
Abstract
RNA editing is a post-transcriptional modification of an RNA nucleotide sequence. Until now, different RNA editing systems were found in the major eukaryotic clades. In the plant kingdom, RNA editing was mainly documented in the mitochondria and chloroplast genomes. However, variation among large taxonomic groups and the evolutionary trajectory in terms of intra- and inter-clades remains unclear. To gain a better understanding of RNA editing evolution, in this study, based on publicly available RNA-seq data across three clades (fern, gymnosperm, and angiosperm), we provided a detailed analysis of chloroplast RNA editing events and discussed its evolution in land plants. A total of 5203 editing sites were determined across 21 species after rigorous screening. We found that the clustering relations of RNA editing sites across 21 species agreed with the phylogenetic tree based on protein sequences approximately, and more editing sites occurred in early diverging lineages for all three clades, implying they shared similar evolutionary trajectories of editing loss. We observed that the average RNA editing level varied among species as well as genes, a lowest RNA editing level (~ 0.42) was detected in Selaginella moellendorffii; the highest editing level (~ 0.88) was detected in the atpA gene. The reduction of cytosine content with evolution detected in our study further suggested that the substitution of the genomic sequence was the significant driver of loss of editing for later-branching plants. Many of the identified sites in our study have not been previously reported and provided a valuable data set for the future research community. Our findings also provide valuable information for the evolution of RNA editing in plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
RNA editing is a post-transcription process through which the nucleotide specified in the genome template is modified to produce a different transcript, thus contributing to the restoration of functional protein and proteomic variation, and providing another mechanism for modulating gene expression (Walkley and Li 2017; Zahn 2017; Small et al. 2020). In the plant kingdom, RNA editing was first documented over a decade ago in the mitochondria of flowering plants (Covello and Gray 1989; Gualberto et al. 1989) and reported in chloroplast two years later (Hoch et al. 1991). There are two types of RNA editing in plants, the most common type is Cytosine-to-Uracil (C-to-U) conversion, and the infrequent type is U-to-C conversion that is reported only in ferns, mosses, and Lycopodiaceae (Gerke et al. 2020). Traditionally, RNA editing seems to occur only in organelle genome-encoded transcripts, and however, a recent study detected U-to-C RNA editing events for nuclear genes in Arabidopsis thaliana (Ruchika et al. 2021). RNA editing predominantly takes place at the first or second positions of codons, thereby affecting the translated regions of protein-coding transcripts. The amino acids specified by the altered codons generated by editing are generally conserved in evolution, suggesting that most RNA editing events can restore the evolutionarily conserved amino acid residues in mRNAs (Ichinose and Sugita 2017). RNA editing thereby is an important process to maintain essential functions of encoded proteins at the RNA level, for example, pigment deficiency in tobacco cybrids is caused by the editing failure of the plastid ATP synthase alpha-subunit (atpA) mRNA (Schmitz-Linneweber et al. 2005). Dynamic response of plant RNA editing to environmental factors was detected in previous studies (Miyata and Sugita 2004, Rodrigues et al. 2017, Xiong et al. 2017, Zhang et al. 2020). Plant RNA editing is regulated in tissue-specific pattern mediated by editosomes were also observed in several recent studies (Fang et al. 2021a, b; Fang et al. 2021a, b). Many factors are involved in plant RNA editing and considered to interact with one another to form a large protein complex, termed as editosome (Shikanai 2015). PLS subfamily members of pentatrico peptide repeat (PPR) proteins function in site recognition of the target cytosine, almost all the PPR proteins are localized in either chloroplasts or mitochondria where those proteins participate in different facets of RNA metabolism such as RNA splicing, RNA stability, and translational initiation (Yagi et al. 2013; Shikanai 2015). Multiple organelle RNA editing factors (MORF) family members are also components of the RNA editosome and are required for RNA editing at multiple editing sites in plants (Yagi et al. 2013; Yan et al. 2018; Xiong et al. 2022).
In the last two decades, RNAs were usually compared with their corresponding DNA templates to detect RNA editing sites, and however, this approach is time-consuming and prone to underestimate the numbers of editing sites. In recent years, the availability of large quantities of RNA sequencing data makes it possible to identify RNA editing sites and quantify their editing level on a large scale. This strategy allows a transcriptome-wide fast detection of editing sites and has enormous potential to deepen our knowledge of transcriptional processes in the plant. Indeed, with the growth of complete plant organellar genomes and related transcriptome data in the last decade, hundreds of editing sites have been identified in more and more plants (Lo Giudice et al. 2018, Lo Giudice et al. 2019; Oldenkott et al. 2020, Shtratnikova et al. 2020). But this strategy is also a challenging task due to its accuracy of mapping the RNA-seq reads against genomic sequence; hence, different bioinformatic strategies have been introduced to improve the detection accuracy of RNA editing sites (Sun et al. 2016; Wang et al. 2016; Zhang et al. 2017; Edera and Sanchez-Puerta 2021; Ichinose and Sugita 2021).
