Hallahan CW, Ehler LA, Liu S, Adelsberger J, Lapointe R, Hwu P, Baseler M, Orenstein JM, Chun TW, Mican JA, Fauci AS (2001) HIV-1 induces phenotypic and functional perturbations of B cells in chronically infectedindividuals. Proc Natl Acad Sci USA 98:10362–10367
Mureithi MW, Cohen K, Moodley R, Poole D, Mncube Z, Kasmar A, Moody DB, Goulder PJ, Walker BD, Altfeld M, Ndung’u T (2011) Impairment of CD1d-restricted natural killer T cells in chronic HIV type 1 clade C infection. AIDS Res Hum Retrovir 27:50150–50159
Cella M, Presti R, Vermi W, Lavender K, Turnbull E, Ochsenbauer-Jambor C, Kappes JC, Ferrari G, Kessels L, Williams I, McMichael AJ, Haynes BF, Borrow P, Colonna M, CHAVI Clinical Core B, NIAID Center for HIV, AIDS Vaccine Immunology (2010) Loss of DNAM-1 contributes to CD8+ T-cell exhaustion in chronic HIV-1 infection. Eur J Immunol 40:949–954
Sachdeva M, Sharma A, Arora SK (2015) Functional impairment of myeloid dendritic cells during advanced stage of HIV-1 infection: role of factors regulating cytokine signaling. PLoS One 10:e0140852
Cardone M, Ikeda KN, Varano B, Gessani S, Conti L (2015) HIV-1-induced impairment of dendritic cell cross talk with γδ T lymphocytes. J Virol 89:4798–4808
Fischer-Smith T, Tedaldi EM, Rappaport J (2008) CD163/CD16 coexpression by circulating monocytes/macrophages in HIV: potential biomarkers for HIV infection and AIDS progression. AIDS Res Hum Retrovir 24:417–421
Pan X, Baldauf HM, Keppler OT, Fackler OT (2013) Restrictions to HIV-1 replication in resting CD4+ T lymphocytes. Cell Res 23:876–885
Zerbato JM, Serrao E, Lenzi G, Kim B, Ambrose Z, Watkins SC, Engelman AN, Sluis-Cremer N (2016) Establishment and reversal of HIV-1 latency in naive and central memory CD4+ T cells in vitro. J Virol 90:8059–8073
Koppensteiner H, Brack-Werner R, Schindler M (2012) Macrophages and their relevance in human immunodeficiency virus type I infection. Retrovirology 9:82
Honeycutt JB, Wahl A, Baker C, Spagnuolo RA, Foster J, Zakharova O, Wietgrefe S, Caro-Vegas C, Madden V, Sharpe G, Haase AT, Eron JJ, Garcia JV (2016) Macrophages sustain HIV replication in vivo independently of T cells. J Clin Invest 126:1353–1366
Kumar A, Herbein G (2014) The macrophage: a therapeutic target in HIV-1 infection. Mol Cell Ther 2:10
Kumar A, Abbas W, Herbein G (2014) HIV-1 latency in monocytes/macrophages. Viruses 6:1837–1860
Araínga M, Edagwa B, Mosley RL, Poluektova LY, Gorantla S, Gendelman HE (2017) A mature macrophage is a principal HIV-1 cellular reservoir in humanized mice after treatment with long acting antiretroviral therapy. Retrovirology 14:17
Honeycutt JB, Thayer WO, Baker CE, Ribeiro RM, Lada SM, Cao Y, Cleary RA, Hudgens MG, Richman DD, Garcia JV (2017) HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat Med 23:638–643
Rinaldo CR (2013) HIV-1 trans infection of CD4(+) T cells by professional antigen presenting cells. Scientifica (Cairo) 2013:164203
Peressin M, Proust A, Schmidt S, Su B, Lambotin M, Biedma ME, Laumond G, Decoville T, Holl V, Moog C (2014) Efficient transfer of HIV-1 in trans and in cis from Langerhans dendritic cells and macrophages to autologous T lymphocytes. AIDS 28:667–677
Crowe S, Zhu T, Muller WA (2003) The contribution of monocyte infection and trafficking to viral persistence, and maintenance of the viral reservoir in HIV infection. J Leukoc Biol 74:635–641
McIlroy D, Autran B, Cheynier R, Wain-Hobson S, Clauvel JP, Oksenhendler E, Debré P, Hosmalin A (1995) Infection frequency of dendritic cells and CD4+ T lymphocytes in spleens of human immunodeficiency virus-positive patients. J Virol 69:4737–4745
Wonderlich ER, Barratt-Boyes SM (2012) A dendrite in every pie: myeloid dendritic cells in HIV and SIV infection. Virulence 3:647–653
Alqudah MAY, Yaseen MMM, Yaseen MSM (2016) HIV-1 strategies to overcome the immune system by evading and invading innate immune system. HIV AIDS Rev 15:1–12
Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, Tesselaar K, Koenderman L (2010) In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 116:625–627
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, Uehata T, Iwasaki H, Omori H, Yamaoka S, Yamamoto N, Akira S (2012) Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12:109–116
Drescher B, Bai F (2013) Neutrophil in viral infections, friend or foe? Virus Res 171:1–7
Kolaczkowska E, Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175
Rot A, von Andrian UH (2004) Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol 22:891–928
Charo IF, Ransohoff RM (2006) The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med 354:610–621
Hayashi F, Means TK, Luster AD (2003) Toll-like receptors stimulate human neutrophil function. Blood 102:2660–2669
Kennedy AD, Willment JA, Dorward DW, Williams DL, Brown GD, DeLeo FR (2007) Dectin-1 promotes fungicidal activity of human neutrophils. Eur J Immunol 37:467–478
Ekman AK, Cardell LO (2010) The expression and function of Nod-like receptors in neutrophils. Immunology 130:55–63
van Kessel KP, Bestebroer J, van Strijp JA (2014) Neutrophil-mediated phagocytosis of Staphylococcus aureus. Front Immunol 5:467
Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L (2000) Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest 80:617–653
Nimmerjahn F, Ravetch JV (2006) Fc receptors: old friends and new family members. Immunity 24:19–28
Repp R, Valerius T, Sendler A, Gramatzki M, Iro H, Kalden JR, Platzer E (1991) Neutrophils express the high affinity receptor for IgG (Fc γ RI, CD64) after in vivo application of recombinant human granulocyte colony-stimulating factor. Blood 78:885–889
Thomas CJ, Schroder K (2013) Pattern recognition receptor function in neutrophils. Trends Immunol 34:317–328
Futosi K, Fodor S, Mócsai A (2013) Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol 17:638–650
Hochreiter-Hufford A, Ravichandran KS (2013) Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol 5:a008748
Smalls-Mantey A, Connors M, Sattentau QJ (2013) Comparative efficiency of HIV-1-infected T cell killing by NK cells, monocytes and neutrophils. PLoS One 8:e74858
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–241
Johansson A, Jesaitis AJ, Lundqvist H, Magnusson KE, Sjölin C, Karlsson A, Dahlgren C (1995) Different subcellular localization of cytochrome b and the dormant NADPH-oxidase in neutrophils and macrophages: effect on the production of reactive oxygen species during phagocytosis. Cell Immunol 161:61–71
Karlsson A, Dahlgren C (2002) Assembly and activation of the neutrophil NADPH oxidase in granule membranes. Antioxid Redox Signal 4:49–60
