
Abstract
Reactive species are frequently formed after viral infections. Antioxidant defences, including enzymatic and non-enzymatic components, protect against reactive species, but sometimes these defences are not completely adequate. An imbalance in the production of reactive species and the body’s inability to detoxify these reactive species is referred to as oxidative stress. The aim of this review is to analyse the role of oxidative stress in the pathogenesis of viral infections and highlight some major therapeutic approaches that have gained importance, with regards to controlling virus-induced oxidative injury. Attention will be focused on DNA viruses (papillomaviruses, hepadnaviruses), RNA viruses (flaviviruses, orthomyxoviruses, paramyxoviruses, togaviruses) and retroviruses (human immunodeficiency virus). In general, viruses cause an imbalance in the cellular redox environment, which depending on the virus and the cell can result in different responses, e.g. cell signaling, antioxidant defences, reactive species, and other processes. Therefore, the modulation of reactive species production and oxidative stress potentially represents a novel pharmacological approach for reducing the consequences of viral pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Reactive species, antioxidant defences and oxidative stress
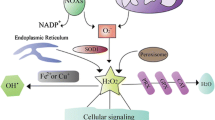
Reactive species (RS) include free radicals containing one or more unpaired electrons in the last electronic layer and species that, although not radicals, also exhibit high reactivity in biological systems [1]. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are produced in cells by means of normal physiological processes or by enzymatic and non-enzymatic mechanisms associated with pathological processes. Examples of RS are the superoxide radical (O •-2 ), hydroxyl radical (OH•), nitric oxide (NO), hydrogen peroxide (H2O2) and peroxynitrite (ONOO-). Molecular oxygen (O2) and NO are the most important mediators between reactive species induced by inflammatory processes, including microbial infections [1, 2].
RS generally play an important role in cellular signaling, the regulation of cytokines, growth factors, transcription, immunomodulation and apoptosis as well as in other processes [3, 4]. However, when there is an overproduction of RS, there might be damage to DNA, lipids and proteins, leading to the loss of cellular integrity and functionality [5]. To prevent and combat this excess RS and maintain cellular homeostasis, both physiologically and in a pathological process, there is an antioxidant defence system that, under physiological conditions, does not allow harmful actions of RS [6–8].
The antioxidant defence system is divided into enzymatic and non-enzymatic aspects. The vast majority of non-enzymatic antioxidants are obtained from the diet and are classified into several classes, including polyphenols. The other classes include vitamins C and E (α-tocopherol), carotenoids, organosulfur compounds, minerals and cofactors that play important roles in the maintenance of human health [9].
Enzymatic antioxidants are produced endogenously, and the major antioxidant enzymes are Superoxide Dismutase (SOD), Catalase (CAT), and Glutathione Peroxidase (GPx). O •-2 is metabolised to H2O2 by enzymes of the SOD family. Higher eukaryotes have three isoforms of SOD: cytoplasmic SOD1 (Cu/Zn-SOD), mitochondrial SOD2 (Mn-SOD) and extracellular SOD3 (Cu/Zn-SOD) [10]. The glutathione redox cycle is complementary to catalase in converting H2O2 to water and oxygen, whereas GPx use glutathione as a reducing agent [11].
The term “oxidative stress” refers to a disturbance in the oxidant-antioxidant balance, leading to potential cellular damage. This imbalance could result from a lack of antioxidant capacity or an overabundance of RS. However, because oxidative stress is involved in multiple systems such as redox signaling pathways, a better definition of oxidative stress is a “disruption/dysregulation of signaling and redox control” [12].
Oxidative stress and virus infections
Peterhans [13] published the first evidence that a virus could induce oxidative stress by increasing the RS levels. The author demonstrated that infection of mouse splenocytes with Sendai virus (a paramyxovirus) induced an increase in chemiluminescence levels, because luminol had been oxidised by RS. It was also shown that virus inactivated with UV light was able to generate RS, whereas virus inactivated by heat would not generate RS, suggesting that the conformation of the viral structure mediates this action. Later, other studies showed that many retroviruses, DNA and RNA viruses could cause cell death by generating RS [14–16].
Regarding its role in the activation of cells, the RS might facilitate or even promote viral replication, depending on the cell type and the virus involved. The effect of RS on cellular functions depends on the amount of RS and how long the cell has been exposed to RS [16–19]. Viruses, in general, vary in the production of RS but share a common pathogenic pathway accentuating the production of RS and antioxidant depletion [20]. Generally, during infection, the virus is detected, encompassed and phagocytosed by inflammatory cells including macrophages, neutrophils and dendritic cells. The pathogens activate the expression of the NADPH oxidase complex and nitric oxide synthase in phagocytic cells, leading to an increased production of RS [21].
The activation of phagocytes induced by viruses is associated with oxidative stress because the RS are released and because activated phagocytes can release pro-oxidant cytokines. Pro-oxidant cytokines such as tumour necrosis factor (TNF) and interleukin-1 (IL-1) promote iron uptake by the reticuloendothelial system, which can accumulate and, through the Fenton and Haber Weiss reactions, generate the hydroxyl radical OH• [22, 23].
Oxidants induced by viral infection include NO, O .-2 , OH• and their by-products (such as H2O2), which might contribute to the modulation of cellular responses, regulation of viral replication, host defences and viral pathogenesis [24]. Because the RS are closely related to the cell, changes in these species in different signaling pathways might modulate gene expression, adhesion, metabolism, cell cycle and death [25, 26]. Therefore, the RS are crucial for the development of viral infections. As obligatory intracellular parasites, viruses depend on the biosynthetic mechanisms of the host for replication. Thus, the redox state of the cell could benefit or harm certain viral infections, depending on the cell type and the virus involved [18, 19, 27, 28].
Initially, RS fight infection and are seen as a protection mechanism for the host cell, which might contribute to the induction of apoptosis [29]. However, with the advancement of viral multiplication, more RS are formed, causing an imbalance in cellular redox homeostasis. Therefore, the oxidative stress caused by viral infections can contribute to several aspects of pathogenesis, including inflammatory responses, cell death, and weight loss, among others [16, 27, 30]. Moreover, this change in the redox state of the host cell could select for certain viral mutants and/or produce mutations and activate transcription factors, such as nuclear Factor kappa B (NF-kB), which increases viral replication [23].
