
Abstract
Similar to the regular enzymatic glycosylation, lysine glycation also attaches a sugar molecule to a peptide, but it does not need the help of an enzyme. It has been found that lysine glycation is involved in various biological processes and is closely associated with many metabolic diseases. Thus, an accurate identification of lysine glycation sites is important to understand its underlying molecular mechanisms. The glycated residues do not show significant patterns, which make both in silico sequence-level predictions and experimental validations a major challenge. In this study, a novel predictor named MDS_GlySitePred is proposed to predict lysine glycation sites by using multidimensional scaling method (MDS) and support vector machine algorithm. As illustrated by the average results of tenfold cross-validation repeated 50 times, MDS_GlySitePred achieves a satisfactory performance with a sensitivity of 95.08%, a specificity of 97.65%, an accuracy of 96.58%, and Matthew’s correlation coefficient of 0.93 on the extensively used benchmark datasets. Experimental results indicate that MDS_GlySitePred significantly outperforms four existing glycation site predictors including NetGlycate, PreGly, Gly-PseAAC, and BPB_GlySite. Therefore, MDS_GlySitePred can be a useful bioinformatics tool for the identification of glycation sites.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Lysine glycation is one of the most common and important post-translational modifications (PTMs), which can potentially affect various biological processes, such as conformation, efficacy, and immunogenicity [1, 2]. Moreover, lysine is one of the essential amino acids in the human body, which can promote human development, enhance immune function, and can improve the functioning of the central nervous system. Because of the low lysine content in cereals and the susceptibility to damage during processing, it is called the first limiting amino acid.
Lysine glycation is a complex multi-step process, beginning with the attachment of reducing sugars to amino groups in cellular proteins, leading to the formation of Schiff’s base as early glycation product [3,4,5]. The advanced glycation end-products are known to facilitate age-related chronic diseases, e.g. atherosclerosis [6], by changing vascular elasticity and thickening vascular walls [7]. Glycation is also observed to promote abnormal amyloid aggregation in aging-related neurodegenerative disorders, such as Alzheimer’s [8] and Parkinson’s [9] diseases. In spite of its essential role, the detection of glycated residues is still solely based on the tedious and time-consuming mass spectrometry technique to measure the monosaccharide modification-induced mass increase in the investigated peptide [10].
Several methods for predicting glycation sites based on protein sequence information have been reported. The neural network predictor named NetGlycate was built by Jonansen et al., which was trained on 89 glycated and 126 non-glycated lysine sites derived from 20 proteins. Later, Liu et al. constructed the model PreGly to predict the glycation sites by extracting the composition of amino acid, 4-interval amino acid pairs, five amino acid physicochemical properties and then selecting effective features through the maximum correlation minimum redundancy (mRMR) algorithm. Xu et al. developed a predictor Gly-PseAAC by combining position-specific amino acid propensity and support vector machine (SVM) algorithm. Recently, Ju et al. used bi-profile Bayes (BPB) feature extraction combined with SVM algorithm to construct a new predictor BPB_GlySite to predict glycosylation sites. While the prediction performance of BPB_GlySite has few improvements over the previous predictors, it is noted that its performance on the Xu training set is not satisfactory, because it obtains the Matthew’s correlation coefficient of 0.3499 only, and thus, requires significant improvement.
In this study, we propose a novel predictor MDS_GlySitePred to improve the prediction performance of glycation sites. To overcome the defective non-uniform distribution of training and test samples, we employed multidimensional scaling (MDS) to cluster the samples [11]. According to different distance radius, the negative samples were divided into three categories, and the positive samples remained unchanged. They characteristics including Parallel correlation pseudo amino acid composition (PC-PseAAC), General parallel correlation pseudo amino acid composition (PC-PseAAC_General), Adapted normal distribution bi-profile Bayes (ANBPB), Double Bi-profile Bayes (DBPB), Bi-profile Bayes (BPB), Top-n-gram, Amino acid composition (AAC), Position-specific di-amino acid propensity (PSDAAP) and Position-specific tri-amino acid propensity (PSTAAP) were extracted from sequence information. By combining the MDS method with the SVM algorithm, and through a tenfold cross-validation test, the MDS method was shown to be superior to the existing prediction in predicting lysine glycation sites. Finally, based on the features combination of PC-PseAAC_General + ANBPB + DBPB + Top-n-gram + AAC, ANBPB + PSDAAP and PC-PseAAC_General + PC-PseAAC + BPB + DBPB + PSTAAP, the importance of the positions around the glycation sites was analyzed. The features analysis shows that the residues around the glycation sites may play the most important role in the prediction of glycation sites. These results may provide useful clues for studying the lysine glycation mechanisms and may facilitate relevant experimental verifications.