Sips M, Krykbaeva M, Diefenbach TJ, Ghebremichael M, Bowman BA, Dugast AS, Boesch AW, Streeck H, Kwon DS, Ackerman ME, Suscovich TJ, Brouckaert P, Schacker TW, Alter G (2016) Fc receptor-mediated phagocytosis in tissues as a potent mechanism for preventive and therapeutic HIV vaccine strategies. Mucosal Immunol 9:1584–1595
Segal AW, Dorling J, Coade S (1980) Kinetics of fusion of the cytoplasmic granules with phagocytic vacuoles in human polymorphonuclear leukocytes. Biochemical and morphological studies. J Cell Biol 85:42–59
Henry RM, Hoppe AD, Joshi N, Swanson JA (2004) The uniformity of phagosome maturation in macrophages. J Cell Biol 164:185–194
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5:e1000639
Desai J, Mulay SR, Nakazawa D, Anders HJ (2016) Matters of life and death. How neutrophils die or survive along NET release and is “NETosis” = necroptosis? Cell Mol Life Sci 73:2211–2219
Bennouna S, Denkers EY (2005) Microbial antigen triggers rapid mobilization of TNF-alpha to the surface of mouse neutrophils transforming them into inducers of high-level dendritic cell TNF-alpha production. J Immunol 174:4845–4851
van Gisbergen KP, Ludwig IS, Geijtenbeek TB, van Kooyk Y (2005) Interactions of DC-SIGN with Mac-1 and CEACAM1 regulate contact between dendritic cells and neutrophils. FEBS Lett 579:6159–6168
van Gisbergen KP, Sanchez-Hernandez M, Geijtenbeek TB, van Kooyk Y (2005) Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J Exp Med 201:1281–1292
Micheletti A, Costantini C, Calzetti F, Camuesco D, Costa S, Tamassia N, Cassatella MA (2013) Neutrophils promote 6-sulfo LacNAc+ dendritic cell (slanDC) survival. J Leukoc Biol 94:705–710
Yang D, de la Rosa G, Tewary P, Oppenheim JJ (2009) Alarmins link neutrophils and dendritic cells. Trends Immunol 30:531–537
Megiovanni AM, Sanchez F, Robledo-Sarmiento M, Morel C, Gluckman JC, Boudaly S (2006) Polymorphonuclear neutrophils deliver activation signals and antigenic molecules to dendritic cells: a new link between leukocytes upstream of T lymphocytes. J Leukoc Biol 79:977–988
Maffia PC, Zittermann SE, Scimone ML, Tateosian N, Amiano N, Guerrieri D, Lutzky V, Rosso D, Romeo HE, Garcia VE, Issekutz AC, Chuluyan HE (2007) Neutrophil elastase converts human immature dendritic cells into transforming growth factor-beta1-secreting cells and reduces allostimulatory ability. Am J Pathol 171:928–937
Eken C, Gasser O, Zenhaeusern G, Oehri I, Hess C, Schifferli JA (2008) Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol 180:817–824
Kasama T, Streiter RM, Standiford TJ, Burdick MD, Kunkel SL (1993) Expression and regulation of human neutrophil-derived macrophage inflammatory protein 1-α. J Exp Med 78:63–72
Kumar V, Sharma A (2010) Neutrophils: Cinderella of innate immune system. Int Immunopharmacol 10:1325–1334
Kasama T, Streiter RM, Lukacs NW, Burdick MD, Kunkel SL (1994) Regulation of neutrophil-derived chemokine expression by IL-10. J Immunol 152:3559–3569
Silva MT (2010) When two is better than one: macrophages and neutrophils work in concert in innate immunity as complementary and cooperative partners of a myeloid phagocyte system. J Leukoc Biol 87:93–106
Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S (2017) Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 38:187–197
Zahran N, Sayed A, William I, Mahmoud O, Sabry O, Rafaat M (2013) Neutrophil apoptosis: impact of granulocyte macrophage colony stimulating factor on cell survival and viability in chronic kidney disease and hemodialysis patients. Arch Med Sci 9:984–989
Mathias B, Szpila BE, Moore FA, Efron PA, Moldawer LL (2015) A review of GM-CSF therapy in sepsis. Medicine (Baltimore) 94:e2044
Nakazawa D, Shida H, Kusunoki Y, Miyoshi A, Nishio S, Tomaru U, Atsumi T, Ishizu A (2016) The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun 67:19–28
Spörri R, Joller N, Hilbi H, Oxenius A (2008) A novel role for neutrophils as critical activators of NK cells. J Immunol 181:7121–7130
Jaeger BN, Donadieu J, Cognet C, Bernat C, Ordoñez-Rueda D, Barlogis V, Mahlaoui N, Fenis A, Narni-Mancinelli E, Beaupain B, Bellanné-Chantelot C, Bajénoff M, Malissen B, Malissen M, Vivier E, Ugolini S (2012) Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J Exp Med 209:565–580
Degli-Esposti MA, Smyth MJ (2005) Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol 5:112
Costantini C, Micheletti A, Calzetti F, Perbellini O, Pizzolo G, Cassatella MA (2010) Neutrophil activation and survival are modulated by interaction with NK cells. Int Immunol 22:827–838
Costantini C, Cassatella MA (2011) The defensive alliance between neutrophils and NK cells as a novel arm of innate immunity. J Leukoc Biol 89:221–233
Bangani N, Nakiwala J, Martineau AR, Wilkinson RJ, Wilkinson KA, Lowe DM (2016) Brief report: HIV-1 infection impairs CD16 and CD35 mediated opsonophagocytosis of Mycobacterium tuberculosis by human neutrophils. J Acquir Immune Defic Syndr 73:263–267
Kuritzkes DR (2000) Neutropenia, neutrophil dysfunction, and bacterial infection in patients with human immunodeficiency virus disease: the role of granulocyte colony-stimulating factor. Clin Infect Dis 30:256–260
Cloke T, Munder M, Bergin P, Herath S, Modolell M, Taylor G, Müller I, Kropf P (2013) Phenotypic alteration of neutrophils in the blood of HIV seropositive patients. PLoS One 8:e72034
Shi X, Sims MD, Hanna MM, Xie M, Gulick PG, Zheng YH, Basson MD, Zhang P (2014) Neutropenia during HIV infection: adverse consequences and remedies. Int Rev Immunol 33:511–536
Keiser P, Higgs E, Smith J (1996) Neutropenia is associated with bacteremia in patients infected with the human immunodeficiency virus. Am J Med Sci 312:118–122
Babadoko AA, Aminu SM, Suleiman AN (2008) Neutropenia and human immunodeficiency virus-1 infection: analysis of 43 cases. Niger J Med 17:57–60
Levine AM, Karim R, Mack W, Gravink DJ, Anastos K, Young M, Cohen M, Newman M, Augenbraun M, Gange S, Watts DH (2006) Neutropenia in human immunodeficiency virus infection: data from the women’s interagency HIV study. Arch Intern Med 166:405–410
Leroi C, Balestre E, Messou E, Minga A, Sawadogo A, Drabo J, Maiga M, Zannou M, Seydi M, Dabis F, Jaquet A, IeDEA West Africa collaboration (2017) Incidence of severe neutropenia in HIV-infected people starting antiretroviral therapy in West Africa. PLoS One 12:e0170753
Rezvani K, Flanagan AM, Sarma U, Constantinovici N, Bain BJ (2001) Investigation of ethnic neutropenia by assessment of bone marrow colony-forming cells. Acta Haematol 105:32–37