RS and lipid peroxidation products may affect viral replication through modulation of the activation state of cells, regulation of host inflammatory and immune responses, and by causing oxidative damage to host tissues and viral components [23, 27, 30–33]. Oxidative damage of infected and adjacent cells may also limit viral spread. However, for most viral infections, the extent to which oxidative damage plays a beneficial role for the host by limiting viral replication is not well understood [33]. Gullberg et al. [28] observed that oxidative stress positively affects viral RNA replication of flaviviruses and alphaviruses and that antioxidant treatment can significantly impair viral RNA replication, altering the amount of capped viral RNA. For human papillomavirus, studies suggest that oxidative stress favors different stages of HPV replication [34]. However, in HCV infection, the relevance of HCV-dependent induction of oxidative stress, with respect to viral genome replication, is controversial [35]. On one hand, there are reports describing an inhibitory effect of elevated RS levels on HCV replication [36, 37] but on the other hand there are reports describing Pycnogenol, a pine extract, which has antioxidant effects and leads to reduced RS levels and impaired HCV replication [38]. Thus, developing a better knowledge of oxidative stress, in particular cellular functions and viral replication, remains a challenge for the following years.
Viruses affect cellular redox balance by increasing oxidants such as superoxide and nitric oxide and inhibit the synthesis of antioxidant enzymes such as SOD, CAT, and GPx. Therefore, given what has been discussed so far, it could be inferred that oxidative stress is related to various aspects of the pathogenesis of various viral aetiologic agents. Evidence of the consequences of oxidative stress on the pathogenesis of some important viruses of different families is reviewed below.
Oxidative stress in papillomavirus infections
Human papilloma virus (HPV)
RS are extremely important in HPV infection because of their roles in mutagenesis, initiation and neoplastic progression [39]. If not adequately regulated by the antioxidant defence system, excess RS can damage lipids, proteins or DNA, inhibiting normal function, which increases chromosome aberrations associated with cell transformation [25, 40]. High RS production was detected in almost all cancers, acting as secondary messengers in intracellular signaling cascades, promoting many aspects of tumour development and progression [41]. Therefore, in HPV infection, oxidative stress could promote cellular transformation, which might facilitate the integration of HPV oncogenes into cellular DNA [34, 42]. Furthermore, studies suggest that oxidative stress is a condition that favors different stages of HPV infection including viral adsorption, viral entry and the initial establishment of viral gene expression [34, 43].
The RS produced by HPV infection can activate cellular signaling pathways, such as those mediated by mitogen-activated protein kinase (MAPK), NF-kB, phosphatidylinositol 3-kinase (PI3K), p53, β-catenin/Wnt and pathways associated with angiogenesis [42, 44]. These changes contribute to viral pathogenesis through modulation of cell growth/proliferation, differentiation, protein synthesis, glucose metabolism, cell survival and inflammation [45]. Moreover, the specific radicals generated, the location of their generation, as well as their local concentrations are important for determining the cellular functions of RS in cancer [41].
Foppoli et al. [43] highlighted that HPV confers to infected cells the ability to survive in an oxidising environment, through various mechanisms, such as the regulation of antioxidant enzymes (CAT, SOD, peroxiredoxin, glutathione S-transferase), protection against oxidation and suppression of apoptosis induced by stress. This redox adaption, through up regulation of anti-apoptotic and antioxidant molecules, allows cancer cells to promote survival and to develop resistance to anti-cancer drugs. However, little is known about how an increase in intracellular oxidative stress levels is sensed and transduced into RS-induced, specific, intracellular signaling to regulate the expression of antioxidant and survival genes [41].
Oxidative stress in hepadnavirus infections
Hepatitis B virus (HBV)
Several studies in the literature indicate that HBV induces oxidative stress in cells, mice or patients and that this stress might precede the development of hepatocellular carcinoma [46–50]. High levels of lipid peroxidation, DNA damage and hepatic transaminase alanine aminotransferase (ALT) were found in patients with chronic hepatitis, suggesting that oxidative stress plays an important role in liver injury induced by HBV [26, 51].
In general, patients infected with HBV show a reduction in Cu/Zn-SOD and GPx and increased levels of malondialdehyde (MDA), indicating oxidative stress during infection. Even after treatment with α-interferon and lamivudine, there is reduced lipid peroxidation and increased antioxidant enzyme levels [26]. Ren et al. [52] showed that the replication of HBV in infected cells induces oxidative stress by increasing the RS production and also showed that transfected cells overexpressing mitochondrial Sirtuin3 (SIRT3) protein decreased RS production (induced by the HBV viral protein, HBx) reducing oxidative damage to infected cells and reducing viral replication.
In addition to non-specific oxidative stress generated by local inflammation in response to viral infection, increasing evidence suggests that HBV-encoded proteins directly regulate cellular RS production and may also inhibit cellular DNA repair pathways. These changes can deleteriously alter intracellular antioxidant defences in HBV infected cells, causing apoptosis and extensive liver damage [51].
Oxidative stress in flavivirus infections
Hepatitis C virus (HCV)
Many studies have shown a role for oxidative stress in the hepatic pathogenesis of HCV. Patients infected with HCV showed elevated levels of various biomarkers of oxidative stress in serum and liver biopsy samples, including 8-hydroxydeoxyguanosine (8-OHdG, an indicator of DNA damage), MDA and thioredoxin [53–55]. Patients with HCV showed higher expression levels of 8-OHdG compared with patients with chronic hepatitis B, suggesting that oxidative damage to DNA is more common in chronic hepatitis C infections [49].
The expression of several structural, non-structural and core proteins of HCV (core protein, NS3, NS5A, E1, E2, NS4B) are associated with the induction of oxidative stress, with consequent damage to DNA contributing to carcinogenesis [51, 56]. The non-structural protein NS5A can activate cellular transcription factors such as NF-kB and STAT-3 by inducing oxidative stress in the cells [57]. As a consequence of endoplasmic reticulum stress, Ca2+ is released and is readily taken up by mitochondria, whereupon it affects the transmembrane potential and induces oxidative stress, exhibited by the rising levels of RS in mitochondria [58–60]. NS5A also cause oxidation of mitochondrial glutathione leading to increased RS [61–63] and resulting in translocation of NF-κB and STAT-3 transcription factors into the nucleus, also leading to oxidative stress [64]. Furthermore, the HCV protein NS5A seems to play a critical role in the activation of p38 MAPK, JNK and AP-1, leading to increased Mn-SOD antioxidant responses. The activation of AP-1 and Mn-SOD by HCV NS5A may be instrumental in the regulation of host oxidant status, and underscores the potential importance of this protein [65, 66].