2 Materials and methods
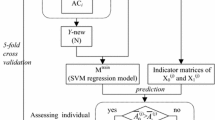
This method comprised four major steps: (1) collecting and processing data, (2) using MDS to cluster training datasets, (3) extracting sequence features, (4) constructing and evaluating models. The conceptual diagram of constructing the prediction model is given in Fig. 1.

The conceptual diagram of constructing the prediction model
2.1 Data collection and pre-processing
The most recently constructed training dataset by Xu et al. [2, 12] and Johansen et al. [13, 14] were used in the present study to provide a comprehensive and unbiased comparison of our methods with existing methods. For convenience, the datasets were named Xu dataset and Johansen dataset, respectively. The proteins in Xu’s training set was retrieved from protein lysine modifications database CPLM [15], and it consisted of 223 experimentally annotated glycation lysine sites and 446 non-glycation lysine sites from 72 proteins. In this study, we retrieved the proteins from NCBI, which were used in Xu dataset to get all negative samples. In this work, pseudo-amino acid were not considered. According to Xu [12] and Ju [2], the window size was set to 15. Thus, every training sample was represented as a peptide segment of length with 7 residues downstream and 7 residues upstream of lysine residue K. At last, the new training dataset contained 215 lysine glycation sites and 1781 lysine non-glycation sites. For Johansen’s benchmark dataset, the same method was used to process, and finally obtain 81 positive samples and 244 negative samples. Finally, amino acid composition (AAC) feature extraction was performed on the negative training set. To avoid linearity, we removed the last column from the 20-dimensional feature, leaving 19 columns of feature vectors for later use in the MDS method.
2.2 Feature extraction and encoding
2.2.1 Amino acid composition (AAC)
The amino acid composition [16, 17] simply represents the frequency of 20 common amino acids in the protein sequence, reflects the global characteristics of the protein sequence, and is a basic protein sequence feature extraction algorithm. The AAC maps the membrane protein sequence to a point in the 20-dimensional Euclidean space and can be defined as a 20-dimensional vector:
where \( x_{i} = f_{i} /\sum\nolimits_{i = 1}^{20} {f_{j} } \), fi is the number of times the first type of amino acid appears in the membrane protein sequence. Obviously, \( \sum\nolimits_{j = 1}^{20} {x_{i} = 1} \). The calculation of amino acid composition is convenient and is the most commonly used sequence feature extraction algorithm in the study of membrane protein classification.
2.2.2 Bi-profile Bayes (BPB)
The bilateral Bayesian feature extraction algorithm proposed by Shao et al. [18, 19] has been widely used to predict various post-translational modification sites [20,21,22]. BPB comprehensively considers the information contained in the two aspects of positive and negative samples. Let \( S = s_{1} s_{2} \ldots s_{n} \) denote a lysine glycosylated sample, where \( s_{j} \left( {j = 1,2, \ldots ,n} \right) \) represents 20 natural amino acids {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y}, and n is the length of the peptide fragment after the amino acid K in the middle position is omitted (i.e., n = 14). Given a protein sequence P (Eq. 1), the BPB feature vector of P is defined:
where P is the posterior probability vector, x1, x2, …, xn represent the posterior probability of each amino acid at each position in positive peptide sequence datasets, \( x_{n + 1} , \ldots ,x_{2n} \) represent the posterior probability of each amino acid at each position in negative peptide sequence datasets. Two position-specific profiles for final model training, including positive position-specific profiles and negative position-specific profiles, were generated by calculating the frequency of each amino acid at each position in the positive, as well as negative datasets.
2.2.3 Double bi-profile bayes (DBPB)
DBPB is an improvement over BPB [23]. BPB is the posterior probability of each single amino acid at each position in the positive and negative datasets, while DBPB is the posterior probability of every di-amino acid at each position in the datasets. Given a protein sequence P (Eq. 1), the DBPB feature vector of P is defined as follows:
where P is the posterior probability vector, \( x_{1} ,x_{2} , \ldots ,x_{n - 1} \) represent the posterior probability of each amino acid pair at each position in positive peptide sequence datasets,\( x_{{\left( {n - 1} \right) + 1}} , \ldots ,x_{{2\left( {n - 1} \right)}} \) represent the posterior probability of each amino acid pair at each position in negative peptide sequence datasets. Two position-specific profiles for final model training, including positive position-specific profiles and negative position-specific profiles, were generated by calculating the frequency of each amino acid pair at each position in the positive, as well as negative datasets.
2.2.4 Adapted normal distribution bi-profile Bayes (ANBPB)
ANBPB [20, 24] is the improvement of BPB in another aspect. Given a protein sequence P (Eq. 1), the ANBPB feature vector of P is defined as:
where p1, p2, …, pn is the posterior probability of each amino acid at each position in positive peptide sequences datasets; \( p_{n + 1} , \ldots ,p_{2n} \) is defined based on the posterior probability of each amino acid at each position in negative peptide sequences datasets. The posterior probability \( p_{1} ,p_{2} , \ldots ,p_{2n } \) is coded by the adapted normal distribution as follows:
where \( \varphi \left( x \right) \) is the standard normal distribution function and the detailed description of the formula is given [20, 24].
