
Abstract
Monoamine oxidases (MAOs) catalyse the oxidation of neurotransmitter amines and a wide variety of primary, secondary and tertiary amine xenobiotics, including therapeutic drugs. While inhibition of MAO activity in the periphery removes protection from biogenic amines and so is undesirable, inhibition in the brain gives vital antidepressant and behavioural advantages that make MAO a major pharmaceutical target for inhibitor design. In neurodegenerative diseases, MAO inhibitors can help to maintain neurotransmitter levels, making it a common feature in novel multi-target combinations designed to combat Alzheimer’s disease, albeit not yet proven clinically. Vital information for inhibitor design comes from an understanding of the structure, mechanism, and kinetics of the catalyst. This review will summarize the kinetic behaviour of MAO A and B and the kinetic evaluation of reversible inhibitors that transiently decrease catalysis. Kinetic parameters and crystal structures have enabled computational approaches to ligand discovery and validation of hits by docking. Kinetics and a wide variety of substrates and inhibitors along with theoretical modelling have also contributed to proposed schemes for the still debated chemical mechanism of amine oxidation. However, most of the marketed MAO drugs are long-lasting irreversible inactivators. The mechanism of irreversible inhibition by hydrazine, cyclopropylamine, and propargylamine drugs will be discussed. The article finishes with some examples of the propargylamine moiety in multi-target ligand design to combat neurodegeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Enzyme catalysis in the biological context is dominated by the need to understand the flux of metabolites in the cell. Kinetic studies indicate how the enzyme works in the cell under the conditions found there, and also facilitate exploration of the chemical mechanism. This review will consider kinetic, mechanistic, and thermodynamic studies that tell us how MAO catalyses the oxidation of amines, how the catalytic turnover is influenced by its substrates, and how it is inhibited by drugs designed to slow the progression of symptoms in neurodegenerative disease. These in vitro studies underpin novel medicinal chemistry approaches to design or discover new lead compounds that (amongst effects on other targets) inhibit the activity of MAO and hence increase the concentrations of its amine substrates that are vital to brain function.
Why inhibit MAO?
Monoamine oxidases (MAOs) catalyse the oxidation of neurotransmitter amines and a wide variety of primary, secondary, and tertiary amine xenobiotics, including therapeutic drugs. The preferred amine substrate for MAO A is serotonin (5-hydroxytryptamine) and that for MAO B is 2-phenylethylamine, while dopamine and kynuramine are oxidized by both (Youdim et al. 2006). MAO A in the gut and placenta and MAO B in the liver and platelets serve a protective role. In the brain, both MAO A and MAO B are found in non-neuronal cells. MAO B predominates in serotonergic neurons, whereas other neurons contain MAO A. To combat depression or prevent neurodegeneration, the desirable target organ is naturally the brain, so the selectivity of the multi-target cholinesterase and MAO inhibitor, ladostigil, to the brain was a helpful step forward in avoiding peripheral side effects. Both MAOs are located on the outside of the mitochondrial outer membrane where MAO may function to protect the mitochondrion from accumulation of deleterious amines. Prevention of MAO activity inside neurons preserves neurotransmitters for the next firing from the terminals and also decreases the formation of hydrogen peroxide (H2O2), a reactive oxygen species, in the vicinity of the mitochondria to which the MAO is attached. Inhibition of MAO (particularly MAO B) in the non-neuronal glial cells ensures that monoamine neurotransmitters that escape from the synaptic junction are deactivated by oxidative deamination. Pharmacologically, inhibition of MAO in the brain increases the global content of amines, resulting in improved neuronal activity and antidepressant effects (Youdim et al. 2006; Fisar 2016). The modulation of brain and behaviour by MAO inhibitors (MAOIs) (Bortolato and Shih 2011) has made the design of new inhibitors a medicinal chemistry challenge in both academia and industry for the last 50 years.
Multi-target designed ligands (MTDL) for complex neurodegeneration
In degenerating brain, it is desirable to maintain the levels of neurotransmitters. MAOIs have been approved adjunctive therapy in Parkinson’s disease (PD) for many years, helping to preserve the diminishing dopamine and so delaying the need to start l-DOPA treatment. Extending the rationale to Alzheimer’s disease (AD), where the licensed treatments are cholinesterase inhibitors (ChEIs), MAO inhibition is an immediate choice for combination into MTDL, along with antioxidative capacity and other neuroprotective properties. The antiPD drug selegiline inhibits MAO B and has neuroprotective properties associated with the propargylamine moiety (Naoi et al. 2011; Magyar et al. 2006; Youdim et al. 2001; Naoi et al. 2016; Weinreb et al. 2011), making that fragment a suitable choice for the combination into MTDL. Many reports of compounds combining MAO inhibition with activity at various other targets have appeared in the last 5 years, for example, (Pisani et al. 2011; Kupershmidt et al. 2012; Guzior et al. 2015; Unzeta et al. 2016; Bautista-Aguilera et al. 2017). Ladostigil, one of the earliest MTDL, has shown only modest benefit in mild cognitive impairment (ClinicalTrials identifier NCT01429623), so the success of the strategy still has to be proven. The need for better drugs remains, so this review will summarize the kinetic behaviour of MAO A and B and the evaluation of inhibitors that decrease the breakdown of monoamine neurotransmitters, to provide an understanding of the structure, mechanism, and kinetics of these flavoprotein catalysts.
MAO kinetics
MAO accelerates the oxidation of amine to imine (Scheme 1) by orienting the substrate toward the N5 of the flavin within the aromatic “cage” of tyrosines (Li et al. 2006). The flavin co-factor oxidizes the amine, probably by hydride transfer (although this is still controversial as discussed below). The resulting in FADH− is re-oxidized by molecular oxygen generating hydrogen peroxide. The imine is hydrolysed non-enzymatically, mostly after its release from the enzyme.

Amine oxidation catalyzed by MAO gives an imine product that is subsequently hydrolysed
Turnover
Kinetic studies show that MAO A and MAO B have different intrinsic catalytic rates (kcat) (Youdim et al. 2006) in addition to the different affinities that result from the structure of the active sites (Binda et al. 2011; Edmondson et al. 2007). It is useful to remember that the affinity of a compound for the active site is measured as the dissociation constant, KD, whereas the kinetic parameter KM, defined as the concentration the gives half the maximum rate (kcat/2), contains terms for both KD and the kcat. Although the specificity constants (kcat/KM) of human MAO A and MAO B for dopamine and noradrenaline are similar, MAO B has a 500 times lower kcat/KM for serotonin (5-HT). The decarboxylation product of phenylalanine, β-phenylethylamine (PEA), that can activate TAAR1 receptors with downstream effects on monoamine systems, reduces human MAO A relatively slowly (rate constant 1 s−1) but reduces bovine MAO B very rapidly at 576 s−1 in the reductive half-reaction (Tan and Ramsay 1993). The slow oxidative half-reactions bring the steady-state rate constants for turnover to only fivefold different. However, the larger amounts of MAO B in rat cortex mean that PEA oxidation by MAO B is 550 times greater than for MAO A in that tissue (Youdim et al. 2006). A comparison of the intrinsic constants for human and rat MAOs with physiological substrates can be found in Tipton et al. (2006).
Kinetic mechanism(s)
The kinetic mechanism of MAO was first identified as ping-pong, where the substrate was oxidized and product released before the re-oxidation of the flavin. However, the different KM values for the second substrate, oxygen, with different amines suggested a more complex mechanism (Fowler and Oreland 1980). Inhibitor studies gave competitive Lineweaver–Burk plots for inhibition of MAO B by d-amphetamine when benzylamine was the substrate but mixed inhibition plots when PEA was the substrate, indicating that reduced enzyme was available to bind d-amphetamine during PEA oxidation but not during benzylamine oxidation (Pearce and Roth 1985). Stopped-flow kinetics for the half-reactions confirmed a binary pathway with PEA but a ternary complex of enzyme, benzylamine or its product, and oxygen (Husain et al. 1982; Ramsay et al. 1987). More recently, detailed steady-state kinetics on MAO B indicate that these alternate binary or ternary pathways for MAO B have impact on the determination of inhibition parameters and on the inhibition pattern observed (McDonald et al. 2010).
Pre-steady-state kinetics to study the reductive and oxidative half-reactions separately indicated that the ternary complex mechanism predominates for MAO A, at least for the substrates studied (Ramsay 1991; Tan and Ramsay 1993). In addition, it was clear that substrates could also bind to the reduced form of the enzyme and some substrates accelerated the re-oxidation of the flavin (Ramsay 1991), giving the more complex alternative kinetic pathways shown in Fig. 1. The increased rate of the oxidative half-reaction when reduced MAO A was pre-equilibrated with substrate prior to the reaction with oxygen in the stopped-flow spectrometer (100 times faster for kynuramine) was the same for all concentrations between 0.1 and 1 mM, indicating that the KD for the Ered–S complex was much lower than for the Eox–S complex (0.58 mM) (Tan and Ramsay 1993). A new study using X-ray crystallography and resonance Raman spectroscopy to study binding of substrates and substrate mimics (inhibitors) to another flavoenzyme, xenobiotic reductase A, revealed key evidence that substrates bound differently to the oxidized and reduced forms of the enzyme, and that substrate but not inhibitor resulted in spectral changes indicating a charge-transfer complex (Werther et al. 2017). The evidence points to alteration of the ground state of the reduced flavin by the proximity of the substrate, accelerating its re-oxidation, possibly a model for the substrate acceleration of FAD re-oxidation in MAO.

Kinetic pathways in MAO catalysis. After the oxidation of substrate and concomitant reduction of the flavin, the enzyme–product complex can be re-oxidized (lower part, k4[O2] via a ternary complex, very slow for some products) or the product (imine) can dissociate immediately (upper part, k5′). The free reduced enzyme can be re-oxidized in a binary reaction with O2 (k4′[O2], a rate of 1 s−1 at air saturation) or can bind new substrate and be re-oxidized in a ternary complex at a faster rate (k4″[O2]). A detailed description of the kinetics in the scheme can be found in Ramsay et al. (2011) and the data behind it in Husain et al. (1982) and Tan and Ramsay (1993)
Turning to the second substrate, oxygen, differences between MAO A and B are seen in the steady-state KM values. For cloned and purified human MAO A with kynuramine as the substrate, KM for oxygen is 0.06 mM, but for both bovine and human MAO B with benzylamine as the substrate, KM is 0.33 mM, slightly higher than the concentration of oxygen in buffer (Ramsay 1998; Newton-Vinson et al. 2000). This means that at normal oxygen concentrations in the cell, MAO B is working at less than half the Vmax. If oxygen concentrations drop, the oxidation of amines will decrease. The KM for oxygen in MAO A purified from human placenta was 0.006 mM, a value reassuring for the protection of the fetus from biogenic amines, but there is no known rationale for the difference from the cloned MAO A. When the steady-state level of reduced enzyme is monitored in turnover experiments in a stopped-flow spectrophotometer, the proportion of reduced enzyme is related to the ratio of the oxidative to reductive rate constants—the slower the oxidation rate relative to the reduction rate, the more MAO is reduced in the steady-state. With kynuramine, the flavin in MAO A remains 95% oxidized at the onset of the steady state, whereas with 5-hydroxytryptamine, it is 78% oxidized (Tan and Ramsay 1993). For bovine MAO B with phenylethylamine, the oxidative half-reaction is rate-limiting (Husain et al. 1982), so that MAO B will be mostly reduced in the steady state. The consequences of the complex kinetics for the brain are first, that the oxygen level will strongly influence the disposal of amines by MAO B, and second, that the redox poise of MAO presents a varied proportion of the two states of the enzyme (oxidized and reduced) with different affinities for a given ligand. Thus, in vitro IC50 values may differ from in vivo values, because the proportion of the two redox states will vary with the amine substrate and with oxygen tension (see more about inhibitors below).
Kinetic isotope effects (KIE)
To probe the mechanism of catalysis, isotope effects have been studied for both MAO A and B. When the hydrogen that is removed from the substrate is replaced by deuterium that has double the mass, the rate is slower. All MAO catalysed reactions show a deuterium isotope effect of 5–10 with α,α-dideutero-benzylamine (Miller and Edmondson 1999b; Walker and Edmondson 1994; Dunn et al. 2008), indicating that the transfer of a hydrogen (the reductive half-reaction) is the slowest part of the process. By varying oxygen concentrations in steady-state assays, it was also shown that the KIE D[kcat/KM(O2)] was 1 for human MAO B (Edmondson and Newton-Vinson 2001).
Theoretical simulation of the H/D KIE for dopamine oxidation in the reductive half-reaction of MAO B calculated a KIE of 12.8 based on a hydride transfer mechanism. Going further into the physical chemistry of the reaction, primary and secondary isotope effects determined at different pH and temperatures provided the experimental evidence for hydrogen tunneling in MAO B (Jonsson et al. 1994). The pH of the medium influences the protonation state of the substrate, as does binding to the enzyme. KIE in the reductive half-reaction of MAO A was higher at low pH due to the influence of the deuterium substitution on the pKa of the amine which drops by almost 2 pH units upon binding (Dunn et al. 2008). In accord with the pH dependence of turnover in steady-state studies (Jones et al. 2007), the bound substrate has a lower pKa, resulting in the catalytically required neutral amine form in the flavin active site. Comparison of the KIE for dideuero-, R-deutero-, and S-deutero-dopamine (the latter giving no change in rate) along with analysis of the products established that it is the R-hydrogen that is removed from dopamine during oxidation by both MAO A and B (Yu et al. 1986).
Insights from structure and mutagenesis
The flavin in MAO is FAD. After trypsin/chymotrypsin digestion, the 8a-S-cysteinyl-FAD pentapeptide liberated is the same for both MAO A and B (Kearney et al. 1971). Using different flavin analogues during expression of MAO B in yeast revealed that the covalent attachment stabilizes the structure, helps to align the cofactor in the active site, and modulates the redox potential upwards (Edmondson and Newton-Vinson 2001).
Sequencing of the cloned human genes revealed 70% identity, with nine conserved cysteine residues (Bach et al. 1988). The FAD attachment was identified as Cys406 in MAO A and Cys397 in MAO B. When riboflavin-deficient mutants were used to explore the covalent flavinylation in both MAO A and B (Miller and Edmondson 1999a), only enzymes with covalently attached flavin were active. When each of the other eight cysteines was mutated to serine and the mutants expressed on COS cells, all retained activity with the same KM for substrate except C374S in MAO A and the Cys–Ser mutants at 156 and 365 in MAO B which were inactive (Wu et al. 1993). It was later found that Cys374/365Ala mutants expressed in yeast were active and a kinetic study confirmed minimal effect on ligand binding. However, the specificity constant (kcat/KM) for the mutant enzyme was 30% lower for five different substrates, indicating an effect on catalysis (Vintem et al. 2005). In MAO B, the equivalent residue Cys365 was alkylated after cyclopropylamine inactivation (Zhong and Silverman 1997). When the crystal structure was obtained, it showed that the location of MAO A Cys374/MAO B Cys365 was on the surface near the entrance cavity rather than in the active site. In contrast to MAO A, MAO B does have one active site cysteine, Cys156. MAO B Cys156 has been implicated in hydrogen bonding for some small ligands.