Reich D, Nalls MA, Kao WH, Akylbekova EL, Tandon A, Patterson N, Mullikin J, Hsueh WC, Cheng CY, Coresh J, Boerwinkle E, Li M, Waliszewska A, Neubauer J, Li R, Leak TS, Ekunwe L, Files JC, Hardy CL, Zmuda JM, Taylor HA, Ziv E, Harris TB, Wilson JG (2009) Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS Genet 5:e1000360
Kourtis AP, Hudgens MG, Kayira D, BAN Study Team (2012) Neutrophil count in African mothers and newborns and HIV transmission risk. N Engl J Med 367:2260
Jan MS, Huang YH, Shieh B, Teng RH, Yan YP, Lee YT, Liao KK, Li C (2006) CC chemokines induce neutrophils to chemotaxis, degranulation, and alpha-defensin release. J Acquir Immune Defic Syndr 41:6–16
Gabrilovich DI, Vassilev V, Nosikov VV, Serebrovskaya LV, Ivanova LA, Pokrovsky VV (1993) Clinical significance of HIV DNA in polymorphonuclear neutrophils from patients with HIV infection. J Acquir Immune Defic Syndr 6:587–591
Biswas P, Mantelli B, Sica A, Malnati M, Panzeri C, Saccani A, Hasson H, Vecchi A, Saniabadi A, Lusso P, Lazzarin A, Beretta A (2003) Expression of CD4 on human peripheral blood neutrophils. Blood 101:4452–4456
Jouve M, Sol-Foulon N, Watson S, Schwartz O, Benaroch P (2007) HIV-1 buds and accumulates in “nonacidic” endosomes of macrophages. Cell Host Microbe 2:85–95
Mazzolini J, Herit F, Bouchet J, Benmerah A, Benichou S, Niedergang F (2010) Inhibition of phagocytosis in HIV-1-infected macrophages relies on Nef-dependent alteration of focal delivery of recycling compartments. Blood 115:4226–4236
Pitrak DL, Tsai HC, Mullane KM, Sutton SH, Stevens P (1996) Accelerated neutrophil apoptosis in the acquired immunodeficiency syndrome. J Clin Invest 98:2714–2719
Baldelli F, Preziosi R, Francisci D, Tascini C, Bistoni F, Nicoletti I (2004) Programmed granulocyte neutrophil death in patients at different stages of HIV infection. AIDS 14:1067–1069
Salmen S, Terán G, Borges L, Goncalves L, Albarrán B, Urdaneta H, Montes H, Berrueta L (2004) Increased Fas-mediated apoptosis in polymorphonuclear cells from HIV-infected patients. Clin Exp Immunol 137:166–172
Salmen S, Montes H, Soyano A, Hernández D, Berrueta L (2007) Mechanisms of neutrophil death in human immunodeficiency virus-infected patients: role of reactive oxygen species, caspases and map kinase pathways. Clin Exp Immunol 150:539–545
Elbim C, Monceaux V, François S, Hurtrel B, Gougerot-Pocidalo MA, Estaquier J (2009) Increased neutrophil apoptosis in chronically SIV-infected macaques. Retrovirology 6:29
Casulli S, Elbim C (2014) Interactions between human immunodeficiency virus type1 and polymorphonuclear neutrophils. J Innate Immun 6:13–20
Hadad N, Levy R, Schlaeffer F, Riesenberg K (2007) Direct effect of human immunodeficiency virus protease inhibitors on neutrophil function and apoptosis via calpain inhibition. Clin Vaccine Immunol 14:1515–1521
Busch M, Beckstead J, Gantz D, Vyas G (1986) Detection of human immunodeficiency virus infection of myeloid precursors in bone marrow samples from AIDS patients (abstract). Blood 68:122a
Zauli G, Furlini G, Vitale M, Re MC, Gibellini D, Zamai L, Visani G, Borgatti P, Capitani S, La Placa M (1994) A subset of human CD34+ hematopoietic progenitors express low levels of CD4, the high-affinity receptor for human immunodeficiency virus-type 1. Blood 84:1896–1905
Aiuti A, Turchetto L, Cota M, Cipponi A, Brambilla A, Arcelloni C, Paroni R, Vicenzi E, Bordignon C, Poli G (1999) Human CD34(+) cells express CXCR4 and its ligand stromal cell-derived factor-1. Implications for infection by T-cell tropic human immunodeficiency virus. Blood 94:62–73
Lee B, Ratajczak J, Doms RW, Gewirtz AM, Ratajczak MZ (1999) Coreceptor/chemokine receptor expression on human hematopoietic cells: biological implications for human immunodeficiency virus-type 1 infection. Blood 93:1145–1156
Deichmann M, Kronenwett R, Haas R (1997) Expression of the human immunodeficiency virus type-1 coreceptors CXCR-4 (fusin, LESTR) and CKR-5 in CD34+ hematopoietic progenitor cells. Blood 89:3522–3528
Redd AD, Avalos A, Essex M (2007) Infection of hematopoietic progenitor cells by HIV-1 subtype C, and its association with anemia in southern Africa. Blood 110:3143–3149
Alexaki A, Wigdahl B (2008) HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PLoS Pathog 4:e1000215
Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell J 4th, Bixby D, Savona MR, Collins KL (2010) HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med 16:446–451
Zauli G, Vitale M, Gibellini D, Capitani S (1996) Inhibition of purified CD34+ hematopoietic progenitor cells by human immunodeficiency virus 1 or gp120 mediated by endogenous transforming growth factor beta 1. J Exp Med 183:99–108
Banda NK, Tomczak JA, Shpall EJ, Sipple J, Akkina RK, Steimer KS, Hami L, Curiel TJ, Singer Harrison G (1997) HIV-gp120 induced cell death in hematopoietic progenitor CD34+ cells. Apoptosis 2:61–68
Calenda V, Graber P, Delamarter JF, Chermann JC (1994) Involvement of HIV nef protein in abnormal hematopoiesis in AIDS: in vitro study on bone marrow progenitor cells. Eur J Haematol 52:103–107
Prost S, Le Dantec M, Augé S, Le Grand R, Derdouch S, Auregan G, Déglon N, Relouzat F, Aubertin AM, Maillere B, Dusanter-Fourt I, Kirszenbaum M (2008) Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARgamma/STAT5 signaling pathway in macaques. J Clin Invest 118:1765–7175
Prakash O, Zhang P, Xie M, Ali M, Zhou P, Coleman R, Stoltz DA, Bagby GJ, Shellito JE, Nelson S (1998) The human immunodeficiency virus type I Tat protein potentiates ethanol-induced neutrophil functional impairment in transgenic mice. Alcohol Clin Exp Res 22:2043–2049
Gibellini D, Clò A, Morini S, Miserocchi A, Ponti C, Re MC (2013) Effects of human immunodeficiency virus on the erythrocyte and megakaryocyte lineages. World J Virol 2:91–101
Isgrò A, Aiuti A, Mezzaroma I, Addesso M, Riva E, Giovannetti A, Mazzetta F, Alario C, Mazzone A, Ruco L, Aiuti F (2002) Improvement of interleukin 2 production, clonogenic capability and restoration of stromal cell function in human immunodeficiency virus-type-1 patients after highly active antiretroviral therapy. Br J Haematol 118:864–874
Isgrò A, Aiuti A, Leti W, Gramiccioni C, Esposito A, Mezzaroma I, Aiuti F (2005) Immunodysregulation of HIV disease at bone marrow level. Autoimmun Rev 4:486–490
Isgrò A, Leti W, De Santis W, Marziali M, Esposito A, Fimiani C, Luzi G, Pinti M, Cossarizza A, Aiuti F, Mezzaroma I (2008) Altered clonogenic capability and stromal cell function characterize bone marrow of HIV-infected subjects with low CD4+ T cell counts despite viral suppression during HAART. Clin Infect Dis 46:1902–1910