Evolutionary studies can help to understand the puzzling nature of RNA editing in plants. More and more recent studies demonstrated that RNA editing is a widespread phenomenon that occurred in various land plants, including the liverworts, mosses, hornworts, lycopods, ferns, and flowering plants (Edera et al. 2018; Ishibashi et al. 2019). However, no instance of RNA editing has yet been detected in algae, suggesting that RNA editing may have evolved in organelles only after the green plants established themselves on the land (Ichinose and Sugita 2017). The frequency of RNA editing sites varies from zero to hundreds across the plant kingdom, among land plants (Takenaka et al. 2013; Smith 2020). The unparalleled variation in RNA editing among fern plastomes was demonstrated in several recent studies (Smith 2020; Fauskee et al. 2021). Yet, variation in the frequency and editing level among gymnosperms and angiosperms remains unclear. Rare comparison study has been conducted in terms of intra- and inter-groups for chloroplast RNA editing except for ferns. To gain a better understanding of RNA editing in plant chloroplast, in this study, we chose diverse plant species that distributed in three main clades (fern, gymnosperm, and angiosperm) and determined thousands of editing sites based on the amount of RNA-seq data. The detailed comparison of RNA editing events provided valuable information for the evolution of plant RNA editing.
Materials and methods
Data acquisition
We selected 21 species across three clades (fern, gymnosperm, and angiosperm) for the detection of chloroplast RNA editing. For each species, the corresponding raw Illumina RNA reads were obtained from Sequence Read Archive (SRA) database at NCBI based on two criteria: (1) paired-end reads that possess higher mapping specificity were preferred; (2) RNA reads obtained from leaves of wild-type individuals were only selected. Besides, for each species, the reference file consisting of chloroplast genome sequences and corresponding gene annotation files were also downloaded from the GenBank database. Detailed information of RNA-seq data and reference files used in our study was listed in Online Resource 1.
Read mapping and SNP calling
The identification process of RNA editing sites can be decomposed into three steps: first read alignment, second the SNP calling, and third detection of RNA editing sites. For each species, to increase sequencing depth, we merged all the replicates into one sample. The quality control of paired-end Illumina sequencing data was evaluated first by NGSQCToolkit (Patel and Jain 2012); low-quality sequence data were filtered out (cutOffQualScore < 20). RNA reads from each species were then mapped to reference using hisat2 software under default parameters (Kim et al. 2015). Afterward, the alignment results were sorted, removed duplicates, indexed, and sorted by using SAMtools (Li et al. 2009). Finally, the resulting BAM file was then used to call DNA/RNA variants using bcftools, VCF files that describe transcriptome variation were generated (Danecek and McCarthy 2017).
Detection of RNA editing sites
For each species, based on the SNP-calling results (in “VCF” format) and genome annotation files (in “tbl” format), RNA editing sites were identified under default parameter values by using the REDO tool (Wu et al. 2018). REDO is a comprehensive application tool for identifying RNA editing events in plant organelles based on variant call format files from RNA sequencing data. REDO works require only three input files: a file that contains the SNP-calling results (records for all sites), the genome sequence file of organelle reference (FASTA format), and its corresponding gene annotation file (feature table file, www.ncbi.nlm.nih.gov/projects/Sequin/table.html). Finally, all raw RNA editing sites were detected, and meanwhile, their corresponding annotation information files were also generated.
Filtration of RNA editing sites
Regarding the high false positive of raw editing sites, we used two different levels of criteria to filter the raw RNA editing sites, one filter criterion (‘Filter1’) is loose: (1) quality control filter (MQ > 255), the low-quality sites are filtered out according to the reads quality; (2) total reads depth filter (DP > 4); (3) Fisher’s exact test filter (p value < 0.05), the significance for a given RNA editing site (alt reads, ref reads) by comparing its expected levels (0, alt reads + ref reads) using the Fisher exact test; (4) multiple alt filter, only the variant with one alt allele is retained. Another filter criterion (‘Filter2’) is strict, besides fulfilling the ‘Filter1,’ we increased the total reads depth up to 10, and sites with more than one altered reads were kept. To minimize the false positives produced by the automated approach, we also manually examined all raw editing sites, only kept the C-to-U and U-to-C editing types, and excluded other mismatches, such as A-to-C and T-to-A. To evaluate the reliability of editing, for each species, we also used PREPACT tool to predict potential RNA editing events supplying with entire chloroplast genomes as input files, with a filter threshold of at least 80% of the references under BLASTX mode (Lenz and Knoop 2013).