During acute liver injury and liver inflammation, RS are generated by Kupffer cells and neutrophils as major toxic mediators to induce cell death [67]. Because these cells are very close to hepatocytes, some RS (for example, H2O2) are able to diffuse into hepatocytes and induce intracellular signaling [68]. In this context, these RS would amplify intracellular effects caused by the viral proteins themselves, although they could also activate intracellular antioxidant defences that could prevent or allow increases in oxidative stress [69].
Generally, in parallel to the RS increase in HCV infection, there is a decrease in antioxidant defence, such as glutathione content, which contributes to the oxidation of important cellular components. These changes can lead to the development of cirrhosis and hepatocellular carcinoma [69–72]. Some clinical studies have shown that the addition of antioxidants can improve liver injury caused by oxidative stress and this could be a potential treatment for HCV infection [71, 72].
Additionally, evidence suggests that HCV non-structural proteins, together with Core, repress hepcidin expression in a RS-dependent manner, altering iron metabolism [73]. Importantly, NS5A-induced RS production may also impact on glucose production [74], demonstrating that oxidative stress induced by HCV impacts on several other HCV-associated pathologies, including diabetes [51].
Japanese encephalitis virus (JEV)
JEV infection can lead to death in approximately 20-30% of infected patients [75, 76]. Neuronal apoptosis and inflammation are generally attributed to JEV-induced cytopathology. However, the extent of cell injury that can be accredited to viral cytopathology remains unclear [24, 77, 78].
Liao et al. [79] showed that infection by JEV induces the generation of the superoxide anion in rat cortical glial cells. Later, Srivastava et al. [80] revealed that RS, such as peroxynitrite, were increased in acute JEV rat infection models. In another study, JEV infection increased levels of SOD in the brains of rats in an attempt to suppress the high levels of superoxide [81]. In addition, in human promonocyte cells, an increase in intracellular RS was observed, in addition to activation of p38 MAPK signaling/ASK1-ERK; both processes that are associated with the apoptosis induced by JEV [82]. Moreover, JEV infection down-regulates thioredoxin expression, which would increase cytoplasmic oxidation and interfere with homeostatic redox balance during infection [28, 82].
Apoptosis induced by JEV has been associated with many important mechanisms, such as endoplasmic reticulum stress, RS generation and activation of NF-kB. JEV infection is also associated with microglial activation, resulting in the production of pro-inflammatory cytokines including IL-1b and IL-18, which is mediated by RS production [83]. Furthermore, RS can react to form peroxynitrite, which triggers the loss of ATP and mitochondrial membrane potential, leading to cytochrome c release from the mitochondria and the activation of caspase 3, causing neuronal apoptosis [24]. Therefore, oxidative stress is closely related to JEV infection in several ways, either through increasing the RS, or causing changes in antioxidant enzyme levels or the activation of major signaling pathways.
Dengue virus (DENV)
In sera of dengue patients, the levels of peroxidation potential, MDA+4-hydroxyalkenals, and SOD activity are significantly higher, whereas the levels of GPx and total hydroperoxides are significantly lower, suggesting that an alteration in redox status could be a result of increased oxidative stress and might play a role in the pathogenesis of the disease [84]. Among patients with dengue fever on different days of infection, Klassen et al. [85] showed an increase in the plasma concentrations of retinol and beta-carotene and a decrease in the glutathione and total antioxidant status. However, when compared to control subjects, patients with dengue fever had lower retinol concentrations in the acute phase of the disease and lower glutathione concentrations 7 d after discharge, suggesting that an imbalance of the antioxidant system might be a response to, or a consequence of, the viral induced inflammation. According to the authors, although levels of vitamin A in patients may be more strongly affected by their dietary intake than by their disease state, the lower plasma concentrations found in patients at the beginning of the disease may be related to the acute infection. Therefore, oxidative stress generated by an imbalance of micronutrient levels becomes significant when disease or infection occurs [16].
Another study assessing oxidative stress in different clinical spectrums of DENV infection (dengue fever, dengue haemorrhagic fever and dengue shock syndrome) showed that the level of oxidative stress was maximal in dengue shock syndrome followed by dengue haemorrhagic fever, and its severity was minimal in dengue fever. Additionally, the thrombocytopenia seen during dengue infections was associated with the extent of lipid peroxidation [86]. Later, Soundravally et al. [87] observed a significant positive correlation between lipid peroxides and TNF-α levels, and the TNF-α/IFN-γ ratio, in severe dengue cases.
Yen et al. [88] showed that DENV infection induced endothelial cell production of RS and apoptotic cell death, which was greatly enhanced by TNF-alpha. Additionally, the development and severity of the haemorrhage were greatly reduced in mice lacking iNOS or p47 (phox), or in mice treated with an oxidase inhibitor, suggesting a critical role for reactive nitrogen and oxygen species in dengue-associated haemorrhaging.
A previous study demonstrated that DENV-2 infection alters host intracellular GSH levels, and that exogenous GSH inhibits viral production by modulating the activity of NF-κB in HepG2 cells [89]. In model animals, mice infected with DENV-2 showed an increase in MDA and the GSSG/GSH ratio and a decrease in the activity of CAT and SOD. This study suggests that exogenous GSH might be a promising therapeutic agent for the prevention of oxidative liver damage during DENV infection [90].
Therefore, many studies show changes in the redox state in DENV infections, changes which contribute to the pathogenesis of the disease. Moreover, some markers of oxidative injury are altered during different stages of infection and might function as markers of disease progression.
Oxidative stress in orthomyxovirus infections
Influenza virus
Studies suggest that oxidative stress might promote lung injury and inflammation after infection with influenza A [91, 92]. RS, such as superoxide and nitric oxide, are released into the extracellular space by inflammatory and airway epithelial cells. These molecules might exacerbate lung injury after influenza virus pneumonia [93]. It has been demonstrated that damage to the lung tissue is a result of virus-induced cytopathic effect and is also due to the cytotoxic effects of excessive inflammation [94–96].
Buffinton et al. [97] evaluated the lungs and bronchoalveolar lavage fluid (BALF) of mice infected with a lethal dose of influenza A/PR8/34 virus and found increased generation of O •-2 in BALF cells during the early stages of infection and increased production of H2O2 in the lungs. The activities of GPx and glutathione reductase remained unaltered, demonstrating that oxidative stress was present in the early stages of influenza infection.
Hennet et al. [98] determined the endogenous concentrations of the antioxidants glutathione and vitamins C and E in the lungs, liver and blood plasma of control mice and mice infected with the influenza A/PR8/34 virus. There was a decrease in the total concentration of glutathione and vitamins C and E, and changes in the concentration of hepatic antioxidants occurred in the early stages of infection. This finding is important because a decrease in the concentrations of antioxidants might contribute to the host becoming more susceptible to the pathogenic effects of other agents.