Gill V, Shattock RJ, Freeman AR, Robinson G, Griffin GE, Gordon-Smith EC, Gibson FM (1996) Macrophages are the major target cell for HIV infection in long-term marrow culture and demonstrate dual susceptibility to lymphocytotropic and monocytotropic strains of HIV-1. Br J Haematol 93:30–37
Canque B, Marandin A, Rosenzwajg M, Louache F, Vainchenker W, Gluckman JC (1995) Susceptibility of human bone marrow stromal cells to human immunodeficiency virus (HIV). Virology 208:779–783
Chelucci C, Federico M, Guerriero R, Mattia G, Casella I, Pelosi E, Testa U, Mariani G, Hassan HJ, Peschle C (1998) Productive human immunodeficiency virus-1 infection of purified megakaryocytic progenitors/precursors and maturing megakaryocytes. Blood 91:1225–1234
Jaresko GS (1999) Etiology of neutropenia in HIV-infected patients. Am J Health Syst Pharm 5:S5–S8
Volberding PA, Baker KR, Levine AM (2003) Human immunodeficiency virus hematology. Hematol Am Soc Hematol Educ Program 2003:294–313
Jacobs JL, Gold JW, Murray HW, Roberts RB, Armstrong D (1985) Salmonella infections in patients with the acquired immunodeficiency syndrome. Ann Intern Med 102:186–188
Puthucheary SD, Ng KP, Hafeez A, Raja NS, Hassan HH (2004) Salmonellosis in persons infected with human immunodeficiency virus: a report of seven cases from Malaysia. Southeast Asian J Trop Med Public Health 35:361–365
Casado JL, Navas E, Frutos B, Moreno A, Martín P, Hermida JM, Guerrero A (1997) Salmonella lung involvement in patients with HIV infection. Chest 112:1197–1201
Wain J, Pham VB, Ha V, Nguyen NM, To SD, Walsh AL, Parry CM, Hasserjian RP, HoHo VA, Tran TH, Farrar J, White NJ, Day NP (2001) Quantitation of bacteria in bone marrow from patients with typhoid fever: relationship between counts and clinical features. J Clin Microbiol 39:1571–1576
Hakawi AM, Alrajhi AA (2006) Tuberculosis of the bone marrow: clinico-pathological study of 22 cases from Saudi Arabia. Int J Tuberc Lung Dis 10:1041–1044
Khandekar MM, Deshmukh SD, Holla VV, Rane SR, Kakrani AL, Sangale SA, Habbu AA, Pandit DP, Bhore AV, Sastry J, Phadke MA, Bollinger RC (2005) Profile of bone marrow examination in HIV/AIDS patients to detect opportunistic infections, especially tuberculosis. Indian J Pathol Microbiol 48:7–12
Getahun H, Gunneberg C, Granich R, Nunn P (2010) HIV infection-associated tuberculosis: The epidemiology and the response. Clin Infect Dis 50:S201–S207
Burke M, Yust I, Katlama C, Vardinon N, Clumeck N, Pinching AJ, Ledergerber B, Gatell JM, Chiesi A, Barton SE, Lundgren JD, Pedersen C (1997) Cytomegalovirus retinitis in patients with AIDS in Europe. AIDS in Europe Study Group. Eur J Clin Microbiol Infect Dis 16:876–882
Sing GK, Ruscetti FW (1995) The role of human cytomegalovirus in haematological diseases. Baillieres Clin Haematol 8:149–163
Pantanowitz L, Omar T, Sonnendecker H, Karstaedt AS (2000) Bone marrow cryptococcal infection in the acquired immunodeficiency syndrome. J Infect 41:92–94
Calore EE, Tanaka PY, Perez NM, de Almeida LV (2004) Bone marrow pathology in AIDS. Pathol Res Pract 200:591–597
Folkvord JM, McCarter MD, Ryder J, Meditz AL, Forster JE, Connick E (2006) Alpha-defensins 1, 2, and 3 are expressed by granulocytes in lymphoid tissues of HIV-1-seropositive and seronegative individuals. J Acquir Immune Defic Syndr 42:529–536
Allers K, Fehr M, Conrad K, Epple HJ, Schürmann D, Geelhaar-Karsch A, Schinnerling K, Moos V, Schneider T (2014) Macrophages accumulate in the gut mucosa of untreated HIV-infected patients. J Infect Dis 209:739–748
Aseffa A, Dietrich MA, Shannon EJ (1997) Effect of thalidomide on apoptosis of lymphocytes and neutrophils. Immunopharmacol Immunotoxicol 19:313–326
Israel DS, Plaisance KI (1991) Neutropenia in patients infected with human immunodeficiency virus. Clin Pharm 10:268–279
Toure S, Gabillard D, Inwoley A, Seyler C, Gourvellec G, Anglaret X (2006) Incidence of neutropenia in HIV-infected African adults receiving co-trimoxazole prophylaxis: a 6-year cohort study in Abidjan, Côte d’Ivoire. Trans R Soc Trop Med Hyg 100:785–790
Dryden-Peterson S, Jayeoba O, Hughes MD, Jibril H, McIntosh K, Modise TA, Asmelash A, Powis KM, Essex M, Shapiro RL, Lockman S (2013) Cotrimoxazole prophylaxis and risk of severe anemia or severe neutropenia in HAART-exposed. HIV-uninfected infants. PLoS One 8:e74171
Smith C, Forster JE, Levin MJ, Davies J, Pappas J, Kinzie K, Barr E, Paul S, McFarland EJ, Weinberg A (2015) Serious adverse events are uncommon with combination neonatal antiretroviral prophylaxis: a retrospective case review. PLoS One 10:e0127062
Richman DD, Fischl MA, Grieco MH, Gottlieb MS, Volberding PA, Laskin OL, Leedom JM, Groopman JE, Mildvan D, Hirsch MS et al (1987) The toxicity of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N Engl J Med 317:192–197
Moh R, Danel C, Sorho S, Sauvageot D, Anzian A, Minga A, Gomis OB, Konga C, Inwoley A, Gabillard D, Bissagnene E, Salamon R, Anglaret X (2005) Haematological changes in adults receiving a zidovudine-containing HAART regimen in combination with cotrimoxazole in Côte d’Ivoire. Antivir Ther 10:615–624
Moyle G, Sawyer W, Law M, Amin J, Hill A (2004) Changes in hematologic parameters and efficacy of thymidine analogue-based, highly active antiretroviral therapy: a meta-analysis of six prospective, randomized, comparative studies. Clin Ther 26:92–97
Bower M, McCall-Peat N, Ryan N, Davies L, Young AM, Gupta S, Nelson M, Gazzard B, Stebbing J (2004) Protease inhibitors potentiate chemotherapy-induced neutropenia. Blood 104:2943–2946
Zon LI, Arkin C, Groopman JE (1987) Haematologic manifestations of the human immune deficiency virus (HIV). Br J Haematol 66:251
Kuritzkes DR, Parenti D, Ward DJ, Rachlis A, Wong RJ, Mallon KP, Rich WJ, Jacobson MA (1998) Filgrastim prevents severe neutropenia and reduces infective morbidity in patients with advanced HIV infection: results of a randomized, multicenter, controlled trial. G-CSF 930101 Study Group. AIDS 12:65–74
Frumkin LR (1997) Role of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in the treatment of patients with HIV infection. Curr Opin Hematol 4:200–206
Hermans P, Rozenbaum W, Jou A, Castelli F, Borleffs J, Gray S, Ward N, Gori A, De Bona A, Ferré C, Loncà M, Lang JM, Ammassari A, Clumeck N (1996) Filgrastim to treat neutropenia and support myelosuppressive medication dosing in HIV infection. G-CSF 92105 Study Group. AIDS 10:1627–1633
Keiser P, Rademacher S, Smith JW, Skiest D, Vadde V (1998) Granulocyte colony-stimulating factor use is associated with decreased bacteremia and increased survival in neutropenic HIV-infected patients. Am J Med 104:48–55