Comparing RNA editing sites
All the filtered RNA editing sites detected in 21 species were used for further statistics and feature analysis, including statistics of editing number, editing type, codon position, amino acid changes, involved genes, and so on. To decipher the distribution of RNA editing frequency across different species, the top 30 genes with the most editing sites across 21 species were selected, and cluster analysis and heatmap plotting were conducted based on the matrix of RNA editing numbers. The CDS sequences of the top 30 genes across 21 species were concatenated and subjected to alignments and phylogenetic tree construction using RAxML (Stamatakis 2014). Meanwhile, the RNA editing level of the top 30 genes was also subjected to statistical analysis. The value of RNA editing level at one site was expressed as the proportion between edited transcripts and total transcripts. If one site was edited, the C/G base (wild type) should be altered to the T/A base (edited type), since one editing site could be detected hundreds of times via sequencing, the number of wild types (C/G) or edited type (T/A) of bases could then be counted at this particular site, then the editing level at one site could then be calculated by the formula: depth of edited bases (T and A)/total read depth of bases. Values of the editing level matrix were normalized by subtracting the row-wise mean from the values in each row of data and multiplying all values in each row of data by the value of standard deviation. For each clade (fern, gymnosperm, and angiosperm), a heatmap was plotted across all of its species using “pheatmap” function in R, respectively, the distance matrix of different samples was calculated using “dist” function with the default Euclidean method, and the hierarchical clustering was computed using “hclust” function.
Comparing cytosines content
Considering protein-coding genes varied among different species, we picked out shared edited genes for the statistics of cytosine content across 21 species. For each shared edited gene, we extracted its CDS sequence and calculated the ratio of cytosine content. For each two of the clades (fern, gymnosperm, and angiosperm), pairwise comparisons of average cytosines content were conducted. A two-tailed Wilcoxon rank-sum test was used.
Illustration of atpA RNA editing sites
The gene sequences of ATP synthase alpha-subunit gene (atpA) across 21 species were collected, the intersection of all the species' RNA editing sites of atpA was concatenated for alignment and annotated, sequence logo of atpA gene was produced by WebLogo (Crooks et al. 2004), alignment was conducted using ClustalW that implemented in MEGA5 under default parameters (Kumar et al. 2018).
Results
Identification of RNA editing sites
A series of species that represent distant evolutionary clades were selected judging by two criteria, one is enough transcriptomic data of leaf tissue in the SRA database, and another is the availability of sequenced chloroplast genome. Hence, 21 species, consisting of 6 ferns, 4 gymnosperms, and 11 angiosperms (Table 1), and corresponding 317 SRA accessions were chosen finally. Detailed information of SRA data and chloroplast genome accessions was listed in Online Resource 1. Mapping results (Online Resource 6a) showed that RNA-seq data volume varied among different species, compared with angiosperms, ferns and gymnosperms have lower mapping depths, especially for Adiantum aleuticum and Histiopteris incisa, indicating that the actual number of RNA editing in ferns and gymnosperms might be underestimated. We also observed that the read density also varied widely among different genomic regions, ranging from less than 30 to more than 800 in a few species, demonstrating varied expression levels of genes or sequence biases. Based on the results of RNA-seq data mapping and SNP calling, an automated bioinformatics pipeline implemented in REDO tool (Wu et al. 2018) was conducted under default thresholds. Consequently, there were a total of 6,011 raw editing sites located chloroplast genome detected in the leaf. Sequence mismatches that accord with RNA editing occasionally appeared, hence, we manually examined all mismatches to eliminate false positives, only kept C-to-U and U-to-C editing types. We used two different levels of criteria to filter the raw variants, one filter criterion (‘Filter1,’ p value < 0.05, DP > 4, one alt allele) is loose. Another filter criterion (‘Filter2’) is strict, besides fulfilling the ‘Filter1,’ we increased the total reads depth up to 10, and only the sites with more than one altered reads were kept. The results showed that ‘Filter1’ reduced the number of RNA editing sites from 6011 to 5203, and the ‘Filter2’ reduced the sites from 6011 to 4433, as shown below. Actually, for the reason of varied RNA-seq data volume among different species, we adopted a relatively lower criteria (‘Filter1’) to keep enough RNA editing sites in ferns in the subsequent analysis. To evaluate the reliability of the results, PREPACT webserver (Lenz et al. 2018) was also used to verify the editing sites. We found that the distribution of the predicted number of RNA editing based on PREPACT agreed with that of our prediction based on RNA-seq data basically (see Fig. 1), reflecting our pipeline offered high performance with reliable results. To describe the attributes of RNA editing sites, we illustrated one example of samples of Adiantum aleuticum (Online Resource 6b), which depicts the reliability of RNA editing sites by REDO tools statistically.