Another study showed that glutathione blocked influenza viral infection in cultures of Madin-Darby canine kidney cells or human small airway epithelial cells. In this context there was protection against the production of active virus particles, inhibition of expression of the viral matrix protein and inhibition of viral-induced caspase activation and Fas upregulation. Moreover, the addition of GSH in the drinking water of BALB/c mice decreased the viral titre in both lung and trachea homogenates, suggesting an anti-influenza activity for glutathione in vitro and in vivo [99].
Shi et al. [100] administered recombinant human catalase (rhCAT) to mice infected by (H1N1) influenza A virus and observed a significant reduction in inflammatory cell infiltration, inflammatory cytokine levels (IL-2, IL-6, TNF-α, IFN-γ), and the mRNA levels of Toll-like receptors TLR-4, TLR-7, as well as NF-κB. This finding indicates a protective effect for rhCAT in viral-induced pneumonia of mice, via the suppression of immune responses.
According to Lin et al. [101], H5N1 virus infection of epithelial lung cells decreased the gene and protein expression of the SOD1 enzyme. Transfection of these cells to overexpress SOD1 significantly inhibited the production of RS by the H5N1 virus and reduced the pro-inflammatory response. It also prevented the phosphorylation of p38 and p65 and the nuclear export of viral ribonucleoprotein and viral replication. H5N1 infection in A549 cells resulted in a significantly greater production of intracellular RS, when compared with H1N1 infections, and, remarkably, decreased the GSH/GSSG ratio when compared with controls.
As observed in these studies, influenza virus infection induces oxidative stress and contributes to viral pathogenesis. Moreover, several studies have shown that the use of antioxidants might have a protective effect against infection by influenza virus.
Oxidative stress in paramyxovirus infections
Respiratory syncytial virus (RSV)
Oxidative stress also plays an important role in the pathogenesis of pulmonary inflammation caused by RSV. Mochizuki et al. [102] evaluated changes in the intracellular glutathione redox state in cultured human airway epithelial cells (A549) and normal human bronchial epithelial cells (NHBE) infected with RSV and found RSV-induced oxidative stress. Furthermore they showed that this stress could induce airway inflammation.
The infection of airway epithelial cells by RSV induces the production of RS and is able to increase lipid peroxidation products whilst decreasing the expression of SOD1, SOD3, CAT and GST, albeit with a slight increase in SOD2. Moreover, there was a verified increase in the activity of SOD and decreases in CAT, GPx and GST [103–105].
In infected mice, RSV induces oxidative stress in the lung, while antioxidant treatment alleviates clinical signs and pulmonary inflammation [106, 107]. Huang et al. [108] showed that mice infected with RSV obtained oxidative stress through increased NO, MDA and OH. levels and decreased GSH and SOD activity, but the administration of melatonin reversed all these effects and inhibited the production of pro-inflammatory cytokines such as TNF-α in the serum of RSV-infected mice. These results suggest that melatonin might be a novel therapeutic agent in virus-induced pulmonary infections.
Hosakote et al. [109] evaluated if synthetic catalytic scavengers could reduce RSV-induced pro-inflammatory gene expression, as well as oxidative cell damage, in airway epithelial cells (AECs). These cells were treated with the salen-manganese complexes EUK-8 or EUK-189, which possess SOD, CAT and GPx activity. This treatment reduced RSV-induced RS formation by increasing cellular antioxidant enzyme activity and the levels of the lipid peroxidation products F(2)-8-isoprostane and MDA. This treatment also inhibited RSV-induced cytokine and chemokine secretion and the activation of the transcription factor NF-κB and interferon regulatory factor-3, suggesting that increasing antioxidant cellular capacity could reduce the oxidative cell damage induced by RSV.
Children with acute bronchiolitis, caused by RSV, present with an induction of oxidative stress caused by the virus. Accordingly, the concentrations of GSSG and GPx increase, and there is a positive correlation between GSSG and the severity of disease [110].
These studies show that RSV is capable of inducing cellular oxidative damage as a result of the imbalance between the production of RS and cellular antioxidant defences.
Oxidative stress in togavirus infections
Chikungunya virus (CHIKV)
Dhanwani et al. [111] showed that new-born mice infected with CHIKV have changes to their apoptotic, inflammatory and stress pathways. There was an increase of inflammatory cytokines, particularly IL-6, TNF-α and IL-1. The antioxidant enzymes CAT and peroxiredoxin-6 were reduced, and there were changes, resulting from infection, in the urea cycle and energy metabolism in the liver and brain. This study showed evidence that the stress response is an important factor in the pathogenesis and inflammation by CHIKV, and tissue injury and apoptosis are the main events.
Another study using the neuroblastoma cell line SH-SY5Y infected with CHIKV showed a decrease in glutathione expression as well as decreased levels of the enzymes SOD, CAT, GPx, GR and GST. The MDA levels increased at all times examined after infection. In addition, there was an increase in the levels of the inflammatory cytokines IL-6, TNF-α and IL-1, showing inflammation in neuronal infection-induced CHIKV. High levels of these cytokines during infection could also activate and aggravate virus-induced cytopathic effects, stress and apoptosis [112].
Patil et al. [113] found changes in oxidative homeostasis in mice infected with CHIKV. These mice had high levels of the inflammatory cytokines iNOS, TNF-α, IL-1α, and IL-1β and high levels of the COX-2 and CCL-3 proteins during the symptomatic stage of disease, followed by normalisation of these levels during the recovery phase of the disease. Joubert et al. [114] showed that the infection of cells and mice by CHIKV induces increased RS production, causing endoplasmic reticulum and oxidative stress. These events act through independent mechanisms to induce autophagy during CHIKV infection.
Oxidative stress in retrovirus infections
Human immunodeficiency virus (HIV)
The presence of oxidative stress during HIV infection is already well established in the literature, and many articles have shown different changes, in various experimental models [115, 116]. It is known that oxidative stress, as well as other viral infections, might promote HIV replication and activate NF-kB, which is necessary for both viral replication and activation of inflammatory cytokines of the immune system [117, 118]. HIV-infected patients and AIDS patients generally have high serum hydroperoxide and MDA levels [119–121].
Most of the work related to HIV shows a reduction of antioxidant enzymes and high levels of oxidants. Malvy et al. [122] Dröge et al. [123] and Fuchs et al. [124] found levels of GSH, cysteine, vitamin C, GPx and SOD to be significantly reduced in plasma and leukocytes in HIV infections and found increased lipid peroxidation, with elevated plasma levels of MDA.