Nielsen SD, Sørensen TU, Aladdin H, Ersbøll AK, Mathiesen L, Ullum H, Gerstoft J, Nielsen JO, Pedersen BK (2000) The effect of long-term treatment with granulocyte colony-stimulating factor on hematopoiesis in HIV-infected individuals. Scand J Immunol 52:298–303
Barbaro G, Di Lorenzo G, Grisorio B, Soldini M, Barbarini G (1997) Effect of recombinant human granulocyte-macrophage colony-stimulating factor on HIV-related leukopenia: a randomized, controlled clinical study. AIDS 11:1453–1461
Angel JB, High K, Rhame F, Brand D, Whitmore JB, Agosti JM, Gilbert MJ, Deresinski S (2000) Phase III study of granulocyte-macrophage colony-stimulating factor in advanced HIV disease: effect on infections, CD4 cell counts and HIV suppression. Leukine/HIV Study Group. AIDS 14:387–395
Mastroianni CM, d’Ettorre G, Forcina G, Lichtner M, Mengoni F, D’Agostino C, Corpolongo A, Massetti AP, Vullo V (2000) Interleukin-15 enhances neutrophil functional activity in patients with human immunodeficiency virus infection. Blood 96:1979–1984
Swaminathan S, Qiu J, Rupert AW, Hu Z, Higgins J, Dewar RL, Stevens R, Rehm CA, Metcalf JA, Sherman BT, Baseler MW, Lane HC, Imamichi T (2016) Interleukin-15 (IL-15) strongly correlates with increasing HIV-1 viremia and markers of inflammation. PLoS One 11:e0167091
Bowers NL, Helton ES, Huijbregts RP, Goepfert PA, Heath SL, Hel Z (2014) Immune suppression by neutrophils in HIV-1 infection: role of PD-L1/PD-1 pathway. PLoS Pathog 10:e1003993
Kubes P, Heit B, van Marle G, Johnston JB, Knight D, Khan A, Power C (2003) In vivo impairment of neutrophil recruitment during lentivirus infection. J Immunol 171:4801–4808
Heit B, Jones G, Knight D, Antony JM, Gill MJ, Brown C, Power C, Kubes P (2006) HIV and other lentiviral infections cause defects in neutrophil chemotaxis, recruitment, and cell structure: immunorestorative effects of granulocyte-macrophage colony-stimulating factor. J Immunol 177:6405–6414
Debaisieux S, Lachambre S, Gross A, Mettling C, Besteiro S, Yezid H, Henaff D, Chopard C, Mesnard JM, Beaumelle B (2015) HIV-1 Tat inhibits phagocytosis by preventing the recruitment of Cdc42 to the phagocytic cup. Nat Commun 6:6211
Michailidis C, Giannopoulos G, Vigklis V, Armenis K, Tsakris A, Gargalianos P (2012) Impaired phagocytosis among patients infected by the human immunodeficiency virus: implication for a role of highly active anti-retroviral therapy. Clin Exp Immunol 167:499–504
Roilides E, Mertins S, Eddy J, Walsh TJ, Pizzo PA, Rubin M (1990) Impairment of neutrophil chemotactic and bactericidal function in children infected with human immunodeficiency virus type 1 and partial reversal after in vitro exposure to granulocyte-macrophage colony-stimulating factor. J Pediatr 117:531–540
Roilides E, Walsh TJ, Pizzo PA, Rubin M (1991) Granulocyte colony-stimulating factor enhances the phagocytic and bactericidal activity of normal and defective human neutrophils. J Infect Dis 163:579–583
Schaumann R, Krosing J, Shah PM (1998) Phagocytosis of Escherichia coli and Staphylococcus aureus by neutrophils of human immunodeficiency virus-infected patients. Eur J Med Res 3:546–548
Roilides E, Holmes A, Blake C, Pizzo PA, Walsh TJ (1993) Impairment of neutrophil antifungal activity against hyphae of Aspergillus fumigatus in children infected with human immunodeficiency virus. J Infect Dis 167:905–911
Coffey MJ, Phare SM, George S, Peters-Golden M, Kazanjian PH (1998) Granulocyte colony-stimulating factor administration to HIV-infected subjects augments reduced leukotriene synthesis and anticryptococcal activity in neutrophils. J Clin Invest 102:663–670
Moore DA, Henderson D, Gazzard BG (1998) Neutrophil adhesion molecules in HIV disease. Clin Exp Immunol 114:73–77
Meddows-Taylor S, Kuhn L, Meyers TM, Tiemessen CT (2001) Altered expression of L-selectin (CD62L) on polymorphonuclear neutrophils of children vertically infected with human immunodeficiency virus type 1. J Clin Immunol 21:286–292
Meddows-Taylor S, Martin DJ, Tiemessen CT (1999) Impaired interleukin-8-induced degranulation of polymorphonuclear neutrophils from human immunodeficiency virus type 1-infected individuals. Clin Diagn Lab Immunol 6:345–351
Vecchiarelli A, Monari C, Palazzetti B, Bistoni F, Casadevall A (2000) Dysregulation in IL-12 secretion by neutrophils from HIV-infected patients. Clin Exp Immunol 121:311–319
Meddows-Taylor S, Kuhn L, Meyers TM, Sherman G, Tiemessen CT (2001) Defective neutrophil degranulation induced by interleukin-8 and complement 5a and down-regulation of associated receptors in children vertically infected with human immunodeficiency virus type 1. Clin Diagn Lab Immunol 8:21–30
Schwartz R, Lu Y, Villines D, Sroussi HY (2010) Effect of human immunodeficiency virus infection on S100A8/A9 inhibition of peripheral neutrophils oxidative metabolism. Biomed Pharmacother 64:572–575
Boros P, Gardos E, Bekesi GJ, Unkeless JC (1990) Change in expression of Fc gamma RIII (CD16) on neutrophils from human immunodeficiency virus-infected individuals. Clin Immunol Immunopathol 54:281–289
Meddows-Taylor S, Martin DJ, Tiemessen CT (1997) Altered expression of Fc gamma RIII (CD16) on polymorphonuclear neutrophils from individuals with human immunodeficiency virus type 1 disease and pulmonary tuberculosis. Clin Diagn Lab Immunol 4:789–791
Muñoz JF, Salmen S, Berrueta LR, Carlos MP, Cova JA, Donis JH, Hernández MR, Torres JV (1999) Effect of human immunodeficiency virus type 1 on intracellular activation and superoxide production by neutrophils. J Infect Dis 180:206–210
Chen TP, Roberts RL, Wu KG, Ank BJ, Stiehm ER (1993) Decreased superoxide anion and hydrogen peroxide production by neutrophils and monocytes in human immunodeficiency virus-infected children and adults. Pediatr Res 34:544–550
Olinger GG, Saifuddin M, Spear GT (2000) CD4-Negative cells bind human immunodeficiency virus type 1 and efficiently transfer virus to T cells. J Virol 74:8550–8557
Gabali AM, Anzinger JJ, Spear GT, Thomas LL (2004) Activation by inflammatory stimuli increases neutrophil binding of human immunodeficiencyvirus type 1 and subsequent infection of lymphocytes. J Virol 78:10833–10836
Yoshida T, Jones VC, Kobayashi M, Li XD, Pollard RB, Suzuki F (2007) Acceleration of R5 HIV replication by polymorphonuclear neutrophils in cultures of macrophages. Immunol Cell Biol 85:215–219
Yoshida T, Kobayashi M, Li XD, Pollard RB, Suzuki F (2009) Inhibitory effect of glycyrrhizin on the neutrophil-dependent increase of R5 HIV replication in cultures of macrophages. Immunol Cell Biol 87:554–558