Comparison of predicted numbers of RNA editing sites by RNA-seq data with PREPACT website server
Distribution of RNA editing across three clades
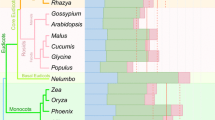
After manual inspection and elimination of mismatches, filtered RNA editing sites across 21 species were screened out, the summary is listed in Table 1, Online Resource 2, and detailed corresponding annotation information was produced simultaneously, as listed in Online Resource 3. All the filtered editing sites were located in 1,038 genes across all species, the average percent of edited genes for the three clades (ferns, gymnosperms, and angiosperms) is 0.85, 0.61, and 0.39, respectively. Compared to the latter two clades, more sites and genes were edited in chloroplast transcripts from ferns remarkably (Fig. 2a). In ferns, Selaginella moellendorffii has the largest number of editing sites, up to 1,316, which is nearly 100-fold more abundant than that of flowering plants, nearly all the chloroplast genes (77 genes, ~ 96%) suffered effective editing. Differ from Selaginella moellendorffii that exclusively belongs to Lycopsida, the other five fern plants are members of Leptosporangiopsida, have relatively smaller numbers of editing sites, represented by Cyrtomium fortunei, which owned the second-largest number of editing sites, with 551 editing sites and 79 edited genes (~ 91%). Whereas for gymnosperms, the average number of editing sites and percent of edited genes were all less than that of ferns. Compared with the other three gymnosperms, Ginkgo biloba has the most editing sites, with 291 editing sites and 68 edited genes (~ 81%). On the opposite end, angiosperms have the lowest average numbers of editing sites, and only a part of genes were effectively edited, lower than 50%. It was noticeable that Liriodendron tulipifera and Nelumbo nucifera distinguished them from other angiosperms with more editing events. A total of 260 editing sites were detected in Liriodendron tulipifera, which is nearly threefold more abundant than that of other angiosperms and gymnosperms except for Ginkgo biloba, the percent of edited genes was up to 75%, well above the average. The numbers of editing sites among the other 9 angiosperms showed no significant differences. The above results illustrated the diversity of RNA editing distribution across the three clades, early diverging clades/species showed higher numbers of editing sites compared with that of later-diverging. The statistics of RNA editing sites showed that 21.97%, 67.16%, and 10.58% of sites were edited at first, second, and third codon positions, respectively (Fig. 2b). In terms of editing types, C-to-U was the dominant editing type (nearly ~ 95.1%), the next is U-to-C type, and other mismatch types were rare (Fig. 2c). The statistics of editing types showed that the majority (~ 95%) of the editing events resulted in non-synonymous codon changes, and the changed amino acids tend to be hydrophobic, the frequency of changes from hydrophilic to hydrophobic was the highest, followed by changes from hydrophobic to hydrophobic (Fig. 2d). The most common amino acid change types were Ser-to-Leu and Pro-to-Leu, serine is hydrophilic, whereas leucine and proline are both hydrophobic. The above results demonstrated that the RNA editing exhibited a selective advantage in the overall increase in hydrophobicity of the resulting proteins, which was also in good agreement with our previous studies (Zhang et al. 2020).

The statistics of identified RNA editing sites across 21 species. a The total number of editing sites for each species. Stacked bars depict numbers of nonsilent editing sites (blue), and silent editing sites (red), respectively. The symbol of each species is represented by its first word of the genus name, such as Oryza – Oryza sativa. b Codon position statistics of RNA editing sites. c Statistics of 12 nucleotide substitution types, each pair is classified by two-color bars (blue and red). d Statistics of amino acid change types
Variability of RNA editing among species and genes
To further explore the evolutionary trajectory of RNA editing for the three clades, for each gene, we summed the number of its editing sites across 21 species (Online Resource 4) and picked out the top 30 genes with the most editing sites for cluster analysis. Based on the matrix of numbers of editing sites across 21 species, a hierarchically clustered heatmap is plotted in Fig. 3. The clustering relations showed that the 21 species were divided into three subgroups. The remarkable abundance of editing events makes two exceptional species (Selaginella moellendorffii and Liriodendron tulipifera) cluster far away from their own clades. We found that not every gene was edited in all the species, by grouping the genes based on their function, genes encoding membrane subunit of the NADH dehydrogenase-like complex (ndh) exhibited larger average numbers of editing sites, this is consistent with previous studies that RNA editing occurred preferentially in genes encoding membrane-bound proteins under strong selection (Mower and Palmer 2006). Whereas ribosomal subunit genes showed lower numbers of editing sites. Due to the well-studied background and abundant editing sites in the plant, subunit of the NADH dehydrogenase-like complex gene (ndhB) is assumed to be a good case for the study of RNA editing evolution, in our study, ndhB was confirmed to possess the most editing sites, with 333 editing sites spread across 17 species. Despite this, there is a biased distribution of RNA editing sites in ndhB among three clades. In ferns, 50 sites were detected in Selaginella moellendorffii, only 4–17 sites in the other five ferns; in angiosperms, there were about 20 editing sites for each of the 11 angiosperm species; in gymnosperms, RNA editing in ndhB was only detected in Ginkgo biloba, for Picea abies and Pinus massoniana, no ndhB gene annotated in their chloroplast genome, whereas for Cycas revoluta, no RNA editing events were detected in its ndhB gene, which may result from loss of editing or too low depth around genomic regions of its ndhB gene. Based on alignments of merged protein sequence for the top 30 genes across 21 species, a phylogenetic tree by Maximum Likelihood method was constructed, as shown in Fig. 3b, which showed that the clustering relations agreed with that of the matrix of numbers of editing sites roughly. The above observation implied the conservation of RNA editing evolution across different clades. Hierarchically clustered heatmaps of numbers of RNA editing sites from all/each clade were shown in Online Resource 6c-f, respectively.