De Rosa et al. [125] also showed diminished levels of glutathione in erythrocytes and T cells associated with disease progression; and after the oral administration of glutathione, NAC and ATP, there was a significant restoration of virus-induced cell injury.
In general, during HIV infection, there is a depletion of antioxidants, which results in a decrease in immune function. The immune cells generally require a higher concentration of antioxidants than other cells to maintain the redox balance and preserve their integrity and function [126]. Therefore, oxidative stress is very important in HIV infection, i.e. reducing the antioxidants CAT and glutathione leads to an excess of H2O2, which increases OH• radicals that signal cell programmed cell death [127]. Excess RS appears to contribute to the progression to AIDS in different ways, including the apoptosis of CD4 cells and alterations in the function of other components of the immune system [16, 19, 128].
Potential therapeutic approaches
As discussed above, a large amount of evidence indicates that oxidative stress plays a complex role in viral diseases, from influencing host cell metabolism to viral replication. Therefore, the targeted use of antioxidants in viral disease therapy can be an effective strategy, capable of acting at different levels of viral infection. Currently, many compounds are available, including minocycline, quercetin, curcumin, butylated hydroxyanisole, melatonin, and exogenous GSH.
An example of research that has addressed antioxidant therapy during viral infection is Mishra et al. [129], who reported that the semisynthetic tetracycline, minocycline, inhibited RS production and neuronal death in mouse neuroblastoma N2a cells infected with JEV. When these same cells infected with JEV were treated with varying doses of curcumin (the main component of turmeric), the cell viability increased and apoptosis, and the cellular RS levels, significantly decreased [130].
Beyond these compounds, Gansukh et al. [131] presented quercetin-7-O-glucoside (Q7G), which exhibited strong antiviral activity against the influenza A and B viruses and acted as an inhibitor of influenza virus-induced symptoms, such as RS production. Gullberg et al. [28] showed that treatment of BHK cells infected with Kunjin virus (KUNV, flavivirus) with the antioxidant agent butylated hydroxyanisole (BHA) significantly decreased RS production, indicating that BHA blocked KUNV-induced oxidative stress. Castro et al. [106] also demonstrated that treatment with BHA significantly attenuated RSV-induced lung oxidative stress by decreasing the MDA and 4-hydroxynonenal levels in RSV-infected mice. Moreover, Huang et al. [108] showed that the administration of melatonin reversed the high NO, MDA, and OH. levels and inhibited the production of pro-inflammatory cytokines, such as TNF-α, in the sera of RSV-infected mice.
These compounds as well as other natural compounds have been studied. However, our own antioxidant components are also used to combat oxidative stress caused by viral infections. In vivo and in vitro studies have shown that the administration of exogenous GSH inhibits DENV-2 viral production by modulating NF-κB activity and reducing the MDA level, GSSG/GSH ratio and RS production [89, 90]. The administration of recombinant human catalase (rhCAT) to mice infected with H1N1 influenza A virus decreased inflammatory cell infiltration, inflammatory cytokine levels and the mRNA levels of the Toll-like receptors and NF-κB [100]. Moreover, transfection of the cells to induce SOD1 overexpression significantly inhibited RS production in response to an H5N1 virus infection and also reduced the pro-inflammatory response as well as viral replication [101].
Thus, we can conclude that many possible antioxidant therapies exist. More therapies are being discovered regularly because oxidative stress is closely associated with viral infections and therapies to combat this stress are therefore very important, especially those based on natural compounds.
Conclusions
In the infected cells of all of the viral agents discussed in this review, there are significant changes to cellular homeostasis. These changes are caused mainly by high levels of oxidative stress biomarkers and depletion of the antioxidant defence system. Increased RS and a reduction in the expression of antioxidant enzymes in infected cells can result in varied responses. In some cells, depending on the type of virus, this process favours viral replication, while in other cells, it inhibits replication. Therefore, it is necessary to research all aspects of oxidative stress, e.g. signaling pathways, antioxidant enzymes, lipid peroxidation, inflammatory responses, RS production etc. to better understand how the host response to viral infection occurs and how viruses act within the cell. This information can then be used to elucidate mechanisms that could be used to help fight, and prevent, certain viral infections.
References
Halliwell B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59:1609–1623
Akaike T (2001) Role of free radicals in viral pathogenesis and mutation. Rev Med Virol 11:87–101
Lander HM (1997) An essential role for free radicals and derived species in signal transduction. FASEB J 11(2):118–124
Gloire G, Legrand-Poels S, Piette J (2006) NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 72(11):1493–1505
Halliwell B, Gutteridge JMC (1999) Free radicals in biology and medicine, 3rd edn. Oxford University Press, Oxford
Nakashima I, Liu W, Akhand AA et al (2003) 4-Hydroxynonenal triggers multistep signal transduction cascades for suppression of cellular functions. Mol Aspects Med 24:231–238
Forman HJ, Dickinson DA (2004) Introduction to serial reviews on 4-hydroxy-2-nonenal as a signaling molecule. Free Radic Biol Med 37:594–596
Armogida M, Nisticò R, Mercuri NB (2012) Therapeutic potential of targeting hydrogen peroxide metabolism in the treatment of brain ischaemia. Br J Pharmacol 166:1211–1224
Ratnam DV, Ankola DD, Bhardwaj V et al (2006) Role of antioxidants in prophylaxis and therapy: a pharmaceutical perspective. J Controll Release 113:189–207
Miao L, St Clair DK (2009) Regulation of superoxide dismutase genes: implications in disease. Free Radic Biol Med 47:344–356