The C-terminus of MAO has 27 residues (residues 498–524 in rat MAO A) that form an alpha-helix embedded in the membrane (Son et al. 2008). C-term truncation to explore the interaction of MAO with the membrane produced active, but unstable enzyme (Weyler 1994; Rebrin et al. 2001). Truncations from residues 498–520 (520 being full length) remain active and membrane-bound. The enzyme truncated at 492, 486, and 481 becomes progressively more soluble but has very low and unstable activity. Interestingly, all these truncated versions give very little alteration in the sensitivity to inactivation by clorgyline and selegiline, indicating that these residues are not part of the active site. The truncation experiments were rationalised when the 1.7 A structure of human MAO B revealed several apolar loops in proximity of the C-terminal helix that provide additional membrane association (Son et al. 2008). Furthermore, molecular dynamics simulations based on the structure of rat MAO A demonstrated strong interactions with the membrane surface (Apostolov et al. 2009). The membrane association influences the catalytic properties, with changes in substrate KM between detergent-solubilized and membrane-bound MAO (Edmondson et al. 2009; Esteban et al. 2014). A recent study examined the kinetic parameters of purified MAO A incorporated into nanodiscs with the same thickness as the phospholipid bilayer. The catalytic efficiency (kcat/KM) increased for substrate oxidation and the Ki for inhibitors decreased 2–4-fold with the nanodisc associated enzyme (Cruz and Edmondson 2007). This experimental observation re-opens the notion in older literature that the phospholipid composition of the membrane might alter MAO activity.
The mutation of the main pair of tyrosines that form the aromatic cage around the substrate near the flavin revealed a strong influence on catalytic efficiency. Substitution of the aromatic cage tyrosines increased the KM for the artificial substrate 1-methyl-4-(1-methyl-1H-pyrrol-2-yl)-1,2,3,6-tetrahydropyridine (MMTP) more in MAO B then in MAO A, and seriously decreased the kcat (Table 1) (Li et al. 2006). The kinetic data for the mutants with a series of substrates, supported by lack of structural effects, suggested that dipole–dipole interactions between the two aligned tyrosines and the amine nitrogen were important for catalysis. Computational studies also support a role for the aromatic cage in substrate binding (Akyuz et al. 2007). The same mutations (tyrosine to phenylalanine and histidine) were used in EPR studies to establish that the radical formed upon one-electron reduction was that of the anionic semiquinone (Ramsay et al. 2005) and not due to a proposed tyrosyl radical (Rigby et al. 2005; Dunn et al. 2010).
Mutagenesis studies were also used to explore the different selectivities of the two enzymes. The determination of the crystal structure of MAO B in 2002 (Binda et al. 2002) and MAO A in 2004 (Ma et al. 2004; De Colibus et al. 2005) provided a boost to understanding ligand binding and the differences between MAO A and B (see below). The structures helped to explain the specificity change induced by reciprocally switching Phe208 and Ile199 in rat MAO A and B, respectively, which was sufficient to switch their substrate and inhibitor preferences (Tsugeno and Ito 1997). However, in the human forms of MAO, the F208I mutant of MAO A showed the same sixfold decrease in the specificity constant kcat/Km with 5-hydroxytryptamine and with β-phenylethylamine, rather than a change in selectivity (Geha et al. 2000). Kinetic studies of inhibition of MAO and MAO mutants in various species showed that whereas the small molecule isatin inhibited all the MAO B enzymes tested with the same Ki, the larger reversible inhibitors 8-(3-chlorostyryl)caffeine, 1,4-diphenyl-2-butene, and trans,trans-farnesol competitively inhibited human and rat MAO B (with Ile at 199) but not MAO A, bovine MAO B or the human MAO B I199F mutant (all with Phe at 199). The crystal structures showed that Ile199 could rotate to accommodate the larger molecules, but Phe199 could not (Hubalek et al. 2005).
The other residue located at the “gate” between the entrance and substrate cavity of MAO B is Tyr326, corresponding to Ile335 in MAO A. Comparing activity with serotonin (as a predominantly MAO A substrate) and phenylethylamine (a predominantly MAO B substrate), the I335Y mutant of MAO A did switch the substrate selectivity albeit with serious loss of activity. This kinetic data provided experimental validation for molecular simulation of the effect of the mutation on the catalysis. Using empirical valence bond methodology, free energy perturbation, and a classical force field to simulate the chemical reaction, it was shown that the mutation increases the free energy barrier for the rate-limiting hydrogen transfer step by slightly more than 1 kcal mol−1 and consequently decreases the rate constant by about an order of magnitude (Oanca et al. 2016). In contrast, for the corresponding switch in MAO B, the Y326I mutant increased the oxidation of serotonin by fourfold and decreased the KM making the kcat/KM higher than that for phenylethylamine. The sensitivities to the selective inhibitors clorgyline and selegiline were also switched (Geha et al. 2001).
In another computational study to define the determinants for binding a 2H-chromene-2-one competitive inhibitor, the contributions of nearby residues to the free energy of binding were quantified. The Phe208/Ile199 and the Ile335/Tyr326 residues contributed 2.3/2.8 and 1.8/1.7 kcal mol−1, respectively, confirming influence on ligand binding (Mangiatordi et al. 2017). However, it was Gln215/Gln206 that gave different contributions: 3.8 vs 1.7 kcal mol−1 in MAO A and B, respectively, confirming a key role for Gln215 in the selectivity towards MAO A of this particular compound. For a different compound, 2-amino-5-(4′-methoxy)-phenylfuran-3,4-dicarbonitrile, it was Asn181 (in addition to steric contribution from Ile335) that was responsible for a tenfold higher affinity to MAO A than B, simply by forming a hydrogen bond with the inhibitor (Juarez-Jimenez et al. 2014). These and similar studies emphasize that although the monoamine oxidases bind a wide and varied range of ligand structures, the interactions in the active sites can be highly specific.
In the only report exploring residues important in the oxidative half-reaction, the conserved lysine that hydrogen bonds (via a water) to N5 of the flavin was mutated in MAO B. Only the K296R mutant was active, but PEA was oxidized at 10% of the WT rate. By varying the oxygen concentration to determine kcat/KM(O2), it was estimated that the rate of oxidation was decreased by about a factor of 6 due to the increased basicity of Arg compared to Lys (Kacar and Edmondson 2006).
MAO chemical mechanism
How the oxidation of an amine with a redox potential of + 1 V using a cofactor with a redox potential of about − 0.2 volts is achieved has intrigued researchers for decades. The answer lies in the interaction with the protein changing not only the energy level of the transition state between substrate and product (Vianello et al. 2012), but also that of the redox cofactor (Fraaije and Mattevi 2000). Although the pKa of dopamine shifts only from 8.9 in water to 8.8 in the active site (Vianello et al. 2012), a group with a pKa at 7.4–7.9 in the enzyme–substrate complex but not the substrate or MAO (Jones et al. 2007; Dunn et al. 2008) is an indication of modulation. For the cofactor, the redox potential in MAO was determined by reductive titration with dithionite in the presence of mediator dyes. For human MAO A, the value for the first electron reduction to the anionic semiquinone was − 159 mV and for the second redox couple from semiquinone to the quinone at pH 7.4 was − 262 mV and similar values were found for bovine MAO B (− 167 and − 275 mV) (Sablin and Ramsay 2001), consistent with the accumulation of semiquinone to about 30% of the total flavin during dithionite reduction. For cloned human MAO B, the one-electron reduction potentials were found to be much closer and slightly positive at + 0.043 V for the first electron reduction and + 0.037 V for the semiquinone–quinone couple (Edmondson et al. 2007). It should be noted that redox equilibrium with the reporter dyes is slow and could be the source of the differences. In the presence of a substrate, no semiquinone is formed, and a higher redox potential of + 200 mV was estimated for human MAO A (Ramsay et al. 1995). In contrast to substrates, inhibitors stabilize the semiquinone form, a clear indication that substrates and inhibitors have different interactions with the co-factor (Ramsay and Hunter 2002; Hynson et al. 2003, 2004). Circular dichroism (CD) spectroscopy that detects alterations in the environment of aromatic residues as a result of ligand binding or redox changes distinguished between substrate and dithionite reduction (a difference seen also in the visible spectrum) and gave different spectral changes depending on the size and substituents of the inhibitor (Hynson et al. 2004). This study revealed changes in the aromatic cage on ligand binding and on reduction, both of which alter the environment of the flavin and its surrounding tyrosines.
Three mechanisms for the oxidation of amines by MAO have been much discussed and remain in contention (Silverman 1995c; Kay et al. 2007; Orru et al. 2013). Evidence for the polar nucleophilic mechanism has been provided by extensive kinetic analyses (based on Hammett correlations) with series of rationally chosen substrates. Experiments on MAO A using para-substituted phenylethylamine derivatives demonstrated that electrophilic substituents increased the rate of oxidation, as expected for the polar nucleophilic mechanism (Nandigama and Edmondson 2000). For MAO B, the opposite effect was found, with the rate slightly decreasing with the more electron-withdrawing substituents to both benzylamine and PEA, although further analysis suggest that steric effects could account for the differences (Walker and Edmondson 1994; Miller and Edmondson 1999b). It has been argued that these conflicting correlations indicate that MAO A and MAO B could use different mechanisms to oxidize the amine to imine. Looking at electronic effects of various benzylamine ring substituents on the kinetics of the reaction, substituents giving high turnover with MAO A were poor substrates for MAO B (Wang and Edmondson 2011; Orru et al. 2013). This result clearly suggests the possibility of different mechanisms in MAO A and B (however, unlikely it seems) and also the possibility that different substrates could be oxidized by different mechanisms depending on the rate constants for the steps involved. However, for both MAO A and B, steric parameters are important. The substitution at the 2′ position of MPTP hinders the planarity required for conjugation of the phenyl ring with the tetrahydropyridine moiety (Youngster et al. 1989). These 2′ derivatives are better substrates than the 4′ substituted derivatives, implying that the 4′ electron-withdrawing substituents do not facilitate catalysis as would be expected for the polar nucleophilic mechanism. The C4a adduct formed with substrate in the initial step of this mechanism has been proposed to make the N5 of the flavin a stronger base to extract the proton (pKa = 25) from the alpha carbon [see (Edmondson et al. 2009) for a detailed discussion]. This two-step process is also supported by the 15N kinetic isotope effect, showing that C–H bond cleavage and the change in the C–N bond order were not completely concerted (MacMillar et al. 2011).
The single electron transfer mechanism has substantial chemical support from experiments based on cyclopropylamine derivatives. Silverman, analysing the kinetics and the products formed, demonstrated that cyclopropyl amines inactivate MAO labelling either a protein thiol group or the flavin depending on the structure of the cyclopropylamine (Silverman 1995c; Vintem et al. 2005). However, no transient flavosemiquinone is seen during catalysis (Miller and Edmondson 1999b). Further evidence against a radical mechanism for normal MAO substrates has been summarized by Fitzpatrick (2010) drawing parallels with other oxidase families.
The simplest mechanism of the three is a hydride transfer. The rates of oxidation of MPTP derivatives mentioned above are consistent with a hydride mechanism (Youngster et al. 1989). In the high-resolution crystal structure of the analogous flavoprotein, d-amino acid oxidase, the alpha carbon was in direct and close alignment to the flavin N5, ready for hydride transfer (Umhau et al. 2000; Fitzpatrick 2010). In the crystal structure of MAO B with farnesol which has an OH group rather than an amine, the alpha carbon is 3.4 Å from the flavin N5 (Hubalek et al. 2005; Binda et al. 2006) which brings the proton to about 2.3 Å, close enough for hydride transfer. The consistency of the calculated activation energies for dopamine and noradrenaline (Vianello et al. 2016) with experimental data for MAO A and MAO B also supports a hydride mechanism.
When calculations based on density functional theory are used to model the MAO reductive half-reaction, various groups come to conflicting conclusions. Using QM/MM calculations, the oxidation of unprotonated benzylamine gave rates in good agreement with experimental values. The electronic structure found during the process was consistent with the asynchronous polar nucleophilic mechanism and ruled out a radical mechanism (Abad et al. 2013). In further work, the author suggested that the protein environment of MAO A gave the mechanism an enhanced polar nucleophilic character compared to that of MAO B (Zenn et al. 2015). In contrast, a relaxed-geometry scan of the alpha–CH bond compressing it in 0.1 Å increments showed no indication of the formation of a stable complex (Vianello et al. 2016), and another QM/MM study suggested direct hydride transfer mechanism for the oxidation of phenethylamine and benzylamine (Akyuz and Erdem 2013). Applying ONIOM methodology to serotonin oxidation, the latter group proposed a hybrid mechanism between hydride and proton transfer where hydride transfer dominates over the proton transfer (Cakir et al. 2016).
Taking all the computational and experimental evidence into consideration, it was proposed that MAO A reacts by H+ abstraction, whereas MAO B works by hydride transfer (Orru et al. 2013). Molecular simulation to calculate the energy required to take the bound dopamine substrate to its transition state in MAO B by the polar nucleophilic mechanism determined a free energy barrier of 44.6 kcal mol−1, but by the hydride mechanism, the energy barrier was only 24.4 kcal mol−1, giving strong thermodynamic argument that MAO B oxidizing dopamine follows the hydride mechanism. For the overall binding and oxidation of dopamine by MAO B, the activation energy via the hydride mechanism of 16.1 kcal mol−1 was in excellent agreement with the experimental value of 16.5 kcal mol−1 (Vianello et al. 2016). For MAO A oxidation of noradrenaline via the hydride transfer reaction, the activation energy was calculated as 20.3 kcal mol−1, only slightly higher than the experimental value of 16.3 kcal mol−1 (Vianello et al. 2016). The debate still continues.
Inhibition
As mentioned earlier, many xenobiotics and therapeutic drugs inhibit MAO with the benefit of raising amines in the brain. Most of the successful drugs are irreversible inhibitors of MAO (Youdim et al. 2006), although selective reversible inhibitors, such as moclobemide, are newer additions to the market. Before considering inhibitors in detail, it is useful to define the major mechanisms of inhibition. These definitions and the kinetic parameters normally measured are shown in Table 2. Irreversible inhibitors form a covalent adduct with the protein, so their effect lasts until new protein is made, a process that takes days for MAO. Reversible inhibitors generally have rapid on and off rates compared to the rate of substrate turnover, so the concentration at the target must be maintained to ensure inhibition. Moclobemide, a reversible inhibitor (Bonnet 2002), requires twice daily administration to give a therapeutic occupancy rate of about 74% for MAO A, whereas a single daily dose (10 mg kg−1) of the irreversible inhibitor tranylcypromine gave 58% occupancy (reviewed in Fowler et al. 2015). Effective antidepressant effect is evident at around 80% for both moclobemide and phenelzine (Chiuccariello et al. 2016).
Kinetics of reversible inhibition
Standard physical methods such as microcalorimetry for the determination of inhibitor dissociation constants (KD) are not suitable for MAO due to instability and aggregation after isolation except in high concentrations of detergent. Reports of immobilised MAO for use as amine-detecting electrodes usually employ the soluble copper amine oxidase or bacterial MAO N. Binding of radio-labelled ligand or direct measurement of changes in the absorbance spectrum as in Fig. 2 (Hynson et al. 2003) or changes in the fluorescence of purified MAO can also be used to determine small molecule KD values, but the kinetic determination of Ki is usually more versatile and convenient.