Distribution of numbers of RNA editing sites in top 30 genes with most editing sites across three clades. a A hierarchical cluster of numbers of RNA editing sites is shown above, the x-axis represents genes, the y-axis represents species, and species from different clades are highlighted by different colors. The total number of editing sites of each gene is shown below correspondingly. b Phylogenetic tree by Maximum Likelihood method based on alignments of merged protein sequence for top 30 genes across three clades, species from different clades are highlighted by different colors
Variability of RNA editing level
RNA editing level was used to measure to what extent the edited transcripts among all transcriptomes for one gene. In this study, we explored the distribution of editing levels among three aspects: codon positions, species, and edited genes. We observed that the distribution of RNA editing levels for three codons did not comply with the normal distribution, featuring a peak around ~ 0.2 and fat tails, as shown in Fig. 4. The comparison between codon positions showed that the average editing level in the second codon position (~ 0.72) is higher than that of the first (~ 0.63) and third codon positions (~ 0.61) significantly, suggesting non-synonymous substitution occurred in the second codon position tend to be effectively edited, it was higher editing level that dominated the landscape of RNA editing. In addition, we also found that the average editing level also varied widely across 21 species, ranging from 0.42 to 0.85 (Fig. 5a), the trend of plotting of filtered sites was consistent with that of raw sites roughly. Selaginella moellendorffii has the lowest editing level (~ 0.42), far below that of other species. In gymnosperms and angiosperms, Ginkgo biloba and Nelumbo nucifera had the lowest editing efficiencies (~ 0.6), respectively. It seemed that abundant editing sites detected in those early branching plants might have a negative impact on their editing level. However, as an ancient plant in angiosperms, Liriodendron tulipifera was one exception, with an editing level up to 0.81. The editing level was also analyzed in each gene individually, we averaged the RNA editing level among the top 30 genes across 21 species, and the result also demonstrated the diversity of distribution, as shown in Fig. 5b, Maturase K gene (matK) has the lowest editing level (~ 0.5); oppositely, editing level of atpA gene was the highest, up to 0.88.

Distribution of RNA editing level per codon position. a Frequency distribution of RNA editing level per codon position, the x-axis represents RNA editing level, and the y-axis represents frequency. b Boxplots of RNA editing level per codon position, the x-axis represents codon position, and the y-axis represents RNA editing level. Asterisks denote significant differences: *p value < 0.05; **p value < 0.01

Distribution of average RNA editing level. a Distribution of average editing level of all identified RNA editing sites in each species. Gray, blue and red plots depict average editing levels for sites of raw, ‘Filter1’ and ‘Filter2,’ respectively. b Distribution of average editing level of top 30 genes, the above shows average editing level of top 30 genes, the below shows numbers of RNA editing sites in top 30 genes correspondingly
Reduced cytosines content with the evolution of plants
Considering the large differences in the scale of RNA editing events along with evolution, we analyzed the nucleic acid base composition of edited genes shared by the 21 species. There were a total of 51 genes shared in all the chloroplast genomes of 21 species. The percent of cytosines for each gene was calculated by the formula: the number of cytosines (C)/total number of bases (A/T/C/G), as listed in Online Resource 5. Afterward, the cytosines content of genes was averaged for each clade, and remarkable significances (p value < 0.05) were detected for pairwise comparisons between every two clades except for gymnosperms-angiosperms, as shown in Figure 6a. The percent of cytosines in ferns was far below that of angiosperms, followed by gymnosperms. The highest average of cytosines content was found in Selaginella moellendorffii (~ 0.26), and the lowest average was found in Glycine max (~ 0.17). Ginkgo biloba and Liriodendron tulipifera have the highest averages (~ 0.19) in their own clade, showing a positive correlation with their high numbers of RNA editing sites. We further compared the cytosines content for each gene across the 21 species, as shown in Figure 6b, which demonstrated that the percent of cytosine dramatically declined roughly along with evolution with a few exceptions, such as 50S ribosomal protein L23 (rpl23) gene. One striking example was photosystem II protein I (psbI) gene, which has the highest percent of cytosines in Selaginella moellendorffii (~ 0.37), and dropped to about 0.18 in other species. The above results indicated that the declination of cytosine content with evolution was the significant driver of the loss of editing sites for later-branching plants.