Zamocky M, Furtmuller PG, Obinger C (2008) Evolution of catalases from bacteria to humans. Antioxid Redox Signal 10:1527–1548
Jones DP (2006) Redefining oxidative stress. Antioxid Redox Signal 8:1865–1879
Peterhans E (1979) Sendai virus stimulates chemiluminescence in mouse spleen cells. Biochem Biophys Res Commun 91:383–392
Peterhans E, Grob M, Burge T et al (1987) Virus-induced formation of reactive oxygen intermediates in phagocytic cells. Free Radic Res Commun 3(1–5):39–46
Muller F (1992) Reactive oxygen intermediates and human immunodeficiency virus (HIV) infection. Free Radic Biol Med 13(6):651–657
Reshi ML, Su Y-C, Hong J-R (2014) RNA viruses: ROS-mediated cell death. Int J Cell Biol 2014:467452-1--467452-16. doi:10.1155/2014/467452
Burdon RH (1995) Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic Biol Med 18:775–794
Albrecht T, Boldogh I, Fons MP (1992) Receptor-initiated activation of cells and their oncogenes by herpes-family viruses. J Investig Dermatol 98(6 Suppl):29S–35S
Pace GW, Leaf CD (1995) The role of oxidative stress in HIV disease. Free Radic Biol Med 19:523–528
Stehbens WE (2004) Oxidative stress in viral hepatitis and AIDS. Exp Mol Pathol 77:121–132
Deramaudt TB, Dill C, Bonay M (2013) Regulation of oxidative stress by Nrf2 in the pathophysiology of infectious diseases. Médecine et maladies infectieuses 43:100–107
Gloenbock DT, Hampton RY, Qusesh RS et al (1991) Lipid Al-like molecule that antagonize the effects of endotoxin on human monocytes. J Biol Chem 266:19490–19498
Schwarz KB (1996) Oxidative stress during viral infection: a review. Free Radic Biol Med 21(5):641–649
Zhang Y, Wanga Z, Chen H et al (2014) Antioxidants: potential antiviral agents for Japanese encephalitis virus infection. Int J Infect Dis 24:30–36
Choi J, Ou JH (2006) Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am J Physiol Gastrointest Liver Physiol 290:G847–G851
Ha H-L, Shin H-J, Feitelson M et al (2010) Oxidative stress and antioxidants in hepatic pathogenesis. World J Gastroenterol 16(48):6035–6043
Peterhans E (1997) Oxidants and antioxidants in viral diseases: disease mechanisms and metabolic regulation. J Nutr 127:962S–965S
Gullberg RC, Jordan Steel J, Moon SL et al (2015) Oxidative stress influences positive strand RNA virus genome synthesis and capping. Virology 475:219–229
Jacobson MD (1996) Reactive oxygen species and programmed cell death. Trends Biochem Sci 21:83–86
Peterhans E (1997) Reactive oxygen species and nitric oxide in viral diseases. Biol Trace Elem Res 56:107–116
Akaike T, Suga M, Maeda H (1998) Free radicals in viral pathogenesis: molecular mechanisms involving superoxide and NO. Proc Soc Exp Biol Med 217:64–73
Beck MA (2000) Nutritionally induced oxidative stress: effect on viral disease. Am J Clin Nutr 71:1676S–1679S
Valyi-Nagy T, Dermody TS (2005) Role of oxidative damage in the pathogenesis of viral infections of the nervous system. Histol Histopathol 20:957–967
Williams VM, Filippova M, Soto U et al (2011) HPV-DNA integration and carcinogenesis: putative roles for inflammation and oxidative stress. Future Virol 6:45–57
Medvedev R, Ploen D, Hildt E (2016) HCV and oxidative stress: implications for HCV life cycle and HCV-associated pathogenesis. Oxid Med Cell Longev 2016:9012580
Huang H, Chen Y, Ye J (2007) Inhibition of hepatitis C virus replication by peroxidation of arachidonate and restoration by vitamin E. Proc Natl Acad Sci USA 104(47):18666–18670
Choi J, Lee KJ, Zheng Y et al (2004) Reactive oxygen species suppress hepatitis C virus RNA replication in human hepatoma cells. Hepatology 39(1):81–89
Ezzikouri S, Nishimura T, Kohara M et al (2015) Inhibitory effects of Pycnogenol® on hepatitis C virus replication. Antiviral Res 113:93–102
Dröge W (2003) Oxidative stress and aging. Adv Exp Med Biol 543:191–200
Perry G, Raina AK, Nunomura A et al (2000) How important is oxidative damage? Lessons from Alzheimer’s disease. Free Radic Biol Med 28:831–834
Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44:479–496
Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192:1–15
Foppoli C, DeMarco F, Cini C et al (2015) Redox control of viral carcinogenesis: the human papillomavirus paradigm. Biochimica et Biophysica Acta 1850:1622–1632
Czaja MJ (2007) Cell signaling in oxidative stress-induced liver injury. Semin Liver Dis 27:378–389
Storz P (2005) Reactive oxygen species in tumor progression. Front Biosci 10:1881–1896 (PubMed:15769673)
Hagen TM, Huang S, Curnutte J et al (1994) Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc Natl Acad Sci USA 91:12808–12812
Demirdag K, Yilmaz S, Ozdarendeli A et al (2003) Levels of plasma malondialdehyde and erythrocyte antioxidant enzyme activities in patients with chronic hepatitis B. Hepatogastroenterology 50:766–770
Bolukbas C, Bolukbas FF, Horoz M et al (2005) Increased oxidative stress associated with the severity of the liver disease in various forms of hepatitis B virus infection. BMC Infect Dis 5:95
Fujita N, Sugimoto R, Ma N et al (2008) Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J Viral Hepat 15:498–507
Gwak GY, Lee DH, Moon TG et al (2008) The correlation of hepatitis B virus PreS mutation with cellular oxidative DNA damage in hepatocellular carcinoma. Hepatogastroenterology 55:2028–2032
Higgs MR, Chouteau P, Lerat H (2014) ‘Liver let die’: oxidative DNA damage and hepatotropic viruses. J Gen Virol 95:991–1004
Ren JH, Chen X, Zhou L et al (2016) Protective role of Sirtuin3 (SIRT3) in oxidative stress mediated by hepatitis B virus X protein expression. PLoS One. doi:10.1371/journal.pone.0150961
Farinati F, Cardin R, Degan P et al (1999) Oxidative DNA damage in circulating leukocytes occurs as an early event in chronic HCV infection. Free Radic Biol Med 27:1284–1291
Sumida Y, Nakashima T, Yoh T et al (2000) Serum thioredoxin levels as an indicator of oxidative stress in patients with hepatitis C virus infection. J Hepatol 33:616–622
Mahmood S, Kawanaka M, Kamei A et al (2004) Immunohistochemical evaluation of oxidative stress markers in chronic hepatitis C. Antioxid Redox Signal 6:19–24