Titration of MAO A (6.6 μM) with befloxatone and calculation of the KD. Left, the selected difference spectra were calculated by subtracting the spectrum for MAO A alone from those equilibrated with befloxatone at 0.38, 0.77, or 1.15 μM, all converted to absorbance for millimolar MAO A. Right, the difference between the absorbance at the 485 nm maximum and at the 503 nm minimum are plotted against the concentration of befloxatone
Fluorescent or bioluminescent ligands have been designed to provide high sensitivity for activity assays and for cell imaging (Valley et al. 2006; Holt and Palcic 2006; Peng et al. 2010; Li et al. 2014a, b) and for in vivo imaging (Li et al. 2016; Kim et al. 2016). Of particular interest is a new probe that releases a fluorophore (4-hydroxy-N-butyl-1,8-naphthalimide) by the catalytic action of MAO A, making it selective for MAO A (Wu et al. 2016). For steady-state in vitro assays, the direct fluorescence of the product from kynuramine oxidation has been used in stopped (single time point) assays to assess several inhibitor series (Matsumoto et al. 1985; Delport et al. 2017). The popular and convenient Amplex Red assay for the product H2O2 allows continuous monitoring of product generation but is a coupled assay so, like all coupled assays, must be used with caution. A major concern is that Amplex Red inhibits MAO A (Ramsay and Tipton 2017), but, additionally, some MAO inhibitors inhibit horseradish peroxidase used in the coupling system (Hroch et al. 2017).
Although all computational methods predict the equilibrium binding or KD, experimental assessment of large numbers of compounds is generally done by measurement of IC50 values avoiding the requirement for definitive equations. The IC50 for a competitive inhibitor is directly related to the Ki as follows:
but for mixed inhibition, the IC50 value has a more complex relationship with two Ki values:
Thus, the relationship between IC50 and Ki depends on the substrate concentration used and on the type of the reversible inhibition (Ramsay and Tipton 2017) (Fig. 3).

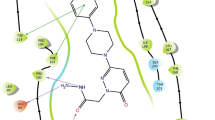
Differential inhibitor binding to oxidized and reduced MAO. a For MAO A with 400 μM 3-phenylpropylamine as substrate, 2-BFI is a competitive inhibitor giving a simple dose–response (closed symbols), IC50 = 50.2 μM. In contrast, for MAO B with 30 μM benzylamine, the dose–response curve for 2-BFI has two components, because it binds to the oxidized form with Ki of 7.9 μM and to the reduced form with a Ki of 326 μM (Ramsay et al. 2011). b Structure of 2-BFI in the entrance cavity of hMAO-B (McDonald et al. 2010)
The type of inhibition by a given compound is determined by varying both substrate and inhibitor. With MAO A, all published reversible inhibitors give competitive inhibition, but with MAO B, mixed inhibition is frequently observed. Kinetic observation of mixed inhibition can come from either differential inhibitor binding to the two redox states present during turnover or from binding at the imidazoline (I2) site which might occur not only to free E but also to the E–S complex, as demonstrated for phenylethylamine and for tranylcypromine (McDonald et al. 2010). These complexities mean that the mechanisms of inhibition should be determined for each series of new compounds. For comparisons across series and for comparison with docking, Ki values are always more informative than IC50 values.
The accurate assessment of reversible inhibitors depends on reliable quantitative assays for determination of initial IC50 values, for which the substrate concentration used must be defined. In all cases, standard inhibitors used as comparators must also be reversible inhibitors. Many published medicinal chemistry articles have used selegiline and clorgyline as comparators without consideration of the time factor that applies to these irreversible inhibitors but not to the reversible one being tested. Where the kinetic mechanism is investigated and the Ki rather than IC50 is determined, the Ki is more likely to reflect the thermodynamic binding from theoretical calculations, particularly for MAO B (McDonald et al. 2010).
The mixed inhibition with MAO B arises from the two forms of the enzyme (oxidized and reduced) that bind the inhibitor, as shown in Fig. 1 (Pearce and Roth 1985; Ramsay et al. 2011). Oxidation of substrates that reduce MAO B at a rate comparable to the rate of re-oxidation of the flavin by oxygen will have free reduced MAO B available for inhibitor binding, and the proportions of reduced enzyme during turnover differ with the substrate (Tan and Ramsay 1993). Inhibitor binding to reduced enzyme can be different from that to the oxidized enzyme: for example, d-amphetamine binds to reduced MAO A with five times lower affinity (Table 2) (Ramsay et al. 2011). The problem can be avoided using a substrate where MAO B is predominantly in the oxidized form during the steady state (as is always the case for MAO A). The only substrate where this has been demonstrated is MPTP, but its neurotoxic properties rule it out as a routine substrate (Tan and Ramsay 1993).
Reversible inhibitors
Optimization of early inhibitors of MAO was based on kinetic parameters determined in ex vivo tissues, for example, the inhibition by d-amphetamine (Mantle et al. 1976; Dorris 1982) or other drugs (McCoubrey 1957; Yang and Neff 1974) before moving to in vivo pharmacology (Youdim et al. 1971; Miller et al. 1980; Riederer and Jellinger 1983; Da Prada et al. 1990). The first pharmacophores for MAO were devised by superimposition of multiple inhibitors using simple computational methods (Efange et al. 1993; Medvedev et al. 1999; Veselovsky et al. 2004). The publication in 2002 (Binda et al. 2002) of the crystal structures with reversible ligands bound opened the way for prediction of modifications of compounds to optimise binding (Reck et al. 2005). Nowadays, cheminformatic 3D-QSAR and Virtual Screening methodologies enable the discovery of hits from millions of compounds (reviewed in Nikolic et al. 2016). The search for potent competitive inhibitors of MAO A and B continues, driven by the market for up-regulating levels of monoamine neurotransmitters to combat depression and neurodegeneration (Carradori and Silvestri 2015; Finberg and Rabey 2016). The MAO A-selective inhibitor, moclobemide, is used as antidepressant (Da Prada et al. 1990), whereas the selective MAO B inhibitor, safinamide (Caccia et al. 2006; Binda et al. 2007), is used against epilepsy. MAO B is increased in neurodegenerative processes due to glial activation and so is a target of interest, particularly for multi-target compounds that address neurodegeneration. Progress in selective inhibitor design for MAO B has been reviewed recently (Carradori and Silvestri 2015).
Figure 4a shows some reversible inhibitors of MAO A (Da Prada et al. 1990; Hynson et al. 2003; Ramsay et al. 2007; Petzer et al. 2012; Heal et al. 2013), mostly with fairly bulky structures that can be accommodated in the single MAO A cavity (Son et al. 2008). The inhibitors of MAO B (Fig. 4b) (Mazouz et al. 1993; Binda et al. 2007, 2012; Desideri et al. 2016; Borroni et al. 2017; Tzvetkov et al. 2017) have a typical linear shape reminiscent of the diphenylbutene molecule in the first crystal structure of MAO B (Binda et al. 2002).

Reversible inhibitors of a MAO A (with Ki values) and b MAO B (with IC50 values)
One of the simplest of the inhibitors is d-amphetamine, the α-carbon methylated analogue of PEA, which is used to treat attention deficit hyperactivity disorder (ADHD) but has complex pharmacology and clinical effects, because it inhibits monoamine reuptake systems as well as MAO (Hutson et al. 2014; Heal et al. 2013). It is a useful reversible competitive inhibitor of MAO A with Ki value of 15 μM (Ramsay 1991; Ramsay and Hunter 2002). With MAO B, the IC50 is about ten times higher and the inhibition is mixed, because both oxidized and reduced MAO B are present during turnover (Pearce and Roth 1985). The reduced MAO B binds the inhibitor less well (Ki = 2.5 mM) than oxidized enzyme (Ki = 0.5 mM, values using either benzylamine or phenylethylamine as the substrate) (Ramsay et al. 2011). Methylation on the nitrogen does not change the Ki, but adding a second methyl to the alpha carbon (phenteramine) decreases the selectivity of binding (Table 3). Interestingly, 3,4-methylenedioxymethamphetamine ((+)-MDMA), methylated both on the α-carbon and on the nitrogen, also inhibited MAO A well (Ki = 22 μM) and gives mixed inhibition of MAO B with an IC50 value of 370 μM (Leonardi and Azmitia 1994). Methylation also affects the activity of the mechanism-based inhibitor, rasagiline, presumably influencing the orientation of the propargylamine group (Sterling et al. 2002).
Some inhibitors of MAO, even reversible inhibitors, show time dependence, probably from slow conformational adjustment after initial binding. Examples requiring at least 5 min to reach equilibrium include the beta-carbolines (Kim et al. 1997) and pirlindole derivatives (Hynson et al. 2003). Most screening studies pre-incubate the test inhibitors with MAO before the assay to avoid this problem. Tight-binding inhibitors present another challenge to accurate determination of Ki values, because the assumption that binding does not decrease the free concentration will not be met. When nanomolar IC50 values are found, then proper analysis for tight binding must be applied (Copeland et al. 1995; Morrison 1969). The commonly used assay coupling formation of H2O2 to a fluorescent dye generally uses about 0.5–1 nM MAO, a desirable affinity for reversible inhibitors in drug design. Using a spectrophotometric assay where the minimum concentration of enzyme was about 30 nM, the Ki of 29 nM for Methylene Blue with MAO A was determined applying tight-binding analysis (Ramsay et al. 2007). An early oxadiazolone series gave IC50 values in the nM range, inhibiting MAO B by a two-step process, initially competitive, followed by slowly reversible tight binding (Mazouz et al. 1993). Other examples include quinolones with IC50 values in the low nM range (Meiring et al. 2013), chromenones (best IC50 3.1 nM) (Pisani et al. 2013), the N-alkylated indazole-5-carboxamide derivatives (N-(3-chloro-4-fluorophenyl)-1-methyl-1H-indazole-5-carboxamide (IC50 hMAO-B 0.662 nM, > 15,000-fold selective versus MAO A) (Tzvetkov et al. 2017). This selectivity is desirable. The antiepileptic drug safinamide is an MAO B inhibitor with a Ki of 0.45 μM, three orders of magnitude better than for MAO A (345 μM) (Binda et al. 2007). Another example is the series of 7-substituted coumarin derivatives assessed for inhibition of MAO and cholinesterases, in which the authors noted that increased MAO B inhibition is seen when a halogen is substituted on the para-position of the benzyl ring, giving an IC50 of 0.5 nM (Joubert et al. 2017). However, the best multi-target compound was the N-benzylpiperidine derivative with only 300 nM IC50 for MAO B, but good micromolar IC50 values for the cholinesterases.
It should be noted that all of these compounds are reversible inhibitors. To claim that they are more potent than an irreversible inhibitor such as selegiline based on one IC50 measurement is wrong. The effect of irreversible inhibitors varies with time. Without pre-incubation, the reversible IC50 for selegiline (measured against substrate at 2 × KM) is about 50 nM, but with 30 min pre-incubation, the IC50 decreases more than tenfold. The comparison aside, a reversible inhibitor with nanomolar potency is potentially a very useful compound. Not only could the compound be suitable for MAO B inhibition, but also it could be a lead fragment to combine with structures inhibiting cholinesterases for a multi-target drug to combat the effects of neurodegeneration.
Reversible inhibitors with nanomolar potency are also useful for positron emission tomography (PET) for in vivo studies of MAO and MAO inhibition in humans. For MAO A, [11C]-harmine (Ki 4 nM) is the main reversible inhibitor used. For MAO B, an oxazolidinone derivative, [11C]-SL25.1188, with an IC50 of 11.8 nM in rat brain (Sara et al. 2010), has now been tested in humans (Rusjan et al. 2014). Most human studies have used the well-established irreversible inhibitors [11C]-clorgyline for MAO A and [11C]-deprenyl for MAO B, to measure the levels of MAO or competition with unlabeled reversible inhibitors (see Fowler et al. 2015).
Computation and reversible inhibitors
Docking has become an integrated part of drug design, progressing from comparison of overlaid ligands as a description of a pharmacophore (Efange and Boudreau 1991) or identification of pockets in homology models where substituents could tune binding (Reck et al. 2005; Bautista-Aguilera et al. 2014a), to fully automated virtual screening to identify the structures of the highest probability of binding to a drug target (Sliwoski et al. 2014; Nikolic et al. 2016). Such cheminformatic approaches are cost-effective ways to identify small pools of lead compounds from huge chemical libraries, particularly for multi-target drug design where the aim is to design one molecule to fit multiple targets, each with different requirements (for example, Bautista-Aguilera et al. 2014a). Molecular docking is the commonly used technique that models the interaction between the ligand and its target. In addition to providing a picture of how the ligand sits in the active site (giving multiple poses ranked for optimal binding), the atomic-level interactions are used to predict Ki values. Despite the prediction capability of the methods, experimental evidence is not always in accordance with the model values (for example, Bautista-Aguilera et al. 2014b). Ultimately, experimental validation of the predicted activity of the target is essential.
Although docking is routinely used in medicinal chemistry papers investigating new series of reversible inhibitors, much more information can be revealed by molecular dynamics, although this demands so much more computational time that dynamic runs are limited to 10–100 ns. The model is prepared from an X-ray crystallographic structure of MAO by removing the crystallized ligand, inserting the cysteine-FAD covalent attachment, adding hydrogens, and retaining the few water molecules common in most of the crystals. A small region near the flavin is selected as flexible and is conformationally relaxed before the simulation of the ligand-binding dynamics starting from the best docking pose. To give one example, molecular dynamics were key to understanding active site interactions as the source of a tenfold better Ki with MAO A than with MAO B for 2-amino-5-(4′-methoxy)-phenylfuran-3,4-dicarbonitrile (as described above). Steric hindrance from Tyr326 in MAO B kept the ligand further away from the flavin than in MAO A preventing optimal hydrogen bonding (Esteban et al. 2014). Using similar computational modelling, binding in different orientations has now been found for several different ligand classes with MAO, as well as for different binding to oxidized and reduced MAO (Basile et al. 2014).
Irreversible inhibition
Unlike reversible enzyme inhibition, irreversible inhibition leads to permanent deactivation of the enzyme. MAO adducts must be removed and replaced with newly synthesized MAO to restore activity. The activity of MAO in rat brain after irreversible inhibition recovered with a half-life of 9 days (Youdim and Tipton 2002). Monitoring levels by positron emission tomography, Fowler et al. observed a wash-out period of 40 days for MAO B (Fowler et al. 2015). The slow turnover of MAO in vivo is a key factor in the success of the irreversible MAOIs as drugs.
The irreversible MAO inhibitors are termed suicide, time-dependent, enzyme-activated, covalent, and mechanism-based inhibitors (Williams and Lawson 1974; Kalgutkar et al. 1995, 2001). The drugs shown in Fig. 5 are all mechanism-based inhibitors. The seven criteria for the designation, discussed in detail by Silverman (Silverman 1995b), are time dependence of inactivation, observation of saturation kinetics, substrate protection, irreversibility, fixed stoichiometry, involvement of an enzyme-catalysed step, and inactivation prior to release of active species. Classes of irreversible MAO inhibitors include propargylamines, cyclopropylamines, hydrazines, amino acetamides, and aryl oxazolidinones. After general consideration of the kinetics of mechanism-based inactivation, the first three classes (Fig. 5), the most studied, will be discussed.