The statistics of cytosine content in 51 shared edited genes across 21 species. a Bar plots of pairwise comparisons of cytosine content between every two clades. A two-tailed Wilcoxon rank-sum test was used. b Line plots of cytosine content for each shared edited gene across 21 species. The average cytosine content of all edited genes for each species is indicated by bold red lines. Three genes (rpl23, psbI, and psbN) are indicated by black arrows, species from different clades are highlighted by different colors
Illustration of atpA RNA editing
We illustrated the RNA editing of atpA gene to help understand the evolution trajectory vividly. All editing sites identified in atpA gene were marked by a yellow color, as shown in Fig. 7, which demonstrated that the numbers of editing sites, as well as cytosine content, declined from ferns to angiosperms. We found that editing sites at the third codon position only occurred in certain species and were poorly conserved, for example, RNA editing in locus-a (Fig. 6) only occurred in Adiantum aleuticum despite the existence of cytosine in other fern members, indicating that synonymous substitutions at third codon position were not conserved. In contrast, RNA editing at the second codon position was relatively conserved, RNA editing in locus-b and -c occurred in all three clades, whereas in locus-d, RNA editing occurred in two members of gymnosperms, and the base types of other 19 species were all corrected to thymine in the genome; furthermore, we found many sites occurred in certain species for the three clades, implying the independent origins of RNA editing across different clades. In extreme cases, when all the cytosines with potential editing capacity of atpA gene were completely edited in fern plants, the overall uracil levels in edited transcripts will increase to the comparable levels of angiosperms and gymnosperms on the whole. However, in ferns’ transcripts, most of the sites were partly edited, and hence, their uracil level is still lower than that of angiosperms and gymnosperms.

Illustration of RNA editing sites from atpA gene across 21 species. a Alignments of RNA editing sites of atpA gene, and edited cytosines are marked by yellow. Codon positions (1, 2, and 3) are labeled below each column of the site with blue, red, and green colors. Locus-a, b, c, and d were indicated at the bottom of codon positions. b Sequence logo of RNA editing sites from atpA gene
Discussion
As a post-transcription process, RNA editing can modify the genome template to produce a different transcript (Ichinose and Sugita 2017). Numerous studies have proved that RNA editing occurred in nearly all plants in the kingdom, RNA editing exhibited dynamics response to different stress factors and development stages, such as flower development and male sterile (Miyata and Sugita 2004, Brenner et al. 2019, Lo Giudice et al. 2019, Zhang et al. 2020, Fang et al. 2021a, b). RNA editing may also cause secondary structure transformation of transcripts (Farre et al. 2012). In this study, to gain a better understanding of RNA editing in plant chloroplast, we collected a large amount of RNA-seq data and performed a series of procedures to investigate RNA editing in 21 diverse plants distributed in three clades. We determined a total of 5203 editing sites located in chloroplast genes across 21 species and quantified their editing level, demonstrating the powerfulness of the bioinformatics approach in studying RNA editing, many of the identified sites have not been previously reported, thus providing valuable data resource for future research. We found that the clustering relation of numbers of RNA editing sites agreed with the phylogenetic tree based on gene sequences approximately, verifying that RNA editing across the plant kingdom is comparatively conservative and accords with laws of evolution roughly. A great deal of variability of RNA editing numbers, as well as the RNA editing level, was detected among species, genes, and codon positions.
In total, the numbers of editing sites declined with the evolution of plants, and editing events occur more often in the early diverging plant than later-branching ones for each clade. As one of the most ancient flowering trees, Liriodendron tulipifera possessed the highest number of editing sites in angiosperms. In gymnosperms and ferns, one gymnosperm plant, Ginkgo biloba, and one fern plant, Selaginella moellendorffii, both exhibited ancient features, such as higher numbers of sites. The above observation was consistent with a previous study, which found that the profile of chloroplast RNA editing of Amborella represented an ancestral RNA editing pattern in angiosperms (Ishibashi et al. 2019). These observations implied that RNA editing may break out in early branching plants from different clades simultaneously and suffer a lot of loss during evolution. We also found that a reasonable percentage of editing sites occurred in certain clades and were lost in other clades whose cytosine already corrected to thymine in the genome. The declination of cytosine content with evolution detected in our study further indicated that the substitution of genomic sequence (C-to-T) was the significant driver of loss of editing for later-branching plants. For certain species, its lack of editing at a few genes may be explained by two reasons, one is the absence of the genes that are annotated in the chloroplast genome, and another is no RNA editing occurred in the genes. We found that new editing sites also occurred in certain higher plants occasionally that lacked in early diverging species, indicating the diversity of RNA editing evolution. Even though the substitution of C-to-T in the genome occurred in all gymnosperms and certain angiosperms, there were still some angiosperms that needed the RNA editing strategy to make functional proteins, such as the locus-c of atpA gene.