2.2.5 Position-specific di-amino acid propensity (PSDAAP)
The posterior probability of every two nearest amino acids at each position in the positive peptide sequence datasets is subtracted from the negative peptide sequence datasets [25, 26]. Given a protein sequence P (Eq. 1), the PSDAAP feature vector of P is defined as follows:
where P is the posterior probability vector, +pi represent the posterior probability of each amino acid pair at each position in positive peptide sequence datasets, −pi represent the posterior probability of each amino acid pair at each position in negative peptide sequence datasets. \( p_{i} = \left( { + p_{i} } \right) - \left( { - p_{i} } \right) \) is the feature vector.
2.2.6 Position-specific tri-amino acid propensity (PSTAAP)
Similar to PSDAAP, the posterior probability of every three nearest amino acids at each position in the positive peptide sequence datasets is subtracted from the negative peptide sequence datasets [25, 26]. Given a protein sequence P (Eq. 1), the PSTAAP feature vector of P is defined as follows:
where P is the posterior probability vector, \( + p_{i} \) represent the posterior probability of each three amino acid pair at each position in positive peptide sequence datasets, −pi represent the posterior probability of each three amino acid pair at each position in negative peptide sequence datasets. \( p_{i} = \left( { + p_{i} } \right) - \left( { - p_{i} } \right) \) is the feature vector.
2.2.7 Parallel correlation pseudo amino acid composition (PC-PseAAC)
PC-PseAAC [27] is an approach merging the global sequence-order information and the contiguous local sequence-order information into the feature vector of the protein sequence. Given a Protein sequence P (Eq. 1), the PC-PseAAC feature vector of P is defined as follows:
where,
where w is the weight factor ranging from 0 to 1, the parameter λ is an integer that represents the highest counted rank (or tier) of the correlation along a protein sequence, fi(i = 1,2,…,20) is the normalized occurrence frequency of the 20 amino acids in the protein P, Θj(j = 1,2,…,20) is called the j-tier correlation factor reflecting the sequence-order correlation among all the jth most contiguous residues along a protein chain, which is defined as follows:
where the correlation function is given by
where \( H_{1} \left( {R_{i} } \right) \) is the hydrophobicity value, H2(Ri) is the hydrophilicity value, and \( M\left( {R_{i} } \right) \) is the side-chain mass of the amino acid Ri. Note that before substituting the values of hydrophobicity, hydrophilicity, and side-chain mass into Eq. 7, they are all subjected to a standard conversion as described by the following equations.
where \( H_{1}^{0} \left( i \right) \), \( H_{1}^{0} \left( i \right),M^{0} \left( i \right) \) are the original hydrophobicity value, the corresponding original hydrophilicity value, the mass of the ith amino acid, respectively. With the wide application of PC-PseAAC, Liu et al. [28] developed a web server “Pse-in-One” that could generate PC-PseAAC. For detailed information on Pse-in-One and its updated version, please refer to [29].
2.2.8 General parallel correlation pseudo amino acid composition (PC-PseAAC-general)
The PC-PseAAC-General approach [30] not only allows users to upload their own indices to generate PC-PseAAC-General feature vectors, but also incorporate comprehensive built-in indices extracted from AA index [31]. Given a protein sequence P (Eq. 1), the PC-PseAAC-General feature vector of P is defined as follows:
where
where w is the weight factor ranging from 0 to 1, the parameter λ is an integer that represents the highest counted rank (or tier) of the correlation along a protein sequence, fi(i = 1,2,…,20) is the normalized occurrence frequency of the 20 amino acids in the protein P, Θj(j = 1,2,…,20) is called the j-tier correlation factor reflecting the sequence-order correlation among all the jth most contiguous residues along with a protein chain, which is defined as follows:
where the correlation function is given by follows:
where µ is the number of physicochemical indices considered; \( H_{u} \left( {R_{i} } \right) \) and \( H_{u} \left( {R_{j} } \right) \) are the uth physicochemical index value of the amino acid Ri and Rj, respectively. Note that before substituting the physicochemical indices values into Eq. 26, they are all subjected to a standard conversion as described by the following equation:
where \( H_{u}^{0} \left( i \right) \) is the uth original physicochemical value of the ith amino acid.
2.2.9 Top-n-gram
Top-n-gram [14] can be viewed as a novel profile-based building block of proteins, containing the evolutionary information extracted from the frequency profiles. The frequency profiles calculated from the multiple sequence alignments given as output by PSI-BLAST [32] are converted into Top-n-grams by combining the n most frequent amino acids in each amino acid frequency profile. The protein sequences are transformed into fixed dimension feature vectors by the number of times of each Top-n-gram occur. For more information about this approach, please refer to [14].