Fu J, Sha BE, Thomas LL (2011) HIV-1-infected peripheral blood mononuclear cells enhance neutrophil survival and HLA-DR expression via increased production of GM-CSF: implications for HIV-1 infection. J Acquir Immune Defic Syndr 56:16–25
Prodger JL, Gray RH, Shannon B, Shahabi K, Kong X, Grabowski K, Kigozi G, Nalugoda F, Serwadda D, Wawer MJ, Reynolds SJ, Liu CM, Tobian AA, Kaul R (2016) Chemokine levels in the penile coronal sulcus correlate with HIV-1 acquisition and are reduced by male circumcision in Rakai, Uganda. PLoS Pathog 12:e1006025
Rajasuriar R, Khoury G, Kamarulzaman A, French MA, Cameron PU, Lewin SR (2013) Persistent immune activation in chronic HIV infection: do any interventions work? AIDS 27:1199–1208
D’Agostino C, Lichtner M, Mastroianni CM, Ceccarelli G, Iannetta M, Antonucci S, Vullo V, Massetti AP (2009) In vivo release of alpha-defensins in plasma, neutrophils and CD8 T-lymphocytes of patients with HIV infection. Curr HIV Res 7:650–655
Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD (2006) PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354
Rosignoli G, Cranage A, Burton C, Nelson M, Steel A, Gazzard B, Gotch F, Imami N (2007) Expression of PD-L1, a marker of disease status, is not reduced by HAART in aviraemic patients. AIDS 21:1379–1381
Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, Ahmed R, Amara RR (2009) Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 458:206–210
Dyavar Shetty R, Velu V, Titanji K, Bosinger SE, Freeman GJ, Silvestri G, Amara RR (2012) PD-1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J Clin Invest 122:1712–1716
Hunt PW, Brenchley J, Sinclair E, McCune JM, Roland M, Page-Shafer K, Hsue P, Emu B, Krone M, Lampiris H, Douek D, Martin JN, Deeks SG (2008) Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis 197:126–133
Campillo-Gimenez L, Casulli S, Dudoit Y, Seang S, Carcelain G, Lambert-Niclot S, Appay V, Autran B, Tubiana R, Elbim C (2014) Neutrophils in antiretroviral therapy-controlled HIV demonstrate hyperactivation associated with a specific IL-17/IL-22 environment. J Allergy Clin Immunol 134:1142–1152
Torre D, Gennero L, Baccino F, Speranza F, Biondi G, Pugliese A (2002) Impaired macrophage phagocytosis of apoptotic neutrophils in patients with human immunodeficiency virus type 1 infection. Clin Vaccine Immunol 9:983
Morquin D, Tuaillon E, Makinson A, Bendriss S, Le Moing V, Reynes J (2016) Impact of T cell activation, HIV replication and hepatitis C virus infection on neutrophil CD64 expression. Cytom B Clin Cytom. doi:10.1002/cyto.b.21385
Mitsumoto-Kaseida F, Murata M, Ura K, Takayama K, Hiramine S, Shimizu M, Toyoda K, Ogawa E, Furusyo N (2017) The expression level of neutrophil CD64 is a useful marker of systemic inflammation associated with HIV infection. AIDS Res Hum Retrovir 33:147–156
Giraldo DM, Hernandez JC, Velilla P, Urcuqui-Inchima S (2016) HIV-1-neutrophil interactions trigger neutrophil activation and Toll-like receptor expression. Immunol Res 64:93–103
Giraldo DM, Hernandez JC, Urcuqui-Inchima S (2016) HIV-1-derived single-stranded RNA acts as activator of human neutrophils. Immunol Res 64:1185–1194
Salmen S, Colmenares M, Peterson DL, Reyes E, Rosales JD, Berrueta L (2010) HIV-1 Nef associates with p22-phox, a component of the NADPH oxidase protein complex. Cell Immunol 263:166–171
Kavoosi G, Ardestani SK, Kariminia A (2009) The involvement of TLR2 in cytokine and reactive oxygen species (ROS) production by PBMCs in response to Leishmania major phosphoglycans (PGs). Parasitology 136:1193–1199
Prince LR, Whyte MK, Sabroe I, Parker LC (2011) The role of TLRs in neutrophil activation. Curr Opin Pharmacol 11:397–403
Jenne CN, Kubes P (2012) NETs tangle with HIV. Cell Host Microbe 12:5–7
Hooper LV (2015) Epithelial cell contributions to intestinal immunity. Adv Immunol 126:129–172
Fournier BM, Parkos CA (2012) The role of neutrophils during intestinal inflammation. Mucosal Immunol 5:354–366
Colgan SP (2015) Neutrophils and inflammatory resolution in the mucosa. Semin Immunol 27:177–183
Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, Chaplin DD, Weaver CT (2013) IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci USA 110:12768–12773
Hooper LV, Macpherson AJ (2010) Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol 10:159–169
Peterson LW, Artis D (2014) Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14:141–153
Obata Y, Takahashi D, Ebisawa M, Kakiguchi K, Yonemura S, Jinnohara T, Kanaya T, Fujimura Y, Ohmae M, Hase K, Ohno H (2012) Epithelial cell-intrinsic Notch signaling plays an essential role in the maintenance of gut immune homeostasis. J Immunol 188:2427–2436
Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT (2013) Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol 4:280
Hoffmanová I, Sánchez D, Hábová V, Anděl M, Tučková L, Tlaskalová-Hogenová H (2015) Serological markers of enterocyte damage and apoptosis in patients with celiac disease, autoimmune diabetes mellitus and diabetes mellitus type 2. Physiol Res 64:537–546
Cenit MC, Olivares M, Codoñer-Franch P, Sanz Y (2015) Intestinal microbiota and celiac disease: cause, consequence or co-evolution? Nutrients 7:6900–6923
Timmons T, Shen C, Aldrovandi G, Rollie A, Gupta SK, Stein JH, Dubé MP (2014) Microbial translocation and metabolic and body composition measures in treated and untreated HIV infection. AIDS Res Hum Retrovir 30:272–277
Louis P, Hold GL, Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12:661–672
Peterson CT, Sharma V, Elmén L, Peterson SN (2015) Immune homeostasis, dysbiosis and therapeutic modulation of the gut microbiota. Clin Exp Immunol 179:363–377
Tojo R, Suárez A, Clemente MG, de los Reyes-Gavilán CG, Margolles A, Gueimonde M, Ruas-Madiedo P (2014) Intestinal microbiota in health and disease: role of bifidobacteria in gut homeostasis. Intestinal microbiota in health and disease: role of bifidobacteria in gut homeostasis. World J Gastroenterol 20:15163–15176
Rai R, Saraswat VA, Dhiman RK (2015) Gut microbiota: its role in hepatic encephalopathy. J Clin Exp Hepatol 5:S29–S36
Festi D, Schiumerini R, Eusebi LH, Marasco G, Taddia M, Colecchia A (2014) Gut microbiota and metabolic syndrome. World J Gastroenterol 20:16079–16094
Bjerknes M, Cheng H (2005) Gastrointestinal stem cells. II. Intestinal stem cells. Am J Physiol Gastrointest Liver Physiol 289:G381–G387
Crosnier C, Stamataki D, Lewis J (2006) Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet 7:349–359
Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459:262–265