RNA editing level also showed variability in three aspects: codon positions, species, and edited genes. It seemed that abundant editing sites detected in early branching plants have a negative impact on their editing levels, such as Selaginella moellendorffii, Ginkgo biloba, and Nelumbo nucifera, these early diverging species had a relatively lower editing level. It is speculated that the efficiency of RNA editing might be at the mercy of the limited expression of RNA editing factors. RNA editing activities also showed a varied degree of conservation among different codon positions, the higher conservation and editing level in the second codon position suggested non-synonymous substitution tended to be effectively edited, and it was a higher editing level that dominated the landscape of RNA editing. To a certain degree, the discrepancy in editing level among genes reflected their importance of function.
Until now, there are two viewpoints about the nature of RNA editing: one is a contribution to variations in proteomic sequence and modulating gene expression; another point thinks that RNA editing in plants might be a repair mechanism to correct genomic point mutations at the post-transcription level, thus increasing the substitution rate that is extremely low in the organellar genome (Takenaka et al. 2014; Tang and Luo 2018). Previous studies revealed that organelle genomes have a slower evolutionary rate than the nuclear genome, thus accumulating many T-to-C mutations that constitute a prerequisite for the generation of plant organellar RNA editing (Barbrook et al. 2010), which could correct those T-to-C mutations to restore the evolutionarily conserved amino acid residues in mRNAs. During the evolution of land plants, most mutations would have been finally corrected to thymine in the genome, especially for higher plants, eliminating the need for editing at certain sites, and thus, the number of editing sites showed a remarkable reduction. But some sites still need to be edited to thymine or remain to be cytosine for coding for different amino acids. Hence, the genome mutations are the driving force behind the evolution of editing sites in plants, and the increasing modification of C-to-T at the genome level might be more accurate to describe the evolution trajectory instead of RNA editing.
Conclusions
In this study, based on a bioinformatics pipeline, we provided a detailed analysis of RNA editing events in chloroplast genomes across three main plant clades and discussed the evolution of RNA editing in land plants. Our study represents a valuable data set for the research community and thus helps understand the puzzling nature of RNA editing in plants.
Data Availability and Material
The datasets analyzed during the current study are available in the NCBI repository (https://www.ncbi.nlm.nih.gov/) and are available as Online Resource 1–6 associated with this manuscript.
References
Barbrook AC, Howe CJ, Kurniawan DP, Tarr SJ (2010) Organization and expression of organellar genomes. Philos Trans Roy Soc London B Biol Sci 365:785–797. https://doi.org/10.1098/rstb.2009.0250
Brenner WG, Mader M, Muller NA, Hoenicka H, Schroeder H, Zorn I, Fladung M, Kersten B (2019) High level of conservation of mitochondrial RNA editing sites among four populus species. Genes Genom Genet 9:709–717. https://doi.org/10.1534/g3.118.200763
Covello PS, Gray MW (1989) Rna editing in plant-mitochondria. Nature 341:662–666. https://doi.org/10.1038/341662a0
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190. https://doi.org/10.1101/gr.849004
Danecek P, McCarthy SA (2017) BCFtools/csq: haplotype-aware variant consequences. Bioinformatics 33:2037–2039. https://doi.org/10.1093/bioinformatics/btx100
Edera AA, Sanchez-Puerta MV (2021) Computational detection of plant RNA editing events. Meth Molec Biol 2181:13–34. https://doi.org/10.1007/978-1-0716-0787-9_2
Edera AA, Gandini CL, Sanchez-Puerta MV (2018) Towards a comprehensive picture of C-to-U RNA editing sites in angiosperm mitochondria. Pl Molec Biol 97:215–231. https://doi.org/10.1007/s11103-018-0734-9
Fang J, Jiang XH, Wang TF, Deng ZY, Zhang AD, Zhang XJ (2021a) Dynamic landscape of mitochondrial Cytidine-to-Uridine RNA editing in tobacco (Nicotiana tabacum) shows its tissue specificity. Pl Cell Tiss Org 148:363–376. https://doi.org/10.1007/s11240-021-02194-0
Fang J, Jiang XH, Wang TF, Zhang XJ, Zhang AD (2021b) Tissue-specificity of RNA editing in plant: analysis of transcripts from three tobacco (Nicotiana tabacum) varieties. Pl Biotechnol Rep 15:471–482. https://doi.org/10.1007/s11816-021-00692-3
Farre JC, Aknin C, Araya A, Castandet B (2012) RNA editing in mitochondrial trans-introns is required for splicing. PLoS ONE 7:e52644. https://doi.org/10.1371/journal.pone.0052644