2.3 Multidimensional scaling (MDS) method
MDS is a multivariate data analysis technique that displays “distance or similarity” data structures in low-dimensional space, which has been widely applied in many applications, such as data visualization [33], object retrieval [34], data clustering [35], and localization [36].
The solution provided by the multidimensional scaling method is that when the similarity (or distance) between pairs of objects in n objects is given, the representation of these objects in the low dimensional space is determined (Perceptual Mapping) and is made as “substantially matched” as possible with the original similarity (or distance) to minimize any distortion caused by dimensionality reduction. Each point arranged in a multidimensional space represents an object, so the distance between two points is highly related to the similarity between them. In other words, two similar objects are represented by two points with similar distances in a multidimensional space, and two dissimilar objects are represented by two points in the multidimensional space that are far apart. Here, we use the dimensionality reduction clustering function of MDS. The relationship among amino acid sequences in a polypeptide is converted to a distance matrix, and each sequence is regarded as a point in a multidimensional space. By MDS dimensionality reduction clustering, the evolutionary relationship among these sequences can be displayed in low-dimensional space [11].
Generally, the classical MDS [37] is a three-step algorithm, including distance matrix construction, inner product matrix computation, and low dimensional representation calculation. Details of these steps are presented as follows:
-
1.
Distance matrix construction. For each vector xi (1 ≤ i ≤ N), the Euclidean distance di,j between xi and xj (1 ≤ j ≤ N) was calculated and thus the distance matrix D = (di,j)N×N was obtained as follows:
$$ D = \left[ {\begin{array}{*{20}c} {d_{1,1} } & {d_{1,2} } & \ldots & {d_{1,N} } \\ {d_{2,1} } & {d_{2,2} } & \ldots & {d_{2,N} } \\ \ldots & \ldots & \ldots & \ldots \\ {d_{N,1} } & {d_{N,2} } & \ldots & {d_{N,N} } \\ \end{array} } \right] $$(24)$$ {\text{d}}_{i,j} = \sum\nolimits_{l = 1}^{K} {\left[ {x_{i} (l) - x_{\text{j}} (l)} \right]}^{2} $$(25)where xi(l) is the lth elements of xi,xj(l) is the lth elements of xj,Distance matrix D is a real symmetric matrix with all 0 diagonal elements.
-
2.
Inner product matrix computation. With the distance matrix D, the inner product matrix B can be determined by
$$ B = - \frac{1}{2}JDJ $$(26)$$ J = E - \frac{1}{N}ee^{T} $$(27)where J is a centralized matrix obtained by the below equation, and E is a unit matrix sized N × N, e is a unit vector sized N × 1, Je = 0, and JT= J.
-
3.
Low dimensional representation calculation. As B is symmetric and positive semi-definite, it can be decomposed as:
$$ B = SVS^{T} $$(28)$$ Z = SV^{{{\raise0.7ex\hbox{$1$} \!\mathord{\left/ {\vphantom {1 2}}\right.\kern-0pt} \!\lower0.7ex\hbox{$2$}}}} $$(29)where V is a diagonal matrix of the eigenvalues of B, and S is a matrix of the corresponding eigenvectors. Consequently, a low dimensional representation G can be generated by taking the first d columns of Z.
Therefore, G is a matrix of size N × d (d < K). Assume that Vd is the diagonal matrix composed of the d largest eigenvalues, and Ud is the N × d matrix composed of the corresponding d norm of orthogonalized characteristic vectors. If \( U_{d} = (\overrightarrow {v}_{1} ,\overrightarrow {{v_{2} }} , \ldots ,\overrightarrow {{v_{d} }} ) \) and \( V_{\text{d}} = {\text{diag}}(\lambda_{1} ,\lambda_{2} , \ldots ,\lambda_{d} ) \), then the coordinate matrix in d-dimensional space is:
2.4 Model construction and evaluation
A support vector machine (SVM) is a set associated with supervised learning methods used for classification and regression based on statistical learning theory. SVM looks for a rule that best maps each member of the training dataset into the correct classification [38, 39], and it has been proved to be a powerful tool in a lot of bioinformatics fields [18, 40,41,42,43]. In this study, the LIBSVM package [44] was applied to build and train a prediction model. The radial basis function \( \left( {RBF} \right) \, K\left( {S_{i} ,S_{j} } \right) = e^{{\left( { - \gamma \left\| {S_{i} - S_{j} } \right\|^{2} } \right)}} \) was used for the kernel function. The grid search was used to search the optimal parameters of SVM. Parameter c was selected from {20, 21,…, 213}, and kernel parameter g was selected from {2−13, 2−12,…, 20}.