Barker N (2014) Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol 15:19–33
Lee SH (2015) Intestinal permeability regulation by tight junction: implication on inflammatory bowel diseases. Intest Res 13:11–18
Mowat A, Viney J (1997) The anatomical basis of intestinal immunity. Immunol Rev 156:145–166
Carter LW (1994) Bacterial translocation: nursing implications in the care of patients with neutropenia. Oncol Nurs Forum 21:857–865
Koh AY, Priebe GP, Pier GB (2005) Virulence of Pseudomonas aeruginosa in a murine model of gastrointestinal colonization and dissemination in neutropenia. Infect Immun 73:2262–2272
Koh AY, Köhler JR, Coggshall KT, Van Rooijen N, Pier GB (2008) Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog 4:e35
Faber J, van Limpt K, Kegler D, Luiking Y, Garssen J, van Helvoort A, Vos AP, Knol J (2011) Bacterial translocation is reduced by a specific nutritional combination in mice with chemotherapy-induced neutropenia. J Nutr 141:1292–1298
Green SI, Ajami NJ, Ma L, Poole NM, Price RE, Petrosino JF, Maresso AW (2015) Murine model of chemotherapy-induced extraintestinal pathogenic Escherichia coli translocation. Infect Immun 83:3243–3256
Colgan SP, Ehrentraut SF, Glover LE, Kominsky DJ, Campbell EL (2013) Contributions of neutrophils to resolution of mucosal inflammation. Immunol Res 55:75–82
Kolls JK, McCray PB Jr, Chan YR (2008) Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol 8:829–835
Valeri M, Raffatellu M (2016) Cytokines IL-17 and IL-22 in the host response to infection. Pathog Dis 74. doi:10.1093/femspd/ftw111
Kim CJ, Nazli A, Rojas OL, Chege D, Alidina Z, Huibner S, Mujib S, Benko E, Kovacs C, Shin LY, Grin A, Kandel G, Loutfy M, Ostrowski M, Gommerman JL, Kaushic C, Kaul R (2012) A role for mucosal IL-22 production and Th22 cells in HIV-associated mucosal immunopathogenesis. Mucosal Immunol 5:670–680
Abi Abdallah DS, Egan CE, Butcher BA, Denkers EY (2011) Mouse neutrophils are professional antigen-presenting cells programmed to instruct Th1 and Th17 T-cell differentiation. Int Immunol 23:317–326
Hansen JJ (2015) Immune responses to intestinal microbes in inflammatory bowel diseases. Curr Allergy Asthma Rep 15:61
Zhang YZ, Li YY (2014) Inflammatory bowel disease: pathogenesis. World J Gastroenterol 20:91–99
Vrakas S, Mountzouris KC, Michalopoulos G, Karamanolis G, Papatheodoridis G, Tzathas C, Gazouli M (2017) Intestinal bacteria composition and translocation of bacteria in inflammatory bowel disease. PLoS One 12:e0170034
Sartor RB, Mazmanian SK (2012) Intestinal microbes in inflammatory bowel diseases. Am J Gastroenterol 1:15–21
Levine AP, Segal AW (2013) What is wrong with granulocytes in inflammatory bowel diseases? Dig Dis 31:321–327
Somasundaram R, Nuij VJ, van der Woude CJ, Kuipers EJ, Peppelenbosch MP, Fuhler GM (2013) Peripheral neutrophil functions and cell signalling in Crohn’s disease. PLoS One 8:e84521
Koelink PJ, Overbeek SA, Braber S, Morgan ME, Henricks PA, Abdul Roda M, Verspaget HW, Wolfkamp SC, te Velde AA, Jones CW, Jackson PL, Blalock JE, Sparidans RW, Kruijtzer JA, Garssen J, Folkerts G, Kraneveld AD (2014) Collagen degradation and neutrophilic infiltration: a vicious circle in inflammatory bowel disease. Gut 63:578–587
Yamada T, Grisham MB (1991) Role of neutrophil-derived oxidants in the pathogenesis of intestinal inflammation. Klin Wochenschr 69:988–994
Kristjánsson G, Venge P, Wanders A, Lööf L, Hällgren R (2004) Clinical and subclinical intestinal inflammation assessed by the mucosal patch technique: studies of mucosal neutrophil and eosinophil activation in inflammatory bowel diseases and irritable bowel syndrome. Gut 53:1806–1812
Al-Sadi R, Ye D, Boivin M, Guo S, Hashimi M, Ereifej L, Ma TY (2014) Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS One 9:e85345
Al-Sadi R, Guo S, Ye D, Ma TY (2013) TNF-α modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am J Pathol 183:1871–1884
Hirao LA, Grishina I, Bourry O, Hu WK, Somrit M, Sankaran-Walters S, Gaulke CA, Fenton AN, Li JA, Crawford RW, Chuang F, Tarara R, Marco ML, Bäumler AJ, Cheng H, Dandekar S (2014) Early mucosal sensing of SIV infection by paneth cells induces IL-1β production and initiates gut epithelial disruption. PLoS Pathog 10:e1004311
Takac B, Mihaljević S, Stefanić M, Glavas-Obrovac L, Kibel A, Samardzija M (2014) Importance of interleukin 6 in pathogenesis of inflammatory bowel disease. Coll Antropol 38:659–664
Bank S, Andersen PS, Burisch J, Pedersen N, Roug S, Galsgaard J, Turino SY, Brodersen JB, Rashid S, Rasmussen BK, Avlund S, Olesen TB, Hoffmann HJ, Thomsen MK, Thomsen V, Frydenberg M, Nexø BA, Sode J, Vogel U, Andersen V (2014) Associations between functional polymorphisms in the NFκB signaling pathway and response to anti-TNF treatment in Danish patients with inflammatory bowel disease. Pharmacogenom J 14:526–534
Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM (2004) TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol 286:G367–G376
Ye D, Ma I, Ma TY (2006) Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol 290:G496–G504
Graham WV, Wang F, Clayburgh DR, Cheng JX, Yoon B, Wang Y, Lin A, Turner JR (2006) Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J Biol Chem 281:26205–26215
Sankaran S, George MD, Reay E, Guadalupe M, Flamm J, Prindiville T, Dandekar S (2008) Rapid onset of intestinal epithelial barrier dysfunction in primary human immunodeficiency virus infection is driven by an imbalance between immune response and mucosal repair and regeneration. J Virol 82:538–545
Batman PA, Kapembwa MS, Belmonte L, Tudor G, Kotler DP, Potten CS, Booth C, Cahn P, Griffin GE (2014) HIV enteropathy: HAART reduces HIV-induced stem cell hyperproliferation and crypt hypertrophy to normal in jejunal mucosa. Clin Pathol 67:14–18
Circu ML, Aw TY (2012) Intestinal redox biology and oxidative stress. Semin Cell Dev Biol 23:729–737
Sumagin R, Parkos CA (2015) Epithelial adhesion molecules and the regulation of intestinal homeostasis during neutrophil transepithelial migration. Tissue Barriers 3:e969100
Hatano H, Somsouk M, Sinclair E, Harvill K, Gilman L, Cohen M, Hoh R, Hunt PW, Martin JN, Wong JK, Deeks SG, Yukl SA (2013) Comparison of HIV DNA and RNA in gut-associated lymphoid tissue of HIV-infected controllers and noncontrollers. AIDS 27:2255–2260
Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden D, Racz P, Markowitz M (2004) Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med 200:761–770
Guadalupe M, Reay E, Sankaran S, Prindiville T, Flamm J, McNeil A, Dandekar S (2003) Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J Virol 77:11708–11717