Sukowati Caecilia HC, El-Khobar Korri E, Ie Susan I et al (2016) Significance of hepatitis virus infection in the oncogenic initiation of hepatocellular carcinoma. World J Gastroenterol 22(4):1497–1512
Gong G, Waris G, Tanveer R et al (2001) Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kB. Proc Natl Acad Sci USA 98:9599–9604
Pahl HL (1999) Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev 0:683–701
Berridge MJ, Bootman MD, Lipp P (1998) Calcium—a life and death signal. Nature (London) 395:645–648
Murphy A, Bredesen G, Cortopaasi C et al (1996) Proc Natl Acad Sci USA 93:9893–9898
Yen HH, Shih KL, Lin TT et al (2012) Decreased mitochondrial deoxyribonucleic acid and increased oxidative damage in chronic hepatitis C. World J Gastroenterol 18(36):5084–5089
Piccoli C, Quarato G, Ripoli M et al (2009) Review HCV infection induces mitochondrial bioenergetic unbalance: causes and effects. Biochim Biophys Acta 1787(5):539–546
Piccoli C, Scrima R, D’Aprile A et al (2006) Review mitochondrial dysfunction in hepatitis C virus infection. Biochim Biophys Acta 1757(9–10):1429–1437
Mercurio F, Manning A (1999) Oncogene 18:6163–6171
Qadri I, Iwahashi M, Capasso JM et al (2004) Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: role of JNK, p38 MAPK and AP-1. Biochem J 378:919–928
Paracha UZ, Fatima K, Alqahtani M et al (2013) Oxidative stress and hepatitis C virus. Virol J 10:251
Jaeschke H (2011) Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. J Gastroenterol Hepatol 26(Suppl 1):173–179
Fisher AB (2009) Redox signaling across cell membranes. Antioxid Redox Signal 11:1349–1356
Waris G, Turkson J, Hassanein T et al (2005) Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication. J Virol 79:1569–1580
Arzumanyan A, Reis HM, Feitelson MA (2013) Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer 13:123–135
Lozano-Sepulveda AS, Bryan-Marrugo OL, Cordova-Fletes C et al (2015) Oxidative stress modulation in hepatitis C virus infected cells. World J Hepatol 7(29):2880–2889
Gabbay E, Zigmond E, Pappo O et al (2007) Antioxidant therapy for chronic hepatitis C after failure of interferon: results of phase II randomized, double-blind placebo controlled clinical trial. World J Gastroenterol 13:5317–5323
Miura K, Taura K, Kodama Y et al (2008) Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 48:1420–1429
Deng L, Shoji I, Ogawa W et al (2011) Hepatitis C virus infection promotes hepatic gluconeogenesis through an NS5A-mediated, FoxO1-dependent pathway. J Virol 85:8556–8568
Chambers TJ, Hahn CS, Galler R et al (1990) Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44:649–688
Burke DS, Lorsomrudee W, Leake CJ et al (1985) Fatal outcome in Japanese encephalitis. Am J Trop Med Hyg 34:1203–1210
Mathur A, Kulshreshtha R, Chaturvedi UC (1989) Evidence for latency of Japanese encephalitis virus in T lymphocytes. J Gen Virol 70:461–465
Raung SL, Kuo MD, Wang YM et al (2001) Role of reactive oxygen intermediates in Japanese encephalitis virus infection in murine neuroblastoma cells. Neurosci Lett 315:9–12
Liao SL, Raung SL, Chen CJ (2002) Japanese encephalitis virus stimulates superoxide dismutase activity in rat glial cultures. Neurosci Lett 324:133–136
Srivastava R, Kalita J, Khan MY et al (2009) Free radical generation by neurons in rat model of Japanese encephalitis. Neurochem Res 34:2141–2146
Kumar S, Misra UK, Kalita J et al (2009) Imbalance in oxidant/ antioxidant system in different brain regions of rat after the infection of Japanese encephalitis virus. Neurochem Int 55:648–654
Yang TC, Lai CC, Shiu SL et al (2010) Japanese encephalitis virus down-regulates thioredoxin and induces ROS-mediated ASK1-ERK/p38 MAPK activation in human promonocyte cells. Microbes Infect 12:643–651
Kaushik DK, Gupta M, Kumawat KL, Basu A (2012) NLRP3 inflammasome: key mediator of neuroinflammation in murine Japanese encephalitis. PLoS One 7:e32270
Gil L, Martínez G, Tápanes R et al (2004) Oxidative stress in adult dengue patients. Am J Trop Med Hyg 71:652–657
Klassen P, Biesalski HK, Mazariegos M et al (2004) Classic dengue fever affects levels of circulating antioxidants. Nutrition 20:542–547
Soundravally R, Sankar P, Bobby Z et al (2008) Oxidative stress in severe dengue viral infection: association of thrombocytopenia with lipid peroxidation. Platelets 19(6):447–454
Soundravally R, Hoti SL, Patil SA et al (2014) Association between proinflammatory cytokines and lipid peroxidation in patients with severe dengue disease around defervescence. Int J Infect Dis 18:68–72
Yen YTCH, Lin YD, Shieh CC et al (2008) Enhancement by tumor necrosis factor alpha of dengue virus-induced endothelial cell production of reactive nitrogen and oxygen species is key to hemorrhage development. J Virol 82(24):12312–12324
Tian YJW, Gao N, Zhang J et al (2010) Inhibitory effects of glutathione on dengue virus production. Biochem Biophys Res Commun 397(3):420–424
Wang J, Chen Y, Gao N et al (2013) Inhibitory effect of glutathione on oxidative liver injury induced by dengue virus serotype 2 infections in mice. PloS One 8(1):e55407
Akaike T, Noguchi Y, Ijiri S et al (1996) Pathogenesis of influenza virus-induced pneumonia: Involvement of both nitric oxide and oxygen radicals. Proc Natl Acad Sci USA 93:2448–2453
Choi AM, Knobil K, Otterbein SL et al (1996) Oxidant stress responses in influenza virus pneumonia: gene expression and transcription factor activation. Am J Physiol 271(3 Pt 1):L383–L391
Suliman HB, Ryan LK, Bishop L et al (2001) Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am J Physiol Lung Cell Mol Physiol 280(1):L69–L78
Kobasa D, Jones SM, Shinya K et al (2007) Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445(7125):319–323
Walsh KB, Teijaro JR, Wilker PR et al (2011) Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci USA 108(29):12018–12023
Ng MP, Lee JC, Loke WM et al (2014) Does influenza A infection increase oxidative damage? Antioxid Redox Signal 21(7):1025–1031. doi:10.1089/ars.2014.5907 (Epub Jul 21)
Buffinton GD, Christen S, Peterhans E et al (1992) Oxidative stress in lungs of mice infected with influenza A virus. Free Radic Res Commun 16(2):99–110
Hennet T, Peterhans E, Stocker R (1992) Alterations in antioxidant defences in lung and liver of mice infected with influenza A virus. J Gen Virol 73(Pt 1):39–46