To verify the effectively predictive performance of the model, the sensitivity (Sn), specificity (Sp), accuracy (Acc) and Matthew’s correlation coefficient (MCC) were employed as follows:
where N+ is the total number of the glycation sites investigated, while \( N_{ - }^{ + } \) is the number of the sites incorrectly predicted as the non-glycation sites, and N− is the total number of the non-glycation sites investigated, while N −+ is the number of the non-glycation sites incorrectly predicted as the glycation sites.
In statistical prediction, the following three cross-validation methods are often used to examine a predictor for its effectiveness in practical application: independent dataset test, subsampling or K-fold cross-validation test, and jackknife test [45]. The jackknife test is the most credible one among these three test methods [46], since the outcome obtained by it is always unique for a given benchmark dataset. Accordingly, the jackknife test has been widely recognized and increasingly used by investigators to examine the quality of various predictors [47,48,49,50,51,52]. However, in this work, we used tenfold cross-validation test instead of the jackknife test because the prediction results in previous works [2, 12] are obtained on tenfold cross-validation. Normally, this procedure is repeated 10 times and the final prediction result is an average of the 10 testing subsets. For obtaining a reliable estimate in this study, the tenfold cross-validation was repeated 50 times.
3 Analysis of results
3.1 Experimental data processing
The MDS method was used to train the negative training set, and a perceptual map was obtained (Fig. 2). From the perceptual map, the training samples were observed to be roughly concentrated in three regions. Therefore, the negative training sets were divided into three categories based on these three regions, and the values (0.26, −0.01), (−0.03, −0.28), and (−0.04, 0.06) were selected as origins, with 0.2, 0.12, and 0.1 as radii, respectively. Thus, three groups of non-Glycated lysine are clustered. The numbers of negative samples are 218, 212, 1063 for the three datasets named Xu dataset1, Xu dataset2, and Xu dataset3, respectively. To avoid overestimating the prediction performance of the model due to redundancy and sequence homology, CD-HIT [53, 54] was used to remove redundancy for group Xu dataset3 negative training samples. For those two samples with similarity ≥ 40%, one of the samples was retained, while 395 negative samples with sequence similarity < 40% were obtained in the de-redundant negative samples. For Johansen’s benchmark dataset, we used the same method to obtain the perceptual map (Fig. 3), and also divided the negative samples into three groups, denoted by Johansen dataset1, Johansen dataset2, and Johansen dataset3. The values (−0.07261, 0.07162), (0.1198, 0.07272) and (0.01023, −0.1557) were assigned as origin, with 0.095, 0.085 and 0.145 as respective radii. Finally, the three groups of lysine are obtained, with 81, 56, 51 sites, respectively. Moreover, because Johansen dataset is smaller than Xu dataset, we made the origin coordinates and radius more precise.

Perceptual graph obtained with Xu dataset

Perceptual graph obtained with Johansen dataset
3.2 Combining features
To find the optimization features that are most conducive to the identification of lysine glycosylation, nine feature extraction strategies, including PC-PseAAC (30), PC-PseAAC-General (30), BPB (28), ANBPB (28), DBPB (26), Top-n-gram (20), AAC (20), PSDAAP (13) and PSTAAP (12) were used. Here, the numbers in parentheses represent the dimension of this feature. In this study, a combination of features was used because the combination of multiple features enhances the training effect of the model. The feature combinations were performed in the order in which these nine feature dimensions are decremented. The performance of the combined feature sets for sorting lysine glycation sites and non-glycation sites was examined by tenfold cross-validation. Take, for example, the combination features in Xu dataset1. Firstly, when only the feature PC-PseAAC-General was used, the prediction accuracy achieved was 91%. However, when PC-PseAAC was added, the accuracy decreased to 90.98%, so the PC-PseAAC was rejected and when ANBPB was directly added, the accuracy rate significantly increased to 94.68%. With these trials, a most suitable combination of PC-PseAAC-General + ANBPB + DBPB + Top-n-gram + AAC was obtained, with the sensitivity of 96.28%, specificity of 99.56% and accuracy of 97.92%. Other training groups were handled and processed in a similar way. The results are presented in Table 1.
The results show that the prediction performance was enhanced by increasing the features number step by step. As shown in Table 1, on Xu’s dataset, the Acc reached 97.92%, 99.77%, 99.02%, respectively. And on Johansen’s dataset, the Acc reached 97.79%, 96.22%, 100%, respectively.
3.3 Performance of MDS_GlySitePred
The MDS_GlySitePred was constructed on Xu dataset because the previous predictors Gly-PseAAC and BPB_GlySite were all trained on the same dataset. To show the three groups of results intuitively, the results of tenfold cross-validation was repeated 50 times as listed in Table 2. As can be seen, the predictor MDS_GlySitePred achieved the best prediction performance with the Sn of 95.08%, Sp of 97.65%, Acc of 96.58%, and MCC of 0.93. The performance of MDS_GlySitePred is obviously superior to the second-best model BPB_GlySite, and especially showed 31.40% better Sn. These results indicate that MDS_GlySitePred is more effective and more reliable in identifying lysine glycation sites from query proteins than BPB_GlySite and Gly-PseAAC. Since the classification algorithms used in MDS_GlySitePred, BPB_GlySite, and Gly-PseAAC are all SVMs, a better performance of MDS_GlySitePred indicates that the MDS method may be used to cluster samples from a different probability distribution.