Mechanism-based MAO inhibitors. a Propargylamines; b cyclopropylamine; c hydrazines
Kinetics of irreversible inhibition
In general, mechanism-based irreversible inhibitors are substrate analogues that are processed by the targeted enzyme to generate highly reactive species—the inhibitor is inactive until MAO acts on it. The reactive product covalently modifies the enzyme and suppresses its catalytic activity (Szewczuk et al. 2007; Silverman 1995a). The oxidation of the amine, therefore, represents the first phase in MAO irreversible inhibition pathway by several types of inactivating compounds (Silverman 1995b; Kalgutkar et al. 2001; Chajkowski-Scarry and Rimoldi 2014). The mechanism of inactivation in its most general and simplistic form can be represented as in Fig. 6 where E and I represent the free enzyme and inhibitor, respectively, E–I represents the enzyme-inhibitor complex, E–I* and E–P are the complexes between the enzyme and the bound oxidized inhibitor or product, respectively, and EI* is the covalent enzyme-inhibitor adduct. The adduct formed divided by product formed at infinite time or the ratio of k4/k3 represents the partition coefficient, the ratio of successful inactivation to modified inhibitor release.

Kinetic scheme for mechanism-based inactivation and the equations for the parameters, where E is enzyme, I is inhibitor, and [pi] is the concentration of product at infinite time
Mechanism-based irreversible inhibition depends not just on the concentration of the inhibitor but also on time. If the IC50 is determined by adding enzyme to substrate and inhibitor at the same time, the parameter obtained will be the Ki for reversible binding. If the inhibitor is pre-incubated with MAO for 30 min before adding substrate, then the IC50 obtained will come from a mix of inactivated and reversibly inhibited MAO and cannot be used to compare different classes of compounds with different inactivation rates (Ramsay and Tipton 2017). Proper information can be obtained from progress curves where the oxidation of substrate is measured in the presence of the irreversible inhibitor (McDonald and Tipton 2012). The normal linear assay rate will become curved as the concentration of active enzyme decreases. The other method commonly used is pre-incubation with the inactivator then diluting the mixture into an assay mix with excess substrate to measure the remaining activity. It should be noted that the concentration of substrate used for the assay should be saturating, because the aim is to measure the remaining active enzyme which is proportional to Vmax. For the mechanism-based inactivation of MAO by its irreversible inhibitors, the latter method is analysed according to Kitz and Wilson (1962), as shown in Fig. 6, to give KI and kinact, where KI is similar in meaning to the KM for the oxidation of the inhibitor, but has a contribution from an additional rate constant (k3 in Fig. 6). The kinact is a measure of the overall rate combining the rate of catalysis by MAO and the rate of the chemical modification step (see Fig. 6). A mechanism-based inhibitor is oxidized by MAO to produce a product that can dissociate like any other product or react with a group on the enzyme to form an adduct. The partition ratio will depend on the reactivity of the product with its target group. If the product is relatively stable or not correctly oriented for the chemical reaction with the enzyme group, then dissociation occurs, and after the flavin is re-oxidized, a new catalytic cycle can begin.
The ratio of product released to adduct formed, the partition ratio, can be calculated from the amount of product formed at infinite time relative to the amount of enzyme or from the rate constant for product formation divided by that for adduct formation (k3/k4 as shown in Fig. 6). The lower the partition ratio, the more efficient is the inactivator. For example, the partition ratio for the MAO A-selective inhibitor clorgyline is close to 1, but the multi-target derivative, ASS234, gives a partition ratio of 7 (Fowler et al. 1982; Juárez-Jiménez et al. 2014). Stopped-flow spectrophotometry was used to follow the rate of reduction of the flavin in the absence of oxygen (bleaching at 495 nm, a measure of amine oxidation) and the rate of adduct formation at 410 nm. For ASS234, the rate of reduction of the flavin was 0.049 s−1, but the rate of adduct formation was 0.0053 s−1 (A. Albreht and R.R. Ramsay, unpublished). Making the assumption that dissociation is faster than the rate of amine oxidation, these rates give a partition ratio of 9, in good agreement with the value from steady-state experiments (Juárez-Jiménez et al. 2014). In contrast to the high efficiency of the propargylamine inhibitors, the old drug phenelzine gives a partition ratio of about 40, calculated from the consumption of oxygen relative to the inactivation (Binda et al. 2008).
The determination of the partition ratio requires measurement of the MAO concentration. This is easily done for purified enzyme either by direct measurement of the FAD present or from the spectrum (Newton-Vinson et al. 2000). For membrane-bound and tissue samples, the MAO concentration can be determined by titration with an inactivator, for example by titration with clorgyline for MAO A. For MAO B, rasagiline inactivates in a single turnover (Hubalek et al. 2004), so could be used to determine the amount of MAO B. Instead of titration, modification of the propargylamine with a spin-labelled propargylamine allows the amount of MAO in membranes to be quantified by electron spin resonance (Upadhyay et al. 2008).
Propargylamines
Propargylamine analogues are a thoroughly studied class of irreversible MAO A and MAO B inhibitors. Their structure can be divided into four sections (Fig. 7) (Swett et al. 1963; Kalir et al. 1981; Weinreb et al. 2010). Section A allows a great deal of structural freedom, although an aromatic moiety increases inhibitor potency. In section B, a motif with more than two carbon atoms greatly increases affinity towards MAO A; otherwise, the inhibitor is MAO B selective. Section C and D are more stringent. Only –NH– and –N-alkyl– give reasonable inhibitor activity in section C. The alkynyl functional group in section D is essential for the covalent adduct formation and it is imperative for it to occupy β-position to the nitrogen.

Structural regions of propargylamine inhibitors
Mechanism of inactivation
The first report on the inhibition of MAO by propargylamine derivatives dates back to the late 1950s (Taylor et al. 1960), but neither the structure of irreversible propargylamine-inhibited MAO adduct to the FAD nor the mechanism of its formation is not fully understood (Edmondson et al. 2009; Pavlin et al. 2013). Some of the first plausible inhibition mechanisms of MAO by propargylamines were proposed by Maycock et al. (Maycock et al. 1976a, b). One possibility involves an enzymatic abstraction of the acetylenic proton from the inhibitor and its subsequent attack on oxidized FAD. The second proposed pathway proceeds through radical intermediates that then collapse to form the cyanine adduct. In the third mechanism, reduced FAD and oxidized inhibitor form the N5 covalent adduct with the enzyme. Later, Nakai et al. (Nakai et al. 1999) employed simplified truncated analogues of isoalloxazine and (−)-deprenyl in quantum chemical investigations of MAO inhibition mechanism. The calculations predicted the formation of two stable cyclic adducts. The O4,N5-adduct was the result of a one-step Diels–Alder cycloaddition reaction and the other was the C4a,N5-adduct, for which the inhibition pathway involved several acyclic intermediates and transition states. In a computational study, Borštnar et al. (Borstnar et al. 2011) proposed the deprotonated acetylenic moiety of propargylamine as the reactive species to attack the electrophilic N5 of the oxidized flavin. This results in an adduct bearing an alkynyl moiety to the α-position to the nitrogen. The proposed mechanism was concluded to present the most plausible inactivation mechanism for MAO, since it requires the least amount of activation-free energy for the reaction, although no argument was given on the free energy cost for formation of the carbanion.
A general mechanism for inactivation of MAO by propargylamines is yet to be agreed upon, but this comes as a no surprise, since even the structure of the covalent adduct differs in the published reports (Binda et al. 2002, 2004; Esteban et al. 2014; Pavlin et al. 2013; Borstnar et al. 2011; Maycock et al. 1976b; Nakai et al. 1999; Gartner et al. 1976; Hubalek et al. 2004; Kalgutkar et al. 2001; Edmondson et al. 2004). We favor a model in which the iminium (allenyl) cation product is trapped near the reduced flavin by favorable cation–π interactions in the “aromatic cage” and by interaction with the negatively charged pyrimidinedione ring. The electrophilic imine then forms a covalent adduct with the flavin via nucleophilic addition.
Selectivity
The tighter substrate cavity of MAO B results in increased steric hindrance and in distinctive amino acid residue interactions with small molecules. This makes MAO B more sensitive to the absolute configuration at chiral centres of enantiomeric substrates and inhibitors (Bocchinfuso and Robinson 1999). Specificity is also governed by hydrophobic and hydrophilic regions of the cavity. Site-directed mutagenesis studies showed that Ile335 in MAO A and Tyr326 in MAO B, both near the entrance of the cavity, are key amino acid residues in determining substrate and inhibitor specificities in human MAO (Ma et al. 2004; Milczek et al. 2011). By changing these two residues, the spatial and chemical architecture of the substrate cavity is altered and MAO B selectivity starts to mimic that of MAO A and vice versa. Despite the common reactive group, there are MAO A-selective drugs such as clorgyline and selective MAO B drugs such as selegiline (l-deprenyl) and rasagiline (Finberg and Rabey 2016), and some that inhibit both isozymes with comparable efficiency, such as pargyline and ladostigil. Thus, selectivity is a function of the whole cavity.
Inactivation kinetics
Various propargylamine inhibitors are not differentiated among themselves merely by their affinity towards MAO, but also by the rate at which they actually inactivate the enzyme. Therefore, the kinetics of inactivation of MAO A or MAO B by different propargylamines also gives insight into inhibitor selectivity (Fowler et al. 1982). Affinity of clorgyline towards MAO A is three orders of magnitude higher compared to the B-form and the rate of inactivation (kinact) for MAO A is 0.76 min−1 compared to 0.06 min−1 for MAO B which explains why this inhibitor is highly MAO A-selective. On the other hand, l-deprenyl shows only 40-fold difference in the affinity of the two forms, but the rates of inactivation for MAO A and MAO B are 0.14 and > 0.99 min−1, respectively, which significantly increases the inhibitor selectivity towards the B-form. Pargyline has only slightly higher affinity for MAO B, but the inactivation rates for both forms are very similar, which makes this inhibitor predominantly non-selective. The new generation of propargylamine multi-target inhibitors for the treatment of AD has moieties for the inhibition of MAO and cholinesterase in a single molecule. A representative of this class, ASS234, gives KI and kinact values of 0.053 µM and 0.133 min−1 for the membrane-bound MAO A, which makes it almost as potent as clorgyline (Esteban et al. 2014).
Cyclopropylamines
Cyclopropylamine class of MAO inhibitors has been around since the 1960s; however, tranylcypromine (trans-2-phenylcyclopropylamine) is the only cyclopropylamine type of inhibitor still used for the treatment of severe forms of depression. Many drugs were removed from the market due to a wide array of unwanted side effects. The inherent non-selective action of cyclopropylamines makes them also potent inhibitors of other important enzymes. For instance, they inhibit cytochrome P450 enzymes (Hanzlik and Tullman 1982; Khan et al. 2013; Salsali et al. 2004), copper amine oxidases (Shepard et al. 2003), prostacyclin synthase, and alcohol dehydrogenase (Talele 2016; Khan et al. 2013). More importantly, cyclopropylamines also show cross reactivity with the histone demethylases [lysine-specific demethylase (LSD) 1 and 2] that play a vital role in regulation of gene expression (Schmidt and McCafferty 2007; Binda et al. 2010; Niwa and Umehara 2017).
Mechanism of inactivation
Extensive effort to determine the mechanism of the irreversible inhibition of MAO by cyclopropylamines in the 1980s led to the most widely accepted pathway that proceeds by the formation of highly reactive radical species which in turn inactivate the enzyme. The inactivation mechanism (Scheme 2) assumes a single electron transfer from the inhibitor to the oxidized flavin in the first step, which yields a flavin radical and a cyclopropylamine radical cation (Silverman and Yamasaki 1984; Vazquez and Silverman 1985). Then, the cyclopropyl ring opens generating a highly reactive primary carbon-centred radical, detected by ESR studies (Qin and Williams 1987), which alkylates the enzyme, rendering it inactive. The covalent imine adduct is unstable and is, in most cases, hydrolysed to give an amine and a ketone. Although the mechanism via radical alkylation by cyclopropylamines is supported by many kinetic and chemical studies, the active site nucleophile that undergoes the reaction of covalent bond formation is still a subject of debate.

Proposed general mechanism of MAO inactivation by cyclopropylamines
The reduced flavin spectrum of N-(1-methylcyclopropy1)benzylamine-inactivated MAO B showed no spectral changes after denaturation with 6 M urea, which indicated covalent attachment of inhibitor to the flavin (Silverman and Yamasaki 1984). Using radioactively labelled 1-[phenyl-l4C]cyclopropylamine, the inhibition of MAO B was shown to proceed via two distinct and separate pathways (Silverman and Zieske 1985, 1986). A size-exclusion chromatographic separation of peptides resulting from a Pronase digest of the inactivated enzyme indicated an irreversible covalent bond formation between the N5 of the flavin and the inhibitor, since the fraction, representative of reduced FAD, also exhibited radioactivity. A second alkylation site was proposed as a cysteine amino acid residue Cys365 (Cys374 for MAO A) (Zhong and Silverman 1997). In contrast, this cysteine inactivation pathway was found to be reversible, although the inactivation rate was sevenfold faster compared to the flavin alkylation. With MAO A, 1-phenylcyclopropylamine binds exclusively to the cofactor. N-Cyclopropyl-α-methylbenzylamine inactivates both MAO A and MAO B through the same mechanism where, in both cases, only the active site cysteines are targets of alkylation and the covalent adducts with the flavin do not form (Silverman and Hiebert 1988). The same was observed when MAO B was inactivated by tranylcypromine (Paech et al. 1980; Silverman 1983), presumably forming a reversible covalent adduct with a sulfhydryl group at the active site, leaving the cofactor untouched. However, more than 20 years later an X-ray crystallographic study of MAO B inactivation by tranylcypromine revealed an irreversible covalent modification of FAD at the position C4a of the isoalloxazine ring at the 2.2 Å resolution (Binda et al. 2003). This finding not only opposed the involvement of a cysteine amino acid residue for this particular inactivator, but also proposed a different position for alkylation on the flavin, whereas only N5 position was previously ever considered as the site for covalent bond formation with some specific derivatives of this class of inhibitors. From the crystal structure, it was clear that the modified Cys365 in MAO B was on the surface of the protein, near the entrance to the active site (Binda et al. 2002). A mutation of the equivalent Cys374 in MAO A to alanine showed a somewhat reduced activity of the enzyme compared to the wild type due to an allosteric effect, but it did not prevent any of the three studied inhibitors (1-phenylcyclopropylamine, 2-phenylcyclopropylamine, and N-cyclo-α-methylbenzylamine) from inactivating the enzyme (Vintem et al. 2005). This indicates that for MAO A, this thiol is not modified. Therefore, it is hard to generalize and predict the site of alkylation in the inactivation of MAO by cyclopropylamines, since they are inhibitor and isoenzyme-dependent.
Selectivity
Cyclopropylamines are considered to be non-selective inhibitors that inactivate MAO A and MAO B with comparable efficiency. Moderate selectivity was obtained by additional functionalization of the known inhibitor 2-phenylcyclopropylamine (Hruschka et al. 2008). trans-2-Fluoro-2-(para-trifluoromethylphenyl)cyclopropylamine showed sevenfold higher affinity (measured as IC50 ratio) towards MAO A, whereas cis-2-fluoro-2-(para-fluorophenyl)cyclopropylamine proved 27-fold more effective towards MAO B. Another weakly MAO B selective analogue LY 54761 has a selectivity ratio of 15 (Murphy et al. 1978). On the other hand, LY 51641 (N-(2-(2-chlorophenoxy)ethyl)cyclopropylamine) could be one of the most selective MAO A inhibitors from the cyclopropylamine family with a selectivity ratio of 1990 (Mefford et al. 1985; Murphy et al. 1987). Its structure is reminiscent of a very selective propargylamine MAO A inhibitor—clorgyline. Like clorgyline, LY 51641 bears a long oxygen containing aliphatic side chain between the inactivating functional group and a phenyl ring, which contains a chloro substituent at ortho-position. These structural motifs seem to play a paramount role in MAO A selectivity. Higher enzyme selectivity ensures a more efficient control over individual MAO inhibition by cyclopropylamines, which also show a high cross reactivity with LSD1 (Binda et al. 2010). By enhancing selectivity, possible unwanted side effects of a particular drug could eventually be reduced. Selectivity for MAO over LSD1 has been achieved (Vianello et al. 2014).