Steele AK, Lee EJ, Manuzak JA, Dillon SM, Beckham JD, McCarter MD, Santiago ML, Wilson CC (2014) Microbial exposure alters HIV-1-induced mucosal CD4+ T cell death pathways ex vivo. Retrovirology 11:14
Dillon SM, Lee EJ, Donovan AM, Guo K, Harper MS, Frank DN, McCarter MD, Santiago ML, Wilson CC (2016) Enhancement of HIV-1 infection and intestinal CD4+ T cell depletion ex vivo by gut microbes altered during chronic HIV-1 infection. Retrovirology 13:5
Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M (2005) Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434:1093–1097
George MD, Sankaran S, Reay E, Gelli AC, Dandekar S (2003) High-throughput gene expression profiling indicates dysregulation of intestinal cell cycle mediators and growth factors during primary simian immunodeficiency virus infection. Virology 312:84–94
Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC (2004) CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med 200:749–759
Page EE, Greathead L, Metcalf R, Clark SA, Hart M, Fuchs D, Pantelidis P, Gotch F, Pozniak A, Nelson M, Boasso A, Gazzard B, Kelleher P (2014) Loss of Th22 cells is associated with increased immune activation and IDO-1 activity in HIV-1 infection. J Acquir Immune Defic Syndr 67:227–235
Ullrich R, Zeitz M, Heise W, L’age M, Höffken G, Riecken EO (1989) Small intestinal structure and function in patients infected with human immunodeficiency virus (HIV): evidence for HIV-induced enteropathy. Ann Intern Med 111:15–21
Batman PA, Kapembwa MS, Miller AR, Sedgwick PM, Lucas S, Sewankambo NK, Serwadda D, Pudney J, Moody A, Harris JR, Griffin GE (1998) HIV enteropathy: comparative morphometry of the jejunal mucosa of HIV infected patients resident in the United Kingdom and Uganda. Gut 43:350–355
Epple HJ, Schneider T, Troeger H, Kunkel D, Allers K, Moos V, Amasheh M, Loddenkemper C, Fromm M, Zeitz M, Schulzke JD (2009) Impairment of the intestinal barrier is evident in untreated but absent in suppressively treated HIV-infected patients. Gut 58:220–227
Nazli A, Chan O, Dobson-Belaire WN, Ouellet M, Tremblay MJ, Gray-Owen SD, Arsenault AL, Kaushic C (2010) Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog 6:e1000852
Delézay O, Yahi N, Tamalet C, Baghdiguian S, Boudier JA, Fantini J (1997) Direct effect of type 1 human immunodeficiency virus (HIV-1) on intestinal epithelial cell differentiation: relationship to HIV-1 enteropathy. Virology 238:231–242
Phillips DM, Bourinbaiar AS (1992) Mechanism of HIV spread from lymphocytes to epithelia. Virology 186:261–273
Asmuth DM, Hammer SM, Wanke CA (1994) Physiological effects of HIV infection on human intestinal epithelial cells: an in vitro model for HIV enteropathy. AIDS 8:205–211
Liu R, Huang L, Li J, Zhou X, Zhang H, Zhang T, Lei Y, Wang K, Xie N, Zheng Y, Wang F, Nice EC, Rong L, Huang C, Wei Y (2013) HIV Infection in gastric epithelial cells. J Infect Dis 208:1221–1230
Horejsh D, Ruckwardt TJ, David Pauza C (2002) CXCR4-dependent HIV-1 infection of differentiated epithelial cells. Virus Res 90:275–286
Tang Y, George A, Nouvet F, Sweet S, Emeagwali N, Taylor HE, Simmons G, Hildreth JE (2014) Infection of female primary lower genital tract epithelial cells after natural pseudotyping of HIV 1: possible implications for sexual transmission of HIV-1. PLoS One 9:e101367
Kohli A, Islam A, Moyes DL, Murciano C, Shen C, Challacombe SJ, Naglik JR (2014) Oral and vaginal epithelial cell lines bind and transfer cell-free infectious HIV-1 to permissive cells but are not productively infected. PLoS One 9:e98077
Taborda NA, Gonzalez SM, Correa LA, Montoya CJ, Rugeles MT (2015) Spontaneous HIV controllers exhibit preserved immune parameters in peripheral blood and gastrointestinal mucosa. J Acquir Immune Defic Syndr 70:115–121
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC (2006) Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12:1365–1371
Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, Barclay GR, Smedley J, Pung R, Oliveira KM, Hirsch VM, Silvestri G, Douek DC, Miller CJ, Haase AT, Lifson J, Brenchley JM (2010) Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog 6:e1001052
Stockmann M, Schmitz H, Fromm M, Schmidt W, Pauli G, Scholz P, Riecken EO, Schulzke JD (2000) Mechanisms of epithelial barrier impairment in HIV infection. Ann N Y Acad Sci 915:293–303
Puerta-Arias JD, Pino-Tamayo PA, Arango JC, González Á (2016) Depletion of neutrophils promotes the resolution of pulmonary inflammation and fibrosis in mice infected with Paracoccidioides brasiliensis. PLoS One 11:e0163985
Nusbaum RJ, Calderon VE, Huante MB, Sutjita P, Vijayakumar S, Lancaster KL, Hunter RL, Actor JK, Cirillo JD, Aronson J, Gelman BB, Lisinicchia JG, Valbuena G, Endsley J (2016) Pulmonary tuberculosis in humanized mice infected with HIV-1. Sci Rep 6:21522
Cassol E, Rossouw T, Malfeld S, Mahasha P, Slavik T, Seebregts C, Bond du Plessis J, Janssen C, Roskams T, Nevens F, Alfano M, Poli G, van der Merwe SW (2015) CD14(+) macrophages that accumulate in the colon of African AIDS patients express pro-inflammatory cytokines and are responsive to lipopolysaccharide. BMC Infect Dis 15:430
Somsouk M, Estes JD, Deleage C, Dunham RM, Albright R, Inadomi JM, Martin JN, Deeks SG, McCune JM, Hunt PW (2015) Gut epithelial barrier and systemic inflammation during chronic HIV infection. AIDS 29:43–51
Paiva CN, Bozza MT (2014) Are reactive oxygen species always detrimental to pathogens? Antioxid Redox Signal 20:1000–1037
Cunningham KE, Turner JR (2012) Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci 1258:34–42
Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, Li Z, Evans DB, Abbruzzese JL, Chiao PJ (2004) NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol 24:7806–7819
Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR (2006) Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci 119:2095–2106
Luettig J, Rosenthal R, Barmeyer C, Schulzke JD (2015) Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers 3:e977176
Acknowledgements
This study was funded by Jordan University of Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that this review manuscript was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Li Wu.
Rights and permissions
About this article

Cite this article
Yaseen, M.M., Abuharfeil, N.M., Yaseen, M.M. et al. The role of polymorphonuclear neutrophils during HIV-1 infection. Arch Virol 163, 1–21 (2018). https://doi.org/10.1007/s00705-017-3569-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3569-9