3.4 Comparison between MDS_GlySitePred with existing prediction methods on Johansen’s dataset
To further assess the effectiveness of MDS_GlySitePred, we compared it with other existing prediction methods, including NetGlycate, PreGly, Gly-PseAAC, and BPB_Gly-Site [2, 12,13,14]. All these predictors have been trained on the same Johansen’s benchmark dataset [2, 12,13,14], therefore, the same tenfold cross-validation test could be implemented. The compared results among the four methods are presented in Table 3. Here too, the MDS_GlySitePred method achieved the best results, with the Sn of 94.44%, Sp of 96.15%, Acc of 95.45% and MCC of 0.91. Moreover, the MDS_GlySitePred significantly outperformed the existing glycation sites predictors on Johansen’s benchmark dataset.
4 Conclusions
In this work, we built a prediction model MDS_GlySitePred for identifying protein glycation site based on multidimensional scaling (MDS) clustering negative samples. To the best of our knowledge, this is the first time MDS has been applied to predict glycation sites. The experimental results show that the MDS is efficient in dealing with samples obeying different probability distribution. We hope that this model will further facilitate the protein glycation studies. As demonstrated in a series of recent publications [47, 55,56,57] on developing new prediction methods, user-friendly and publicly accessible web-servers will significantly enhance their impact [48, 58,59,60,61,62,63,64,65,66,67,68,69,70]. Hence, our future course of action will be to provide a web-server for the prediction method presented in this paper.
References
Miller AK, Hambly DM, Kerwin BA, Treuheit MJ, Gadgil HS (2011) Characterization of site-specific glycation during process development of a human therapeutic monoclonal antibody. J Pharm Sci 100(7):2543–2550
Ju Z, Sun JH, Li YJ, Wang L (2017) Predicting lysine glycation sites using bi-profile bayes feature extraction. Comput Biol Chem 71:98–103
Lapolla A, Fedele D, Martano L, Arico’ NC, Garbeglio M, Traldi P, Seraglia R, Favretto D (2001) Advanced glycation end products: a highly complex set of biologically relevant compounds detected by mass spectrometry. J Mass Spectrom 36(4):370–378
Odani H, Iijima K, Nakata M, Miyata S, Kusunoki H, Yasuda Y, Hiki Y, Irie S, Maeda K, Fujimoto D (2001) Identification of N-omega-carboxymethylarginine, a new advanced glycation endproduct in serum proteins of diabetic patients: possibility of a new marker of aging and diabetes. Biochem Biophys Res Commun 285(5):1232–1236
Cho SJ, Roman G, Yeboah F, Konishi Y (2007) The road to advanced glycation end products: a mechanistic perspective. Curr Med Chem 14(15):1653–1671
Baynes JW (2001) The role of AGEs in aging: causation or correlation. Exp Gerontol 36(9):1527–1537
Ahmed N, Thornalley PJ (2003) Quantitative screening of protein biomarkers of early glycation, advanced glycation, oxidation and nitrosation in cellular and extracellular proteins by tandem mass spectrometry multiple reaction monitoring. Biochem Soc Trans 31:1417–1422
Nicolls MR (2004) The clinical and biological relationship between type II diabetes mellitus and Alzheimer’s disease. Curr Alzheimer Res 1(1):47–54
Munch G, Gerlach M, Sian J, Wong A, Riederer P (1998) Advanced glycation end products in neurodegeneration: more than early markers of oxidative stress? Ann Neurol S85–S88
Tang YR, Chen YZ, Canchaya CA, Zhang ZD (2007) GANNPhos: a new phosphorylation site predictor based on a genetic algorithm integrated neural network. Protein Eng Des Sel 20(8):405–412
Tang ZJ, Huang ZQ, Zhang XQ, Lao H (2017) Robust image hashing with multidimensional scaling. Signal Process 137:240–250
Xu Y, Li L, Ding J, Wu LY, Mai GQ, Zhou FF (2017) Gly-PseAAC: identifying protein lysine glycation through sequences. Gene 602:1–7
Johansen MB, Kiemer L, Brunak S (2006) Analysis and prediction of mammalian protein glycation. Glycobiology 16(9):844–853
Liu B, Wang XL, Lin L, Dong QW, Wang X (2008) A discriminative method for protein remote homology detection and fold recognition combining top-n-grams and latent semantic analysis. BMC Bioinform 9
Liu ZX, Wang YB, Gao TS, Pan ZC, Cheng H, Yang Q, Cheng ZY, Guo AY, Ren J, Xue Y (2014) CPLM: a database of protein lysine modifications. Nucleic Acids Res 42(D1):D531–D536
Lee TY, Chen SA, Hung HY, Ou YY (2011) Incorporating distant sequence features and radial basis function networks to identify ubiquitin conjugation sites. Plos One 6(3)
Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM (2010) Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78(2):365–380
Song J, Tan H, Shen H, Mahmood K, Bdyd SE, Webb GI, Whisstock JC (2010) Cascleave: towards more accurate prediction of caspase substrate cleavage sites
Shao J, Xu D, Tsai SN, Wang Y, Ngai SN (2009) Computational identification of protein methylation sites through bi-profile bayes feature extraction
Jia CZ, Liu T, Wang ZP (2013) O-GlcNAcPRED: a sensitive predictor to capture protein O-GlcNAcylation sites. Mol BioSyst 9(11):2909–2913
Jia CZ, He WY, Yao YH (2017) OH-PRED: prediction of protein hydroxylation sites by incorporating adapted normal distribution bi-profile Bayes feature extraction and physicochemical properties of amino acids. J Biomol Struct Dyn 35(4):829–835
Jia CZ, Liu TA, Chang AK, Zhai YY (2011) Prediction of mitochondrial proteins of malaria parasite using bi-profile Bayes feature extraction. Biochimie 93(4):778–782
Jia CZ, Zuo Y, Zou Q (2018) O-GlcNAcPRED-II: an integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 34(12):2029–2036
Jia CZ, Lin X, Wang ZP (2014) Prediction of protein S-nitrosylation sites based on adapted normal distribution bi-profile Bayes and Chou’s pseudo amino acid composition. Int J Mol Sci 15(6):10410–10423
Xu Y, Wen X, Shao XJ, Deng NY, Chou KC (2014) iHyd-PseAAC: predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. Int J Mol Sci 15(5):7594–7610
Xu Y, Ding J, Wu LY, Chou KC (2013) iSNO-PseAAC: predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. Plos One 8(2)
Chou KC (2001) Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins-Struct Funct Genet 43(3):246–255
Liu B, Liu FL, Wang XL, Chen JJ, Fang LY, Chou KC (2015) Pse-in-one: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res 43(W1):W65–W71
Liu B, Wu H, Chou KC (2017) Pse-in-one 2.0: an improved package of web servers for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nat Sci 9:67–91
Cao DS, Xu QS, Liang YZ (2013) Propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics 29(7):960–962
Kawashima S, Pokarowski P, Pokarowska M, Kolinski A, Katayama T, Kanehisa M (2008) AAindex: amino acid index database, progress report 2008. Nucleic Acids Res 36:D202–D205
Altschul S, Madden T, Schaffer A, Zhang JH, Zhang Z, Miller W, Lipman D (1998) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. FASEB J 12(8):A1326–A1326
Klock H, Buhmann JM (2000) Data visualization by multidimensional scaling: a deterministic annealing approach. Pattern Recognit 33(4):651–669
Lian Z, Godil A, Sun X, Zhang H (2010) Non-rigid 3D shape retrieval using multidimensional scaling and bag-of-features. In: Proceedings of IEEE 17th international conference on image processing CrossRefView record in scopus 3181–3184
Hsia C, Lee K, Chuang C, Chiu YH (2010) Multidimensional scaling for fast speaker clustering. In: Proceedings of the 7th international symposium on Chinese Spoken language processing. CrossRefView Record in Scopus 296–299
Jiang WY, Xu CQ, Pei L, Yu WX (2016) Multidimensional scaling-based TDOA localization scheme using an auxiliary line. IEEE Signal Proc Lett 23(4):546–550
France SL, Carroll JD (2011) Two-way multidimensional scaling: a review. IEEE Trans Syst Man Cybern C 41(5):644–661
Tung CW, Ho SY (2008) Computational identification of ubiquitylation sites from protein sequences. BMC Bioinfor 9
Vapnik VN (1999) An overview of statistical learning theory. IEEE Trans Neural Netw 10(5):988–999
Shao JL, Xu D, Hu LD, Kwan YW, Wang YF, Kong XY, Ngai SM (2012) Systematic analysis of human lysine acetylation proteins and accurate prediction of human lysine acetylation through bi-relative adapted binomial score Bayes feature representation. Mol BioSyst 8(11):2964–2973
Wee LJK, Simarmata D, Kam YW, Ng LFP, Tong JC (2010) SVM-based prediction of linear B-cell epitopes using Bayes feature extraction. BMC Genomics 11
Song L, Li DP, Zeng XX, Wu YF, Guo L, Zou Q (2014) nDNA-prot: identification of DNA-binding proteins based on unbalanced classification. BMC Bioinform 15
Li DP, Ju Y, Zou Q (2016) Protein folds prediction with hierarchical structured SVM. Curr Proteomics 13(2):79–85
Chang CC, Lin CJ (2011) LIBSVM: a library for support vector machines. ACM Trans Intell Syst Tecnol 2(3):27
Zhang CT (1995) Review: prediction of protein structural classes. Crit Rev Biochem Mol Biol 30:275–349
Chou KC (2011) Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol 273(1):236–247