Inactivation kinetics
The most representative and studied cyclopropylamine inhibitor—tranylcypromine—is used in the clinical practice as a racemate; however, the d-enantiomer was shown to be ten times more potent MAO inhibitor in vivo and several orders of magnitude more potent in vitro relative to its l-enantiomer (Fuentes et al. 1976; Paech et al. 1980; Reynolds et al. 1980). Tranylcypromine inactivation rates measured for MAO A and MAO B are 0.78 and 0.26 min−1, and Ki values are 7.7 and 3.8 µM, respectively (Malcomson et al. 2015). Many cyclopropylamine analogues have been prepared in search of a suitable lead in drug development with kinact and KI values in the range 0.01–4 min−1 and 0.07–1750 µM, respectively (Malcomson et al. 2015; Silverman and Hoffman 1981; Kalgutkar et al. 1995). Some compounds can show high inhibitory action such as cis-N-benzyl-2-methoxycyclopropylamine with a kinact/KI ratio of 440 and 1600 for MAO A and MAO B, respectively, but they generally exhibit a poor isozyme selectivity which is in the range of one order of magnitude.
Hydrazines
Iproniazid, a hydrazine derivative, was one of the first antidepressants ever marketed in late 1950s soon after the MAO inhibiting properties of hydrazines were recognized (Zeller and Barsky 1952; Zeller and Sarkar 1962). Later, to avoid its high toxicity, iproniazid was replaced by another hydrazine analogue—phenelzine (phenylethylhydrazine). Hydrazines are generally recognized as non-selective MAO A and MAO B inhibitors. Moreover, they also inhibit other groups of enzymes: metalloenzymes such as ribonucleotide reductase (Mure et al. 2005), transferases such as gamma-aminobutyric acid transaminase (Baker et al. 1991) and other amine oxidases such as lysine-specific histone demethylase 1 (LSD1) (Prusevich et al. 2014; Culhane et al. 2010) and primary amine oxidase (previously known as semicarbazide-sensitive amine oxidase) (Lizcano et al. 1996). Consequently, acute and chronic administration of phenelzine to mice shows not only elevated levels of MAO substrates (neurotransmitters serotonin, norepinephrine, and dopamine) and a decrease in their metabolites (3,4-dihydroxyphenylacetic acid and 5-hydroxyindoleacetic acid) but also an increase in amino acids such as alanine and γ-aminobutyric acid (Griebel et al. 1998; Parent et al. 2002). The levels of amino acids in mice brain start to deplete after 48 h, whereas the increased concentration of neurotransmitters is maintained even after 2 weeks. This shows that not only does phenelzine inhibit different classes of enzymes but also that the MAO irreversible inhibition effect is more long-lasting.
Mechanism of inactivation
As with other irreversible inhibitors, hydrazines are initially oxidized by MAO to an active form, in this case diazene intermediates. Through the loss of N2 and a hydrogen atom, diazenes are then converted into highly reactive radical species that covalently bind to the isoalloxazine moiety of the FAD, inactivating MAO (Kalgutkar et al. 2001). Under anaerobic conditions the inactivation of MAO is stopped at the diazene stage and the formation of the covalent adduct does not occur (Binda et al. 2008). It is assumed that molecular O2 is crucial for the formation of the alkylating (arylating) radical (Huang and Kosower 1967; Kosower 1971; Binda et al. 2008). A similar mechanism was also proposed for a hydrazine-type LSD1 inhibitor where diazene is oxidized to a highly reactive primary diazonium species that inactivates the enzyme activity (Culhane et al. 2010). On the other hand, diazenes were shown to react with free FAD as well as with enzyme-bound flavin in the absence of O2 (Nagy et al. 1979), which indicated that the re-oxidation of FADH2 to FAD was paramount for final covalent adduct formation. Although the exact inhibition pathway is still debated, the requirement of O2 for MAO inactivation clearly sets hydrazine inhibitors apart from propargylamine and cyclopropylamine inhibitors where activity of MAO is quenched even under anaerobic conditions.
By studying reactions of model flavin systems with phenylhydrazine and benzylhydrazine, it was shown that the covalent bond formation occurs at the C4a position of the isoalloxazine ring (Nagy et al. 1979; Kim et al. 1995). However, a more recent X-ray crystallographic study proves that benzylhydrazine and phenyethylhydrazine form N5 covalent adducts when incubated with MAO B (Binda et al. 2008). Trace amounts of dialkylated enzyme were also detected by mass spectrometry but only one out of the two inhibitor molecules was proposed to associate with FAD. Thus, catalytic activity of the enzyme could additionally be quenched by an alternative pathway in which substrate cannot enter the active site as a result of alkylation of a nearby amino acid residue (Binda et al. 2008; Cesura and Pletscher 1992). The same phenomenon was also demonstrated for benzylhydrazine inactivation of dopamine β-hydroxylase (Fitzpatrick and Villafranca 1986).
Apart from being good inhibitors, hydrazine derivatives with a methylene group in the alpha position to the hydrazine functional group also act as good conventional MAO substrates which are eventually converted into aldehyde products via hydrazone intermediates (Tipton 1971; Tipton and Spires 1971; Patek and Hellerman 1974; Yu and Tipton 1989). In these cases, the inhibitor can undergo a substantial number of catalytic turnovers (leading to high partition ratios) before inhibition of MAO occurs. On inactivation of MAO by phenylethylhydrazine (phenelzine), 35–40 mol of molecular O2 per one mol of enzyme were required, whereas an equivalent of 7 mol of O2 was consumed in the inactivation by phenylhydrazine (Binda et al. 2008). The difference in molecular O2 consumption between phenylethylhydrazine and phenylhydrazine is due to a higher catalytic turnover for phenylethylhydrazine where O2 is required for the re-oxidation of FADH2 to complete the regular catalytic cycle after product leaves the active site. Therefore, variations in molecular O2 consumption between compounds can be considered as a relative indication of how effectively can inhibitors be transformed into regular oxidation products (aldehydes) and should be proportional to the partition ratio.
Selectivity and inactivation kinetics
Low selectivity of hydrazine-type irreversible inhibitors results from their similar affinities and inactivation rates for MAO A and MAO B. Arylhydrazines phenylethylhydrazine, benzylhydrazine, and phenylhydrazine all have Ki values in the micromolar range (15–205 µM) with the one exception of benzylhydrazine which has a very low affinity for MAO A (Ki = 2 mM) (Binda et al. 2008). Conversely, this inhibitor shows an inactivation rate of 3.1 min−1, a rate faster than all other inhibitor—enzyme pairs studied (kinact = 0.1–1.3 min−1). Thus, the potency of these inhibitors is comparable and non-selective.
The efficiency of hydrazine inhibitors can be potentiated by enhancement of the C–H bond cleavage, which is in the alpha position to the hydrazine moiety and leads to an oxidation product (Yu and Tipton 1989). Alkylation at this position, which presumably hinders hydrazone formation, makes pheniprazine (1-methyl-2-phenylethylhydrazine) a tenfold more potent inhibitor of bovine MAO compared to its non-methylated analogue—phenelzine (Patek and Hellerman 1974). Even further enhancement of pheniprazine efficacy can be induced by cyanide which acts as an enhancer of binding (Ramadan et al. 2007). With MAO A and MAO B from rat and ox liver, potassium cyanide decreased the Ki of pheniprazine 5–10-fold, but there were no observable differences in inactivation rates for either of the isoenzymes studied.
Overall, non-selectivity, low target incorporation, and high potential for toxicity via non-specific interactions make the hydrazine moiety less suitable than propargylamine or cyclopropylamine groups for use in multi-target drug design.
MAO inhibition in multi-target compounds
Many irreversible inhibitors of MAO A and MAO B have been developed into drugs for the treatment of age-related neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s disease and neuropsychiatric disorders such as schizophrenia, depression, anxiety, and aggression (Yu 1994; Youdim et al. 2006; Garcia-Miralles et al. 2016). Understanding the details of the chemical mechanism of MAO inhibition enables the development of new generation drugs by rational design and functionalization of the inhibitor molecule. From the preceding sections, it is clear that high binding affinity is key to selectivity through specific interactions in the active sites. For irreversible inhibitors, the high rate of the chemical step also depends on the orientation of the substrate to the N5 of the flavin and on retention of the reactive product to achieve that step. Electron donating and electron-withdrawing functional groups can strongly influence the behaviour of electron density in a molecule through hyperconjugation, inductive, and resonance effects, which can, in turn, reflect in the inhibitor potency. Large or bulky substituents can represent steric hindrance that prevents the inhibitor molecule acquiring the optimum orientation for successful compound oxidation. Nevertheless, carefully chosen and positioned substituents within the inactivating molecule can affect its binding affinity towards MAO A or MAO B which, alongside the rate of inactivation, governs enzyme selectivity. However, as evident with the reversible inhibitors, ensuring a high selectivity of an irreversible inhibitor for either of the two isozymes through design is anything but trivial.
The propargyl moiety has been incorporated into several new compounds designed to tackle the complex pathology of neurodegeneration. Many compounds that target not only MAO (selective for one isoform) but also reversibly inhibit the cholinesterases have been designed such as tacrine–coumarin hybrids (Xie et al. 2015) donepezil-propargylamine hybrids (PF1901N and ASS234) (Bolea et al. 2011; Marco-Contelles et al. 2016), or ladostigil derivatives, a propargyl-aminoindan–carbamate combination (Sterling et al. 2002). Antioxidant capacity or neuroprotective properties can also be added. In one recent novel development, binding to the H3 receptor was successfully achieved. The patented molecule, contilisant, an indole derivative, has inhibitory activity towards acetyl/butyrylcholinesterases and monoamine oxidases A/B as well as the histamine H3 receptor (H3R). Contilisant was also found to have antioxidative properties, to penetrate the blood–brain barrier, and to mitigate lipopolysaccharide-induced cognitive deficits in mice (Bautista-Aguilera et al. 2017).
Conclusion
In conjunction with crystal structures as starting points, computational modelling has advanced to the point of experimenting to improve the trajectory and rate of binding, to prolong binding, and even to explore individual parts of the catalytic cycle, opening a new era in enzymology. Experimentally, good kinetic analysis is still the key to understanding the effect of inhibitor binding to an enzyme, shortening the list of optimised lead compounds to hand to pharmacologists to minimise expensive failure during translation to the clinic.