Cai J, Chen Y, Seth S et al (2003) Inhibition of influenza infection by glutathione. Free Radic Biol Med 34(7):928–936
Shi X, Shi Z, Huang H et al (2014) Ability of recombinant human catalase to suppress inflammation of the murine lung induced by influenza A. Inflammation 37(3):809–817. doi:10.1007/s10753-013-9800-2
Lin X, Wang R, Zou W et al (2016) The influenza virus H5N1 infection can induce ROS production for viral replication and host cell death in A549 cells modulated by human Cu/Zn superoxide dismutase (SOD1) overexpression. Viruses 8:13. doi:10.3390/v8010013
Mochizuki H, Todokoro M, Arakawa H (2009) RS virus-induced inflammation and the intracellular glutathione redox state in cultured human airway epithelial cells. Inflammation 32(4):252–264. doi:10.1007/s10753-009-9128-0
Casola A, Burger N, Liu T et al (2001) Oxidant tone regulates RANTES gene transcription in airway epithelial cells infected with respiratory syncytial virus: role in viral-induced interferon regulatory factor activation. J Biol Chem 276:19715–19722
Liu T, Shawn C, Brasier AR et al (2004) Reactive oxygen species mediate virus-induced STAT activation: role of tyrosine phosphatases. J Biol Chem 279:2461–2469
Hosakote YM, Liu T, Castro SM et al (2009) Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am J Respir Cell Mol Biol 41:348–357
Castro SM, Guerrero-Plata A, Suarez-Real G et al (2006) Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am J Respir Crit Care Med 174:1361–1369
Hosakote YM, Jantzi PD, Esham DL et al (2011) Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med 183:1550–1560
Huang SH, Cao XJ, Liu W et al (2010) Inhibitory effect of melatonin on lung oxidative stress induced by respiratory syncytial virus infection in mice. J Pineal Res 48(2):109–116. doi:10.1111/j.1600-079X.2009.00733.x (Epub Jan 8)
Hosakote YM, Komaravelli N, Mautemps N et al (2012) Antioxidant mimetics modulate oxidative stress and cellular signaling in airway epithelial cells infected with respiratory syncytial virus. Am J Physiol Lung Cell Mol Physiol 303(11):L991–L1000. doi:10.1152/ajplung.00192.2012
Moreno-Solís G, de la Torre-Aguilar MJ, Torres-Borrego J et al (2015) Oxidative stress and inflamatory plasma biomarkers in respiratory syncytial virus bronchiolitis. Clin Respir J. doi:10.1111/crj.12425
Dhanwani R, Khan M, Alam SI et al (2011) Differential proteome analysis of Chikungunya virus infected new-born mice tissues reveal implication of stress, inflammatory and apoptotic pathways in disease pathogenesis. Proteomics 11:1936–1951
Dhanwani R, Khan M, Bhaskar AS et al (2012) Characterization of Chikungunya virus infection in human neuroblastoma SH-SY5Y cells: role of apoptosis in neuronal cell death. Virus Res 163(2012):563–572
Patil DR, Hundekar SL, Arankalle VA (2012) Expression profile of immune response genes during acute myopathy induced by chikungunya virus in a mouse model. Institut Pasteur Microbes Infect 14:457–469
Joubert PE, Werneke SW, de la Calle C et al (2012) Chikungunya virus-induced autophagy delays caspase-dependent cell death. J Exp Med 209(5):1029–1047
Nabel G, Baltimore D (1987) An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326(6114):711–713
Fuchs J, Ochsendorf F, Schofer H et al (1991) Oxidative imbalance in HIV infected patients. Med Hypotheses 36(1):60–64
Staal FJT, Roederer M, Herzenberg LA et al (1990) Intracellular thiols regulate activation of nuclear factor κB and transcription of human immunodeficiency virus. Proc Natl Acad Sci USA 87(24):9943–9947
Meyer M, Pahl HL, Baeuerle PA (1994) Regulation of the transcription factors NF-κB and AP-1 by redox changes. Chem-Biol Interact 91(2–3):91–100
Sonnerborg A, Carlin G, Akerlund B et al (1988) Increased production of malondialdehyde in patients with HIV infection”. Scand J Infect Dis 20(3):287–290
Revillard J-P, Vincent CMA, Favier AE et al (1992) Lipid peroxidation in human immunodeficiency virus infection. J Acquired Immune Defic Syndromes 5(6):637–638
Favier A, Sappey C, Leclerc P et al (1994) Antioxidant status and lipid peroxidation in patients infected with HIV. Chem-Biol Interact 91(2–3):165–180
Malvy DJM, Richard M-J, Arnaud J et al (1994) Relationship of plasma malondialdehyde, vitamin E and antioxidant micronutrients to human immunodeficiency virus-1 seropositivity. Clin Chim Acta 224:89–94
Dröge W, Eck HP, Mihm S (1994) Oxidant-antioxidant status in human immunodeficiency virus infection. Oxygen Radic Biol Syst Methods Enzymol 233:594–601
Fuchs J, Emerit I, Levy A et al (1995) Clastogenic factors in plasma of HIV-1 infected patients. Free Radic Biol Med 19:843–848
De Rosa SC, Zaretsky MD, Dubs JG et al (2000) N-acetylcysteinereplenishes glutathione in HIV infection. Eur J Clin Investig 30:915–929
De La Fuente M, Miquel J, Catalan MP et al (2002) The amount of thiolic antioxidant ingestion related to improve several immune functions is higher in aged than in adult mice. Free Radic Res 36:119–126
Leff JA, Oppegard MA, Curiel TJ et al (1992) Progressive increases in serum catalase activity in advancing human immunodeficiency virus infection. Free Radic Biol Med 13(2):143–149
Greenspan HC (1994) Aruoma OI (1994) Oxidative stress and apoptosis in HIV infection: a role for plant-derived metabolites with synergistic antioxidant activity. Immunol Today 15(5):209–213
Mishra MK, Ghosh D, Duseja R et al (2009) Antioxidant potential of minocycline in Japanese encephalitis virus infection in murine neuroblastoma cells: correlation with membrane fluidity and cell death. Neurochem Int 54:464–470
Dutta K, Ghosh D, Basu A (2009) Curcumin protects neuronal cells from Japanese encephalitis virus-mediated cell death and also inhibits infective viral particle formation by dysregulation of ubiquitin–proteasome system. J Neuroimmune Pharmacol 4:328–337
Gansukh E, Kazibwe Z, Pandurangan M et al (2016) Probing the impact of quercetin-7-O-glucoside on influenza virus replication influence. Phytomedicine 23:958–967
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Camini, F.C., da Silva Caetano, C.C., Almeida, L.T. et al. Implications of oxidative stress on viral pathogenesis. Arch Virol 162, 907–917 (2017). https://doi.org/10.1007/s00705-016-3187-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-3187-y