Jia JH, Liu Z, Xiao X, Liu BX, Chou KC (2015) iPPI-Esml: an ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into PseAAC. J Theor Biol 377:47–56
Jiao YS, Du PF (2017) Predicting protein submitochondrial locations by incorporating the positional-specific physicochemical properties into Chou’s general pseudo- amino acid compositions. J Theor Biol 416:81–87
Ali F, Hayat M (2015) Classification of membrane protein types using voting feature interval in combination with Chou’s pseudo amino acid composition. J Theor Biol 384:78–83
Kumar R, Srivastava A, Kumari B, Kumar M (2015) Prediction of beta-lactamase and its class by Chou’s pseudo-amino acid composition and support vector machine. J Theor Biol 365:96–103
Behbahani M, Mohabatkar H, Nosrati M (2016) Analysis and comparison of lignin peroxidases between fungi and bacteria using three different modes of Chou’s general pseudo amino acid composition. J Theor Biol 411:1–5
Rahimi M, Bakhtiarizadeh MR, Mohammadi-Sangcheshmeh A (2017) OOgenesis_Pred: a sequence-based method for predicting oogenesis proteins by six different modes of Chou’s pseudo amino acid composition. J Theor Biol 414:128–136
Huang Y, Niu BF, Gao Y, Fu LM, Li WZ (2010) CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 26(5):680–682
Li W, Godzik A (2006) CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659
Jia JH, Liu Z, Xiao X, Liu BX, Chou KC (2016) iSuc-PseOpt: identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal Biochem 497:48–56
Jia JH, Liu Z, Xiao X, Liu BX, Chou KC (2016) iPPBS-opt: a sequence-based ensemble classifier for identifying protein-protein binding sites by optimizing imbalanced training datasets. Molecules 21(1)
Liu Z, Xiao X, Qiu WR, Chou KC (2015) iDNA-methyl: identifying DNA methylation sites via pseudo trinucleotide composition. Anal Biochem 474:69–77
Wei LY, Luan SS, Nagai LAE (2018) Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics
Su R, Wu HC, Xu B (2018) Developing a multi-dose computational model for drug-induced hepatotoxicity prediction based on toxicogenomics data. IEEE/ACM Trans Comput Biol Bioinform
Wei LY, Zhou C, Chen HG (2018) ACPred-FL: a sequence-based predictor based on effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 34(23):4007–4016
Wei LY, Xing PW, Shi GT (2017) Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE/ACM Trans Comput Biol Bioinform
Wei LY, Ding YJ, Su R (2018) Prediction of human protein subcellular localization using deep learning. J Parallel Distrib Comput 117:212–217
Wei LY, Su R, Wang B (2018) Integration of deep feature representations and handcrafted features to improve the prediction of N-6-methyladenosine sites. Neurocomputing 324:3–9
Chou KC (2015) Impacts of bioinformatics to medicinal chemistry. Med Chem 11(3):218–234
Cheng X, Zhao SG, Xiao X, Chou KC (2017) iATC-mISF: a multi-label classifier for predicting the classes of anatomical therapeutic chemicals. Bioinformatics 33(3):341–346
Qiu WR, Sun BQ, Xiao X, Xu ZC, Chou KC (2016) iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics 32(20):3116–3123
Chen W, Tang H, Ye J, Lin H, Chou KC (2016) iRNA-PseU: identifying RNA pseudouridine sites. Mol Ther-Nucl Acids 5
Chen W, Feng PM, Yang H, Ding H, Lin H, Chou KC (2017) iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget 8(3):4208–4217
Zhang CJ, Tang H, Li WC, Lin H, Chen W, Chou KC (2016) iOri-human: identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget 7(43):69783–69793
Chen JJ, Long R, Wang XL, Liu B, Chou KC (2016) dRHP-PseRA: detecting remote homology proteins using profile-based pseudo protein sequence and rank aggregation. Sci Rep 6
Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant (Number 71271034), the National Social Science Foundation of China under Grant (15CGL031), the Fundamental Research Funds for the Central Universities under Grant (Number 3132016306, 3132018160), the Program for Dalian High Level Talent Innovation Support under Grant (2015R063), the National Natural Science Foundation of Liaoning Province under Grant (20180550307), and the National Scholarship Fund of China for Studying Abroad.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, T., Yin, Q., Song, R. et al. Multidimensional scaling method for prediction of lysine glycation sites. Computing 101, 705–724 (2019). https://doi.org/10.1007/s00607-019-00710-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00607-019-00710-x