References
Abad E, Zenn RK, Kaestner J (2013) Reaction mechanism of monoamine oxidase from QM/MM calculations. J Phys Chem B 117(46):14238–14246. https://doi.org/10.1021/jp4061522
Akyuz MA, Erdem SS (2013) Computational modeling of the direct hydride transfer mechanism for the MAO catalyzed oxidation of phenethylamine and benzylamine: ONIOM (QM/QM) calculations. J Neural Transm 120:937–945. https://doi.org/10.1007/s00702-013-1027-8
Akyuz MA, Erdem SS, Edmondson DE (2007) The aromatic cage in the active site of monoamine oxidase B: effect on the structural and electronic properties of bound benzylamine and p-nitrobenzylamine. J Neural Transm 114(6):693–698
Apostolov R, Yonezawa Y, Standley DM, Kikugawa G, Takano Y, Nakamura H (2009) Membrane attachment facilitates ligand access to the active site in monoamine oxidase A. Biochemistry 48(25):5864–5873
Bach AWJ, Lan NC, Johnson DL, Abell CW, Bembenek ME, Kwan SW, Seeburg PH, Shih JC (1988) cDNA cloning of human-liver monoamine oxidase-A and oxidase-B—molecular basis of differences in enzymatic properties. PNAS USA 85(13):4934–4938
Baker GB, Wong JT, Yeung JM, Coutts RT (1991) Effects of the antidepressant phenelzine on brain levels of gamma-aminobutyric acid (GABA). J Affect Disord 21(3):207–211
Basile L, Pappalardo M, Guccione S, Milardi D, Ramsay RR (2014) Computational comparison of imidazoline association with the 12 binding site in human monoamine oxidases. J Chem Inf Model 54(4):1200–1207
Bautista-Aguilera OM et al (2014a) Design, synthesis, pharmacological evaluation, QSAR analysis, molecular modeling and ADMET of novel donepezil-indolyl hybrids as multipotent cholinesterase/monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. Eur J Med Chem 75:82–95. https://doi.org/10.1016/j.ejmech.2013.12.028
Bautista-Aguilera OM et al (2014b) N-Methyl-N-((1-methyl-5-(3-(1-(2-methylbenzyl)piperidin-4yl)propoxy)-1H- indol-2-yl)methyl)prop-2-yn-1-amine, a new cholinesterase and monoamine oxidase dual inhibitor. J Med Chem 57(24):10455–10463. https://doi.org/10.1021/jm501501a
Bautista-Aguilera O et al (2017) Multitarget-directed ligands combining cholinesterase and monoamine oxidase Inhibition with H3R antagonism for neurodegenerative diseases. Angew Chem 56:12765–12769. https://doi.org/10.1002/anie.201706072
Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A (2002) Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol 9(1):22–26
Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A (2003) Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc Natl Acad Sci USA 100(17):9750–9755
Binda C, Hubalek F, Li M, Herzig Y, Sterling J, Edmondson DE, Mattevi A (2004) Crystal structures of monoamine oxidase B in complex with four inhibitors of the N-propargylaminoindan class. J Med Chem 47(7):1767–1774
Binda C, Hubalek F, Li M, Castagnoli N, Edmondson DE, Mattevi A (2006) Structure of the human mitochondrial monoamine oxidase B—new chemical implications for neuroprotectant drug design. Neurology 67(7):S5–S7
Binda C, Wang J, Pisani L, Caccia C, Carotti A, Salvati P, Edmondson DE, Mattevi A (2007) Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J Med Chem 50(23):5848–5852
Binda C, Wang J, Li M, Hubalek F, Mattevi A, Edmondson DE (2008) Structural and mechanistic studies of arylalkylhydrazine inhibition of human monoamine oxidases A and B. Biochemistry 47(20):5616–5625
Binda C et al (2010) Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J Am Chem Soc 132(19):6827–6833
Binda C, Mattevi A, Edmondson DE (2011) Structural properties of human monoamine oxidases A AND B. In: Youdim MBH, Riederer P (eds) Monoamine oxidases and their inhibitors. Int Rev Neurobiol 100:1–11. https://doi.org/10.1016/b978-0-12-386467-3.00001-7
Binda C, Aldeco M, Geldenhuys WJ, Tortorici M, Mattevi A, Edmondson DE (2012) Molecular insights into human monoamine oxidase B inhibition by the glitazone antidiabetes drugs. Med Chem Lett 3(1):39–42
Bocchinfuso R, Robinson JB (1999) The stereoselectivity of inhibition of rat liver mitochondrial MAO-A and MAO-B by the enantiomers of 2-phenylpropylamine and their derivatives. Eur J Med Chem 34(4):293–300. https://doi.org/10.1016/S0223-5234(99)80080-4
Bolea I, Juarez-Jimenez J, de los Rios C, Chioua M, Pouplana R, Luque FJ, Unzeta M, Marco-Contelles J, Samadi A (2011) Synthesis, biological evaluation, and molecular modeling of donepezil and N-(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl-N-methylprop-2-yn-1-ami ne hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. J Med Chem 54(24):8251–8270. https://doi.org/10.1021/jm200853t
Bonnet U (2002) Moclobemide: evolution, pharmacodynamic, and pharmacokinetic properties. CNS Drug REV 8(3):283–308
Borroni E et al (2017) Sembragiline: a novel, selective monoamine oxidase type B inhibitor for the treatment of Alzheimer’s disease. J Pharmacol Exp Ther 362(3):413–423. https://doi.org/10.1124/jpet.117.241653
Borstnar R, Repic M, Krzan M, Mavri J, Vianello R (2011) Irreversible inhibition of monoamine oxidase B by the antiparkinsonian medicines rasagiline and selegiline: a computational study. Eur J Org Chem 32:6419–6433. https://doi.org/10.1002/ejoc.201100873
Bortolato M, Shih JC (2011) Behavioral outcomes of monoamine oxidase deficiency: Preclinical and clinical evidence. In: Youdim MBH, Riederer P (eds) Monoamine oxidases and their inhibitors. Int Rev Neurobiol 100:13–42. https://doi.org/10.1016/b978-0-12-386467-3.00002-9
Caccia C, Maj R, Calabresi M, Maestroni S, Faravelli L, Curatolo L, Salvati P, Fariello RG (2006) Safinamide—from molecular targets to a new anti-Parkinson drug. Neurology 67(7):S18–S23
Cakir K, Erdem SS, Atalay VE (2016) ONIOM calculations on serotonin degradation by monoamine oxidase B: insight into the oxidation mechanism and covalent reversible inhibition. Org Biomol Chem 14(39):9239–9252. https://doi.org/10.1039/c6ob01175f
Carradori S, Silvestri R (2015) New frontiers in selective human MAO-B inhibitors. J Med Chem 58(17):6717–6732. https://doi.org/10.1021/jm501690r
Cesura AM, Pletscher A (1992) The new generation of monoamine oxidase inhibitors. Prog Drug Res (Fortschritte der Arzneimittelforschung Progres des recherches pharmaceutiques) 38:171–297
Chajkowski-Scarry S, Rimoldi JM (2014) Monoamine oxidase A and B substrates: probing the pathway for drug development. Future Med Chem 6(6):697–717. https://doi.org/10.4155/fmc.14.23
Chiuccariello L et al (2016) Monoamine oxidase-A occupancy by moclobemide and phenelzine: implications for the development of monoamine oxidase inhibitors. Int J Neuropsychopharmacol. https://doi.org/10.1093/ijnp/pyv078
Copeland RA, Lombardo D, Giannaras J, Decicco CP (1995) Estimating K-i values for tight-binding inhibitors from dose–response plots. Bioorg Med Chem Lett 5(17):1947–1952. https://doi.org/10.1016/0960-894x(95)00330-v
Cruz F, Edmondson DE (2007) Kinetic properties of recombinant MAO-A on incorporation into phospholipid nanodisks. J Neural Transm 114(6):699–702
Culhane JC, Wang D, Yen PM, Cole PA (2010) Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J Am Chem Soc 132(9):3164–3176. https://doi.org/10.1021/ja909996p
Da Prada M et al (1990) From moclobemide to Ro 19-6327 and Ro 41-1049: the development of a new class of reversible, selective MAO-A and MAO-B inhibitors. J Neural Transm 29:279–292
De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A (2005) Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. PNAS USA 102(36):12684–12689
Delport A, Harvey BH, Petzer A, Petzer JP (2017) The monoamine oxidase inhibition properties of selected structural analogues of methylene blue. Toxicol Appl Pharmacol 325:1–8. https://doi.org/10.1016/j.taap.2017.03.026
Desideri N, Proietti Monaco L, Fioravanti R, Biava M, Yanez M, Alcaro S, Ortuso F (2016) (E)-3-Heteroarylidenechroman-4-ones as potent and selective monoamine oxidase-B inhibitors. Eur J Med Chem 117:292–300. https://doi.org/10.1016/j.ejmech.2016.03.081
Dorris RL (1982) A simple method for screening monoamine oxidase (MAO) inhibitory drugs for type preference. J Pharmacol Methods 7(2):133–137
Dunn RV, Marshall KR, Munro AW, Scrutton NS (2008) The pH dependence of kinetic isotope effects in monoamine oxidase A indicates stabilization of the neutral amine in the enzyme-substrate complex. FEBS J 275(15):3850–3858. https://doi.org/10.1111/j.1742-4658.2008.06532.x
Dunn RV, Munro AW, Turner NJ, Rigby SEJ, Scrutton NS (2010) Tyrosyl radical formation and propagation in flavin dependent monoamine oxidases. ChemBioChem 11(9):1228–1231
Edmondson DE, Newton-Vinson P (2001) The covalent FAD of monoamine oxidase: structural and functional role and mechanism of the flavinylation reaction. Antioxid Redox Signal 3(5):789–806
Edmondson DE, Mattevi A, Binda C, Li M, Hubalek F (2004) Structure and mechanism of monoamine oxidase. Curr Med Chem 11(15):1983–1993
Edmondson DE, Binda C, Mattevi A (2007) Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch Biochem Biophys 464(2):269–276
Edmondson DE, Binda C, Wang J, Upadhyay AK, Mattevi A (2009) Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 48(20):4220–4230
Efange SM, Boudreau RJ (1991) Molecular determinants in the bioactivation of the dopaminergic neurotoxin N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). J Comput Aided Mol Des 5(5):405–417
Efange SMN, Michelson RH, Tan AK, Krueger MJ, Singer TP (1993) Molecular-size and flexibility as determinants of selectivity in the oxidation of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine analogs of monoamine oxidase-A and oxidase-B. J Med Chem 36(9):1278–1283
Esteban G, Allan J, Samadi A, Mattevi A, Unzeta M, Marco-Contelles J, Binda C, Ramsay RR (2014) Kinetic and structural analysis of the irreversible inhibition of human monoamine oxidases by ASS234, a multi-target compound designed for use in Alzheimer’s disease. Biochem Biophys Acta 1844(6):1104–1110
Finberg J, Rabey J (2016) Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front Pharmacol 7:340. https://doi.org/10.3389/fphar.2016.00340
Fisar Z (2016) Drugs related to monoamine oxidase activity. Prog Neuro Psychopharmacol Biol Psychiatry 69:112–124. https://doi.org/10.1016/j.pnpbp.2016.02.012
Fitzpatrick PF (2010) Oxidation of amines by flavoproteins. Arch Biochem Biophys 493(1):13–25
Fitzpatrick PF, Villafranca JJ (1986) The mechanism of inactivation of dopamine beta-hydroxylase by hydrazines. J Biol Chem 261(10):4510–4518
Fowler CJ, Oreland L (1980) The nature of the substrate-selective interaction between rat liver mitochondrial monoamine-oxidase and oxygen. Biochem Pharmacol 29(16):2225–2233
Fowler CJ, Mantle TJ, Tipton KF (1982) The nature of the inhibition of rat-liver monoamine-oxidase type-A and type-B by the acetylenic inhibitors clorgyline, l-deprenyl and pargyline. Biochem Pharmacol 31(22):3555–3561
Fowler JS, Logan J, Shumay E, Alia-Klein N, Wang G-J, Volkow ND (2015) Monoamine oxidase: radiotracer chemistry and human studies. J Label Compd Radiopharm 58(3):51–64. https://doi.org/10.1002/jlcr.3247
Fraaije MW, Mattevi A (2000) Flavoenzymes: diverse catalysts with recurrent features. Trends Biochem Sci 25(3):126–132
Fuentes JA, Oleshansky MA, Neff NH (1976) Comparison of apparent antidepressant activity of (−) and (+) tranylcypromine in an animal-model. Biochem Pharmacol 25(7):801–804. https://doi.org/10.1016/0006-2952(76)90150-7
Garcia-Miralles M, Ooi J, Ferrari Bardile C, Tan LJ, George M, Drum CL, Lin RY, Hayden MR, Pouladi MA (2016) Treatment with the MAO-A inhibitor clorgyline elevates monoamine neurotransmitter levels and improves affective phenotypes in a mouse model of Huntington disease. Exp Neurol 278:4–10. https://doi.org/10.1016/j.expneurol.2016.01.019
Gartner B, Hemmerich P, Zeller EA (1976) Structure of flavin adducts with acetylenic substrates—chemistry of monoamine-oxidase and lactate oxidase Inhibition. Eur J Biochem 63(1):211–221
Geha RM, Chen K, Shih JC (2000) Phe(208) and Ile(199) in human monoamine oxidase A and B do not determine substrate and inhibitor specificities as in rat. J Neurochem 75(3):1304–1309
Geha RM, Rebrin I, Chen K, Shih JC (2001) Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. J Biol Chem 276(13):9877–9882
Griebel G, Curet O, Perrault G, Sanger DJ (1998) Behavioral effects of phenelzine in an experimental model for screening anxiolytic and anti-panic drugs: correlation with changes in monoamine-oxidase activity and monoamine levels. Neuropharmacol 37(7):927–935. https://doi.org/10.1016/S0028-3908(98)00077-X
Guzior N, Wieckowska A, Panek D, Malawska B (2015) Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr Med Chem 22(3):373–404
Hanzlik RP, Tullman RH (1982) Suicidal Inactivation of cytochrome-P-450 by cyclopropylamines—evidence for cation-radical intermediates. J Am Chem Soc 104(7):2048–2050
Heal DJ, Smith SL, Gosden J, Nutt DJ (2013) Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol (Oxford, England) 27(6):479–496. https://doi.org/10.1177/0269881113482532
Holt A, Palcic MM (2006) A peroxidase-coupled continuous absorbance plate-reader assay for flavin monoamine oxidases, copper-containing amine oxidases and related enzymes. Nat Protoc 1:2498–2505
Hroch L et al (2017) Synthesis and evaluation of frentizole-based indolyl thiourea analogues as MAO/ABAD inhibitors for Alzheimer’s disease treatment. Bioorg Med Chem 25(3):1143–1152. https://doi.org/10.1016/j.bmc.2016.12.029
Hruschka S, Rosen TC, Yoshida S, Kirk KL, Froehlich R, Wibbeling B, Haufe G (2008) Fluorinated phenylcyclopropylamines. Part 5: effects of electron-withdrawing or -donating aryl substituents on the inhibition of monoamine oxidases A and B by 2-aryl-2-fluoro-cyclopropylamines. Bioorg Med Chem 16(15):7148–7166
Huang P-KC, Kosower EM (1967) Properties of phenyldiimide. J Am Chem Soc 89(15):3910–3911. https://doi.org/10.1021/ja00991a047
Hubalek F, Binda C, Li M, Herzig Y, Sterling J, Youdim MB, Mattevi A, Edmondson DE (2004) Inactivation of purified human recombinant monoamine oxidases A and B by rasagiline and its analogues. J Med Chem 47(7):1760–1766. https://doi.org/10.1021/jm0310885
Hubalek F, Binda C, Khalil A, Li M, Mattevi A, Castagnoli N, Edmondson DE (2005) Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J Biol Chem 280(16):15761–15766
Husain M, Edmondson DE, Singer TP (1982) Kinetic-studies on the catalytic mechanism of liver monoamine oxidase. Biochemistry 21(3):595–600
Hutson PH, Tarazi FI, Madhoo M, Slawecki C, Patkar AA (2014) Preclinical pharmacology of amphetamine: implications for the treatment of neuropsychiatric disorders. Pharmacol Ther 143(3):253–264. https://doi.org/10.1016/j.pharmthera.2014.03.005
Hynson RMG, Wouters J, Ramsay RR (2003) Monoamine oxidase A inhibitory potency and flavin perturbation are influenced by different aspects of pirlindole inhibitor structure. Biochem Pharmacol 65(11):1867–1874
Hynson R, Kelly S, Price N, Ramsay R (2004) Conformational changes in monoamine oxidase A in response to ligand binding or reduction. Biochem Biophys Acta 1672(1):60–66. https://doi.org/10.1016/j.bbagen.2004.02.011
Jones T, Balsa D, Unzeta M, Ramsay R (2007) Variations in activity and inhibition with pH: the protonated amine is the substrate for monoamine oxidase, but uncharged inhibitors bind better. J Neural Transm 114(6):707–712. https://doi.org/10.1007/s00702-007-0675-y
Jonsson T, Edmondson DE, Klinman JP (1994) Hydrogen tunneling in the flavoenzyme monoamine-oxidase-B. Biochemistry 33(49):14871–14878
Joubert J, Foka GB, Repsold BP, Oliver DW, Kapp E, Malan SF (2017) Synthesis and evaluation of 7-substituted coumarin derivatives as multimodal monoamine oxidase-B and cholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur J Med Chem 125:853–864. https://doi.org/10.1016/j.ejmech.2016.09.041
Juarez-Jimenez J, Mendes E, Galdeano C, Martins C, Silva D, Marco-Contelles J, Carreiras M, Luque F, Ramsay R (2014) Exploring the structural basis of the selective inhibition of monoamine oxidase A by dicarbonitrile aminoheterocycles: role of Asn181 and Ile335 validated by spectroscopic and computational studies. Biochem Biophys Acta 1844(2):389–397. https://doi.org/10.1016/j.bbapap.2013.11.003
Juárez-Jiménez J, Mendes E, Galdeano C, Martins C, Silva DB, Marco-Contelles J, Carreiras MdC, Luque FJ, Ramsay RR (2014) Exploring the structural basis of the selective inhibition of monoamine oxidase A by dicarbonitrile aminoheterocycles: role of Asn181 and Ile335 validated by spectroscopic and computational studies. Biochim Biophys Acta (Proteins Proteom) 1844:389–397. https://doi.org/10.1016/j.bbapap.2014.03.006
Kacar B, Edmondson D (2006) Studies on the role of lysine-296 in human mitochondrial monoamine oxidase B catalysis. FASEB J 20(4):A478
Kalgutkar AS, Castagnoli N, Testa B (1995) Selective inhibitors of monoamine-oxidase (MAO-A and MAO-B) as probes of its catalytic site and mechanism. Med Res Rev 15(4):325–388
Kalgutkar AS, Dalvie DK, Castagnoli N, Taylor TJ (2001) Interactions of nitrogen-containing xenobiotics with monoamine oxidase (MAO) isozymes A and B: sAR studies on MAO substrates and inhibitors. Chem Res Toxicol 14(9):1139–1162
Kalir A, Sabbagh A, Youdim MB (1981) Selective acetylenic ‘suicide’ and reversible inhibitors of monoamine oxidase types A and B. Br J Pharmacol 73(1):55–64
Kay CWM, El Mkami H, Molla G, Pollegioni L, Ramsay RR (2007) Characterization of the covalently bound anionic flavin radical in monoamine oxidase a by electron paramagnetic resonance. J Am Chem Soc 129(51):16091–16097
Kearney EB, Salach JI, Walker WH, Seng RL, Kenney W, Zeszotek E, Singer TP (1971) Covalently-bound flavin of hepatic monoamine oxidase.1. Isolation and sequence of a flavin peptide and evidence for binding at 8 alpha-position. Eur J Biochem 24(2):321–327
Khan MNA, Suzuki T, Miyata N (2013) An overview of phenylcyclopropylamine derivatives: biochemical and biological significance and recent developments. Med Res Rev 33(4):873–910
Kim J-M, Hoegy SE, Mariano PS (1995) Flavin chemical models for monoamine oxidase inactivation by cyclopropylamines, alpha-silylamines, and hydrazines. J Am Chem Soc 117(1):100–105. https://doi.org/10.1021/ja00106a012
Kim H, Sablin S, Ramsay R (1997) Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch Biochem Biophys 337(1):137–142. https://doi.org/10.1006/abbi.1996.9771
Kim D, Baik SH, Kang S, Cho SW, Bae J, Cha MY, Sailor MJ, Mook-Jung I, Ahn KH (2016) Close correlation of monoamine oxidase activity with progress of alzheimer’s disease in mice, observed by in vivo two-photon imaging. ACS Cent Sci 2(12):967–975. https://doi.org/10.1021/acscentsci.6b00309
Kitz R, Wilson IB (1962) Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem 237:3245–3249
Kosower EM (1971) Monosubstituted diazenes (diimides). Surprising intermediates. Acc Chem Res 4(6):193–198. https://doi.org/10.1021/ar50042a001
Kupershmidt L, Amit T, Bar-Am O, Youdim MBH, Weinreb O (2012) The novel multi-target iron chelating-radical scavenging compound M30 possesses beneficial effects on major hallmarks of Alzheimer’s disease. Antioxid Redox Signal 17(6):860–877. https://doi.org/10.1089/ars.2011.4279
Leonardi ET, Azmitia EC (1994) MDMA (ecstasy) inhibition of MAO type A and type B: comparisons with fenfluramine and fluoxetine (Prozac). Neuropsychopharmacol 10(4):231–238. https://doi.org/10.1038/npp.1994.26
Li M, Binda C, Mattevi A, Edmondson DE (2006) Functional role of the “aromatic cage” in human monoamine oxidase B: structures and catalytic properties of Tyr435 mutant proteins. Biochemistry 45(15):4775–4784
Li XF, Yu JJ, Zhu Q, Qian LH, Li L, Zheng YG, Yao SQ (2014a) Visualization of monoamine oxidases in living cells using “Turn-ON” fluorescence resonance energy transfer probes. Analyst 139(23):6092–6095. https://doi.org/10.1039/c4an01195c
Li XF, Zhang HT, Xie YS, Hu Y, Sun HY, Zhu Q (2014b) Fluorescent probes for detecting monoamine oxidase activity and cell imaging. Org Biomol Chem 12(13):2033–2036. https://doi.org/10.1039/c3ob42326c
Li LL, Li K, Liu YH, Xu HR, Yu XQ (2016) Red emission fluorescent probes for visualization of monoamine oxidase in living cells. Sci Rep. https://doi.org/10.1038/srep31217
Lizcano JM, Fernandez de Arriba A, Tipton KF, Unzeta M (1996) Inhibition of bovine lung semicarbazide-sensitive amine oxidase (SSAO) by some hydrazine derivatives. Biochem Pharmacol 52(2):187–195
Ma JC, Yoshimura M, Yamashita E, Nakagawa A, Ito A, Tsukihara T (2004) Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J Mol Biol 338(1):103–114
MacMillar S, Edmondson DE, Matsson O (2011) Nitrogen kinetic isotope effects for the monoamine oxidase B-catalyzed oxidation of benzylamine and (1,1-(2)H(2))benzylamine: nitrogen rehybridization and CH bond cleavage are not concerted. J Am Chem Soc 133(32):12319–12321
Magyar K, Palfi M, Jenei V, Szoko E (2006) Deprenyl: from chemical synthesis to neuroprotection. J Neural Transm Suppl 71:143–156
Malcomson T, Yelekci K, Borrello MT, Ganesan A, Semina E, De Kimpe N, Mangelinckx S, Ramsay RR (2015) cis-Cyclopropylamines as mechanism-based inhibitors of monoamine oxidases. FEBS J 282(16):3190–3198. https://doi.org/10.1111/febs.13260
Mangiatordi GF et al (2017) A rational approach to elucidate human monoamine oxidase molecular selectivity. Eur J Pharm Sci 101:90–99. https://doi.org/10.1016/j.ejps.2017.02.008
Mantle TJ, Tipton KF, Garrett NJ (1976) Inhibition of monoamine oxidase by amphetamine and related compounds. Biochem Pharmacol 25(18):2073–2077
Marco-Contelles J, Unzeta M, Bolea I, Esteban G, Ramsay RR, Romero A, Martnez-Murillo R, Carreiras MC, Ismaili L (2016) ASS234, as a new multi-target directed propargylamine for Alzheimer’s disease therapy. Front Neurosci. https://doi.org/10.3389/fnins.2016.00294
Matsumoto T, Suzuki O, Furuta T, Asai M, Kurokawa Y, Nimura Y, Katsumata Y, Takahashi I (1985) A sensitive fluorometric assay for serum monoamine-oxidase with kynuramine as substrate. Clin Biochem 18(2):126–129
Maycock AL, Abeles RH, Salach JI, Singer TP (1976a) The action of acetylenic inhibitors on mitochondrial monoamine oxidase: structure of the flavin site in the inhibited enzyme. In: Bernheim MLC (ed) Monoamine oxidase and its inhibition, Ciba foundation symposium, vol 439. Wiley, pp 33–47. https://doi.org/10.1002/9780470720219.ch3
Maycock AL, Abeles RH, Salach JI, Singer TP (1976b) Structure of covalent adduct formed by interaction of 3-dimethylamino-1-propyne and flavin of mitochondrial amine oxidase. Biochemistry 15(1):114–125
Mazouz F, Gueddari S, Burstein C, Mansuy D, Milcent R (1993) 5- 4-(Benzyloxy)phenyl -1,3,4-oxadiazol-2(3h)-one derivatives and related analogs—new reversible, highly potent, and selective monoamine-oxidase type-B inhibitors. J Med Chem 36(9):1157–1167
McCoubrey A (1957) Inhibition of monoamine oxidase by 1-phenylethylamines. Biochem Pharmacol 2(4):264–269. https://doi.org/10.1016/0006-2952(59)90039-5
McDonald AG, Tipton KF (2012) Enzymes: irreversible inhibition. In: eLS. John Wiley & Sons Ltd, Chichester. https://doi.org/10.1002/9780470015902.a0000601.pub2
McDonald GR, Olivieri A, Ramsay RR, Holt A (2010) On the formation and nature of the imidazoline I(2) binding site on human monoamine oxidase-B. Pharmacol Res 62(6):475–488
Medvedev AE et al (1999) inhibition of monoamine oxidase by pirlindole analogues: 3D-QSAR analysis. Neurobiology 7(2):151–158
Mefford IN, Roth KA, Jurik SM, Collman V, McIntire S, Tolbert L, Barchas JD (1985) Epinephrine accumulation in rat brain after chronic administration of pargyline and LY 51641—comparison with other brain amines. Brain Res 339(2):342–345
Meiring L, Petzer JP, Petzer A (2013) Inhibition of monoamine oxidase by 3,4-dihydro-2(1H)-quinolinone derivatives. Bioorg Med Chem Lett 23(20):5498–5502. https://doi.org/10.1016/j.bmcl.2013.08.071
Milczek EM, Binda C, Rovida S, Mattevi A, Edmondson DE (2011) The ‘gating’ residues Ile199 and Tyr326 in human monoamine oxidase B function in substrate and inhibitor recognition. FEBS J 278(24):4860–4869
Miller JR, Edmondson DE (1999a) Influence of flavin analogue structure on the catalytic activities and flavinylation reactions of recombinant human liver monoamine oxidases A and B. J Biol Chem 274(33):23515–23525
Miller JR, Edmondson DE (1999b) Structure-activity relationships in the oxidation of para- substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry 38(41):13670–13683
Miller HH, Shore PA, Clarke DE (1980) In vivo monoamine oxidase inhibition by d-amphetamine. Biochem Pharmacol 29(10):1347–1354
Morrison JF (1969) Kinetics of reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim Biophys Acta 185:269–286. https://doi.org/10.1016/0005-2744(69)90420-3
Mure M et al (2005) Role of the interactions between the active site base and the substrate Schiff base in amine oxidase catalysis. Evidence from structural and spectroscopic studies of the 2-hydrazinopyridine adduct of Escherichia coli amine oxidase. Biochemistry 44(5):1568–1582. https://doi.org/10.1021/bi047988k
Murphy DL, Donnelly CH, Richelson E, Fuller RW (1978) N-Substituted cyclopropylamines as inhibitors of MAO-A and MAO-B forms. Biochem Pharmacol 27(13):1767–1769
Murphy DL, Sunderland T, Garrick NA, Aulakh CS, Cohen RM (1987) Selective amine oxidase inhibitors: basic to clinical studies and back. In: Dahl ML, Gram LF, Potter LF (eds) Clinical pharmacology in psychiatry. Springer, Berlin, pp 135–146
Nagy J, Kenney WC, Singer TP (1979) Reaction of phenylhydrazine with trimethylamine dehydrogenase and with free flavins. J Biol Chem 254(8):2684–2688
Nakai S, Yoneda F, Yamabe T, Fukui K (1999) Inhibition mechanism of flavin by deprenyl as an acetylenic irreversible inhibitor. Theor Chem Acc 102(1–6):147–160
Nandigama RK, Edmondson DE (2000) Structure-activity relations in the oxidation of phenethylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry 39(49):15258–15265
Naoi M, Maruyama W, Inaba-Hasegawa K, Akao Y (2011) Type A monoamine oxidase regulates life and death of neurons in neurodegeneration and neuroprotection. In: Monoamine oxidases and their inhibitors. Int Rev Neurobiol 100:85–106
Naoi M, Riederer P, Maruyama W (2016) Modulation of monoamine oxidase (MAO) expression in neuropsychiatric disorders: genetic and environmental factors involved in type A MAO expression. J Neural Transm 123(2):91–106. https://doi.org/10.1007/s00702-014-1362-4
Newton-Vinson P, Hubalek F, Edmondson DE (2000) High-level expression of human liver monoamine oxidase B in Pichia pastoris. Protein Expr Purif 20(2):334–345
Nikolic K, Mavridis L, Djikic T, Vucicevic J, Agbaba D, Yelekci K, Mitchell JBO (2016) Drug design for CNS diseases: polypharmacological profiling of compounds using cheminformatic, 3D-QSAR and virtual screening methodologies. Front Neurosci. https://doi.org/10.3389/fnins.2016.00265
Niwa H, Umehara T (2017) Structural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylases. Epigenetics 12(5):340–352. https://doi.org/10.1080/15592294.2017.1290032
Oanca G, Purg M, Mavri J, Shih JC, Stare J (2016) Insights into enzyme point mutation effect by molecular simulation: phenylethylamine oxidation catalyzed by monoamine oxidase A. Phys Chem Chem Phys 18(19):13346–13356. https://doi.org/10.1039/c6cp00098c
Orru R, Aldeco M, Edmondson DE (2013) Do MAO A and MAO B utilize the same mechanism for the C–H bond cleavage step in catalysis? Evidence suggesting differing mechanisms. J Neural Transm 120(6):847–851. https://doi.org/10.1007/s00702-013-0991-3
Paech C, Salach JI, Singer TP (1980) Suicide inactivation of monoamine-oxidase by trans-phenylcyclopropylamine. J Biol Chem 255(7):2700–2704
Parent MB, Master S, Kashlub S, Baker GB (2002) Effects of the antidepressant/antipanic drug phenelzine and its putative metabolite phenylethylidenehydrazine on extracellular gamma-aminobutyric acid levels in the striatum. Biochem Pharmacol 63(1):57–64. https://doi.org/10.1016/S0006-2952(00)00244-6
Patek DR, Hellerman L (1974) Mitochondrial monoamine-oxidase—mechanism of inhibition by phenylhydrazine and by aralkylhydrazines—role of enzymatic oxidation. J Biol Chem 249(8):2373–2380
Pavlin M, Mavri J, Repic M, Vianello R (2013) Quantum-chemical approach to determining the high potency of clorgyline as an irreversible acetylenic monoamine oxidase inhibitor. J Neural Transm 120(6):875–882. https://doi.org/10.1007/s00702-013-1016-y
Pearce LB, Roth JA (1985) Human-brain monoamine-oxidase type-B—mechanism of deamination as probed by steady-state methods. Biochemistry 24(8):1821–1826
Peng L, Zhang G, Zhang D, Wang Y, Zhu D (2010) A direct continuous fluorometric turn-on assay for monoamine oxidase B and its inhibitor-screening based on the abnormal fluorescent behavior of silole. Analyst 135(7):1779–1784
Petzer A, Harvey BH, Wegener G, Petzer JP (2012) Azure B, a metabolite of methylene blue, is a high-potency, reversible inhibitor of monoamine oxidase. Toxicol App Pharmacol 258(3):403–409
Pisani L, Catto M, Leonetti F, Nicolotti O, Stefanachi A, Campagna F, Carotti A (2011) Targeting monoamine oxidases with multipotent ligands: an emerging strategy in the search of new drugs against neurodegenerative diseases. Curr Med Chem 18(30):4568–4587
Pisani L et al (2013) Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur J Med Chem 70:723–739. https://doi.org/10.1016/j.ejmech.2013.09.034
Prusevich P et al (2014) A selective phenelzine analogue inhibitor of histone demethylase LSD1. ACS Chem Biol 9(6):1284–1293. https://doi.org/10.1021/cb500018s
Qin XZ, Williams F (1987) ESR studies on the radical cation mechanism of the ring opening of cyclopropylamines. J Am Chem Soc 109(2):595–597. https://doi.org/10.1021/ja00236a055
Ramadan ZB, Wrang ML, Tipton KF (2007) Species differences in the selective inhibition of monoamine oxidase (1-methyl-2-phenylethyl)hydrazine and its potentiation by cyanide. Neurochem Res 32(10):1783–1790. https://doi.org/10.1007/s11064-007-9309-x
Ramsay RR (1991) Kinetic mechanism of monoamine oxidase-A. Biochemistry 30(18):4624–4629
Ramsay RR (1998) Substrate regulation of monoamine oxidases. J Neural Transm-Suppl 52:139-147
Ramsay RR, Hunter DJB (2002) Inhibitors alter the spectrum and redox properties of monoamine oxidase A. BBA Proteins Proteom 1601(2):178–184
Ramsay RR, Tipton KF (2017) Assessment of enzyme inhibition: a review with examples from the development of monoamine oxidase and cholinesterase inhibitory drugs. Molecules (Basel, Switzerland). https://doi.org/10.3390/molecules22071192
Ramsay RR, Koerber SC, Singer TP (1987) Stopped-flow studies on the mechanism of oxidation of N-methyl-4-phenyltetrahydropyridine by bovine liver monoamine oxidase-B. Biochemistry 26(11):3045–3050
Ramsay RR, Sablin SO, Singer TP (1995) Redox properties of the flavin cofactor of monoamine oxidases A and B and their relationship to the kinetic mechanism. Prog Brain Res 106:33–39
Ramsay RR, Upadhyay AK, Li M, Edmondson DE (eds) (2005) Optical and EPR spectroscopic studies on the anionic flavin radical in MAO B and its Y435 mutant forms. Flavins and flavoproteins 2005. ArchiTect Inc., Tokyo
Ramsay RR, Dunford C, Gillman PK (2007) Methylene blue and serotonin toxicity: inhibition of monoamine oxidase A (MAO A) confirms a theoretical prediction. Br J Pharmacol 152(6):946–951
Ramsay RR, Olivieri A, Holt A (2011) An improved approach to steady-state analysis of monoamine oxidases. J Neural Transm 118(7):1003–1019
Rebrin I, Geha RM, Chen K, Shih JC (2001) Effects of carboxyl-terminal truncations on the activity and solubility of human monoamine oxidase B. J Biol Chem 276(31):29499–29506
Reck F, Zhou F, Girardot M, Kern G, Eyermann CJ, Hales NJ, Ramsay RR, Gravestock MB (2005) Identification of 4-substituted 1,2,3-triazoles as novel oxazolidinone antibacterial agents with reduced activity against monoamine oxidase A. J Med Chem 48(2):499–506
Reynolds GP, Rausch WD, Riederer P (1980) Effects of tranylcypromine stereoisomers on monamine oxidation in man. Br J Clin Pharmacol 9(5):521–523
Riederer P, Jellinger K (1983) Neurochemical insights into monoamine oxidase inhibitors, with special reference to deprenyl (selegiline). Acta Neurol Scand Suppl 95:43–55
Rigby SEJ, Hynson RMG, Ramsay RR, Munro AW, Scrutton NS (2005) A stable tyrosyl radical in monoamine oxidase A. J Biol Chem 280(6):4627–4631
Rusjan PM, Wilson AA, Miler L, Fan I, Mizrahi R, Houle S, Vasdev N, Meyer JH (2014) Kinetic modeling of the monoamine oxidase B radioligand [(1)(1)C]SL25.1188 in human brain with high-resolution positron emission tomography. J Cereb Blood Flow Metab 34(5):883–889. https://doi.org/10.1038/jcbfm.2014.34
Sablin SO, Ramsay RR (2001) Substrates but not inhibitors alter the redox potentials of monoamine oxidases. Antioxid Redox Signal 3(5):723–729
Salsali M, Holt A, Baker GB (2004) Inhibitory effects of the monoamine oxidase inhibitor tranylcypromine on the cytochrome P450 enzymes CYP2C19, CYP2C9, and CYP2D6. Cell Mol Neurobiol 24(1):63–76
Santillo MF (2014) Inhibition of monoamine oxidase (MAO) by alpha-ethylphenethylamine and N,alpha-diethylphenethylamine, two compounds related to dietary supplements. Food Chem Toxicol 74:265–269. https://doi.org/10.1016/j.fct.2014.10.009
Sara W, Valette H, Peyronneau M-A, Bramoullé Y, Coulon C, Curet O, George P, Dollé F, Bottlaender M (2010) [11C]SL25.1188, a new reversible radioligand to study the monoamine oxidase type B with PET: preclinical characterisation in nonhuman primate. Synapse 64(1):61–69. https://doi.org/10.1002/syn.20703
Schmidt DMZ, McCafferty DG (2007) trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 46(14):4408–4416
Shepard EM, Heggem H, Juda GA, Dooley DM (2003) Inhibition of six copper-containing amine oxidases by the antidepressant drug tranylcypromine. BBA Proteins Proteom 1647(1–2):252–259
Silverman RB (1983) Mechanism of inactivation of monoamine-oxidase by trans-2-phenylcyclopropylamine and the structure of the enzyme-inactivator adduct. J Biol Chem 258(24):14766–14769
Silverman RB (1995a) Mechanism-based enzyme inactivators. Enzyme Kinet Mech 249:240–283
Silverman RB (1995b) Mechanism-based enzyme inactivators. In: Enzyme kinetics and mechanism, Pt D. Methods Enzymol 249:240–283
Silverman RB (1995c) Radical ideas about monoamine-oxidase. Acc Chem Res 28(8):335–342. https://doi.org/10.1021/ar00056a003
Silverman RB, Hiebert CK (1988) Inactivation of monoamine oxidase-A by the monoamine oxidase-B inactivators 1-phenylcyclopropylamine, 1-benzylcyclopropylamine, and N-cyclopropyl-alpha-methylbenzylamine. Biochemistry 27(22):8448–8453
Silverman RB, Hoffman SJ (1981) N-(1-Methyl)cyclopropylbenzylamine—a novel inactivator of mitochondrial monoamine-oxidase. Biochem Biophys Res Commun 101(4):1396–1401
Silverman RB, Yamasaki RB (1984) Mechanism-based inactivation of mitochondrial monoamine-oxidase by N-(1-methylcyclopropyl)benzylamine. Biochemistry 23(6):1322–1332. https://doi.org/10.1021/bi00301a046
Silverman RB, Zieske PA (1985) Mechanism of inactivation of monoamine-oxidase by 1-phenylcyclopropylamine. Biochemistry 24(9):2128–2138
Silverman RB, Zieske PA (1986) Identification of the amino-acid bound to the labile adduct formed during inactivation of monoamine-oxidase by 1-phenylcyclopropylamine. Biochem Biophys Res Commun 135(1):154–159
Sliwoski G, Kothiwale S, Meiler J, Lowe EW Jr (2014) Computational methods in drug discovery. Pharmacol Rev 66(1):334–395. https://doi.org/10.1124/pr.112.007336
Son SY, Ma A, Kondou Y, Yoshimura M, Yamashita E, Tsukihara T (2008) Structure of human monoamine oxidase A at 2.2-angstrom resolution: The control of opening the entry for substrates/inhibitors. PNAS USA 105(15):5739–5744
Sterling J et al (2002) Novel dual inhibitors of AChE and MAO derived from hydroxy aminoindan and phenethylamine as potential treatment for Alzheimer’s disease. J Med Chem 45(24):5260–5279. https://doi.org/10.1021/jm020120c
Swett LR, Martin WB, Taylor JD, Everett GM, Wykes AA, Gladish YC (1963) Structure-activity relations in the pargyline series. Ann N Y Acad Sci 107:891–898
Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA (2007) Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry 46(23):6892–6902
Talele TT (2016) The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules. J Med Chem 59(19):8712–8756. https://doi.org/10.1021/acs.jmedchem.6b00472
Tan AK, Ramsay RR (1993) Substrate-specific enhancement of the oxidative half-reaction of monoamine-oxidase. Biochemistry 32(9):2137–2143
Taylor JD, Wykes AA, Gladish YC, Martin WB (1960) New inhibitor of monoamine oxidase. Nature 187:941–942. https://doi.org/10.1038/187941a0
Tipton KF (1971) Reaction of monoamine oxidase with phenethylhydrazine. Biochem J 121(3):P33
Tipton KF, Spires IPC (1971) Kinetics of phenethylhydrazine oxidation by monoamine oxidase. Biochem J 125(2):521–524. https://doi.org/10.1042/bj1250521
Tipton KF, Davey G, Motherway M (2006) Monoamine oxidase assays. In: Current protocols in toxicology 30:4.21:4.21.1–4.21.42
Tsugeno Y, Ito A (1997) A key amino acid responsible for substrate selectivity of monoamine oxidase A and B. J Biol Chem 272(22):14033–14036. https://doi.org/10.1074/jbc.272.22.14033
Tzvetkov NT, Stammler HG, Neumann B, Hristova S, Antonov L, Gastreich M (2017) Crystal structures, binding interactions, and ADME evaluation of brain penetrant N-substituted indazole-5-carboxamides as subnanomolar, selective monoamine oxidase B and dual MAO-A/B inhibitors. Eur J Med Chem 127:470–492. https://doi.org/10.1016/j.ejmech.2017.01.011
Umhau S, Pollegioni L, Molla G, Diederichs K, Welte W, Pilone M, Ghisla S (2000) The X-ray structure of d-amino acid oxidase at very high resolution identifies the chemical mechanism of flavin-dependent substrate dehydrogenation. PNAS USA 97(23):12463–12468. https://doi.org/10.1073/pnas.97.23.12463
Unzeta M, Esteban G, Bolea I, Fogel WA, Ramsay RR, Youdim MBH, Tipton KF, Marco-Contelles J (2016) Multi-target directed donepezil-like ligands for Alzheimer’s disease. Front Neurosci. https://doi.org/10.3389/fnins.2016.00205
Upadhyay AK, Borbat PP, Wang J, Freed JH, Edmondson DE (2008) Determination of the oligomeric states of human and rat monoamine oxidases in the outer mitochondrial membrane and octyl beta-d-glucopyranoside micelles using pulsed dipolar electron spin resonance spectroscopy. Biochemistry 47(6):1554–1566
Valley MP et al (2006) A bioluminescent assay for monoamine oxidase activity. Anal Biochem 359(2):238–246. https://doi.org/10.1016/j.ab.2006.09.035
Vazquez ML, Silverman RB (1985) Revised mechanism for inactivation of mitochondrial monoamine-oxidase by N-cyclopropylbenzylamine. Biochemistry 24(23):6538–6543
Veselovsky AV, Ivanov AS, Medvedev AE (2004) Computer modelling and visualization of active site of monoamine oxidases. Neurotoxicology 25(1–2):37–46
Vianello R, Repic M, Mavri J (2012) How are biogenic amines metabolized by monoamine oxidases? Eur J Org Chem 36:7057–7065
Vianello P et al (2014) Synthesis, biological activity and mechanistic insights of 1-substituted cyclopropylamine derivatives: a novel class of irreversible inhibitors of histone demethylase KDM1A. Eur J Med Chem 86:352–363. https://doi.org/10.1016/j.ejmech.2014.08.068
Vianello R, Domene C, Mavri J (2016) The use of multiscale molecular simulations in understanding a relationship between the structure and function of biological systems of the brain: the application to monoamine oxidase enzymes. Front Neurosci. https://doi.org/10.3389/fnins.2016.00327
Vintem APB, Price NT, Silverman RB, Ramsay RR (2005) Mutation of surface cysteine 374 to alanine in monoamine oxidase A alters substrate turnover and inactivation by cyclopropylamines. Bioorg Med Chem 13(10):3487–3495
Walker MC, Edmondson DE (1994) Structure-activity-relationships in the oxidation of benzylamine analogs by bovine liver mitochondrial monoamine-oxidase-B. Biochemistry 33(23):7088–7098
Wang J, Edmondson DE (2011) H-2 kinetic isotope effects and pH dependence of catalysis as mechanistic probes of rat monoamine oxidase A: comparisons with the human enzyme. Biochemistry 50(35):7710–7717. https://doi.org/10.1021/bi200951z
Weinreb O, Amit T, Bar-Am O, Youdim MBH (2010) Rasagiline: a novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog Neurobiol 92(3):330–344
Weinreb O, Amit T, Riederer P, Youdim MBH, Mandel SA (2011) Neuroprotective profile of the multitarget drug rasagiline in Parkinson’s disease. In: Youdim MBH (ed) Monoamine oxidases and their inhibitors. Int Rev Neurobiol 100:127–149
Werther T, Wahlefeld S, Salewski J, Kuhlmann U, Zebger I, Hildebrandt P, Dobbek H (2017) Redox-dependent substrate-cofactor interactions in the Michaelis-complex of a flavin-dependent oxidoreductase. Nat Commun 8:16084. https://doi.org/10.1038/ncomms16084
Weyler W (1994) Functional expression of C-terminally truncated human monoamine-oxidase type-a in Saccharomyces cerevisiae. J Neural Transm Suppl 41:3–15
Williams CH, Lawson J (1974) Monoamine oxidase. II. Time-dependent inhibition by propargylamines. Biochem Pharmacol 23(3):629–636
Wu HF, Chen K, Shih JC (1993) Site-directed mutagenesis of monoamine oxidase-A and oxidase-B—role of cysteines. Mol Pharmacol 43(6):888–893
Wu XF, Li LH, Shi W, Gong QY, Li XH, Ma HM (2016) Sensitive and selective ratiometric fluorescence probes for detection of intracellular endogenous monoamine oxidase A. Anal Chem 88(2):1440–1446. https://doi.org/10.1021/acs.analchem.5b04303
Xie SS, Wang XB, Jiang N, Yu WY, Wang KDG, Lan JS, Li ZR, Kong LY (2015) Multi-target tacrine-coumarin hybrids: cholinesterase and monoamine oxidase B inhibition properties against Alzheimer’s disease. Eur J Med Chem 95:153–165. https://doi.org/10.1016/j.ejmech.2015.03.040
Yang HY, Neff NH (1974) The monoamine oxidases of brain: selective inhibition with drugs and the consequences for the metabolism of the biogenic amines. J Pharmacol Exp Ther 189(3):733–740
Youdim MBH, Tipton KF (2002) Rat striatal monoamine oxidase-B inhibition by l-deprenyl and rasagiline: its relationship to 2-phenylethylamine-induced stereotypy and Parkinson’s disease. Parkinsonism Relat Disord 8(4):247–253. https://doi.org/10.1016/s1353-8020(01)00011-6
Youdim MB, Collins GG, Sandler M (1971) Monoamine oxidase: multiple forms and selective inhibitors. Biochem J 121:32P–34P
Youdim MBH, Wadia A, Tatton W, Weinstock M (2001) The anti-Parkinson drug rasagiline and its cholinesterase inhibitor derivatives exert neuroprotection unrelated to MAO inhibition in cell culture and in vivo. In: Slikker W, Trembly B (eds) Neuroprotective agents. Ann N Y Acad Sci, vol 939, pp 450–458
Youdim MBH, Edmondson D, Tipton KF (2006) The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci 7(4):295–309
Youngster SK, McKeown KA, Jin YZ, Ramsay RR, Heikkila RE, Singer TP (1989) Oxidation of analogs of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by monoamine oxidase-A and oxidase-B and the inhibition of monoamine oxidases by the oxidation products. J Neurochem 53(6):1837–1842
Yu PH (1994) Pharmacological and clinical implications of MAO-B inhibitors. Gen Pharmacol 25(8):1527–1539
Yu PH, Tipton KF (1989) Deuterium-isotope effect of phenelzine on the inhibition of rat-liver mitochondrial monoamine-oxidase activity. Biochem Pharmacol 38(23):4245–4251
Yu PH, Bailey BA, Durden DA, Boulton AA (1986) Stereospecific deuterium substitution at the alpha-carbon position of dopamine and its effect on oxidative deamination catalyzed by MAOA and MAOB from different tissues. Biochem Pharmacol 35(6):1027–1036
Zeller EA, Barsky J (1952) In vivo inhibition of liver and brain monoamine oxidase by 1-isonicotinyl-2-isopropyl hydrazine. Proc Soc Exper Bio Med 81(2):459–461
Zeller EA, Sarkar S (1962) Amine oxidases. XIX. Inhibition of monoamine oxidase by phenylcyclopropylamines and iproniazid. J Biol Chem 237:2333–2336
Zenn RK, Abad E, Kastner J (2015) Influence of the environment on the oxidative deamination of p-substituted benzylamines in monoamine oxidase. J Phys Chem B 119(9):3678–3686. https://doi.org/10.1021/jp512470a
Zhong BY, Silverman RB (1997) Identification of the active site cysteine in bovine liver monoamine oxidase B. J Am Chem Soc 119(28):6690–6691
Acknowledgements
The authors thank COST Action CM1103.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ramsay, R.R., Albreht, A. Kinetics, mechanism, and inhibition of monoamine oxidase. J Neural Transm 125, 1659–1683 (2018). https://doi.org/10.1007/s00702-018-1861-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-018-1861-9