Fauskee BD, Sigel EM, Pryer KM, Grusz AL (2021) Variation in frequency of plastid RNA editing within Adiantum implies rapid evolution in fern plastomes. Amer J Bot 108:820–827. https://doi.org/10.1002/ajb2.1649
Gerke P, Szovenyi P, Neubauer A, Lenz H, Gutmann B, McDowell R, Small I, Schallenberg-Rudinger M, Knoop V (2020) Towards a plant model for enigmatic U-to-C RNA editing: the organelle genomes, transcriptomes, editomes and candidate RNA editing factors in the hornwort Anthoceros agrestis. New Phytol 225:1974–1992. https://doi.org/10.1111/nph.16297
Gualberto JM, Lamattina L, Bonnard G, Weil JH, Grienenberger JM (1989) RNA editing in wheat mitochondria results in the conservation of protein sequences. Nature 341:660–662. https://doi.org/10.1038/341660a0
Hoch B, Maier RM, Appel K, Igloi GL, Kossel H (1991) Editing of a chloroplast mRNA by creation of an initiation codon. Nature 353:178–180. https://doi.org/10.1038/353178a0
Ichinose M, Sugita M (2017) RNA editing and its molecular mechanism in plant organelles. Genes (Basel) 8:5. https://doi.org/10.3390/genes8010005
Ichinose M, Sugita M (2021) Substitutional RNA editing in plant organelles. Meth Molec Biol 2181:1–12. https://doi.org/10.1007/978-1-0716-0787-9_1
Ishibashi K, Small I, Shikanai T (2019) Evolutionary model of plastidial RNA editing in angiosperms presumed from genome-wide analysis of Amborella trichopoda. Pl Cell Physiol 60:2141–2151. https://doi.org/10.1093/pcp/pcz111
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nature Meth 12:357–360. https://doi.org/10.1038/nmeth.3317
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Molec Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lenz H, Knoop V (2013) PREPACT 2.0: Predicting C-to-U and U-to-C RNA Editing in organelle genome sequences with multiple references and curated RNA editing annotation. Bioinform Biol Insights 7:1–19. https://doi.org/10.4137/BBI.S11059
Lenz H, Hein A, Knoop V (2018) Plant organelle RNA editing and its specificity factors: enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinf 19:255. https://doi.org/10.1186/s12859-018-2244-9
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome S, Genome Project Data Processing (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Lo Giudice C, Pesole G, Picardi E (2018) REDIdb 3.0: a comprehensive collection of RNA editing events in plant organellar genomes. Frontiers Pl Sci 9:482. https://doi.org/10.3389/fpls.2018.00482
Lo Giudice C, Hernandez I, Ceci LR, Pesole G, Picardi E (2019) RNA editing in plants: a comprehensive survey of bioinformatics tools and databases. Pl Physiol Biochem 137:53–61. https://doi.org/10.1016/j.plaphy.2019.02.001
Miyata Y, Sugita M (2004) Tissue- and stage-specific RNA editing of rps 14 transcripts in moss (Physcomitrella patens) chloroplasts. J Pl Physiol 161:113–115. https://doi.org/10.1078/0176-1617-01220
Mower JP, Palmer JD (2006) Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Molec Genet Genomics 276:285–293. https://doi.org/10.1007/s00438-006-0139-3
Oldenkott B, Burger M, Hein AC, Jorg A, Senkler J, Braun HP, Knoop V, Takenaka M, Schallenberg-Rudinger M (2020) One C-to-U RNA editing site and two independently evolved editing factors: testing reciprocal complementation with DYW-type PPR Proteins from the Moss Physcomitrium (Physcomitrella) patens and the Flowering Plants Macadamia integrifolia and Arabidopsis. Pl Cell 32:2997–3018. https://doi.org/10.1105/tpc.20.00311
Patel RK, Jain M (2012) NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7:e30619. https://doi.org/10.1371/journal.pone.0030619
Rodrigues NF, da Fonseca GC, Kulcheski FR, Margis R (2017) Salt stress affects mRNA editing in soybean chloroplasts. Genet Molec Biol 40:200–208. https://doi.org/10.1590/1678-4685-Gmb-2016-0055
Ruchika C, Okudaira MS, Tsukahara T (2021) Genome-wide identification of U-To-C RNA editing events for nuclear genes in Arabidopsis thaliana. Cells 10:635. https://doi.org/10.3390/cells10030635
Schmitz-Linneweber C, Kushnir S, Babiychuk E, Poltnigg P, Herrmann RG, Maier RM (2005) Pigment deficiency in nightshade/tobacco cybrids is caused by the failure to edit the plastid ATPase alpha-subunit mRNA. Pl Cell 17:1815–1828. https://doi.org/10.1105/tpc.105.032474
Shikanai T (2015) RNA editing in plants: machinery and flexibility of site recognition. Bba-Bioenergetics 1847:779–785. https://doi.org/10.1016/j.bbabio.2014.12.010
Shtratnikova VY, Schelkunov MI, Penin AA, Logacheva MD (2020) Mitochondrial genome of the nonphotosynthetic mycoheterotrophic plant Hypopitys monotropa, its structure, gene expression and RNA editing. PeerJ 8:e9309. https://doi.org/10.7717/peerj.9309
Small ID, Schallenberg-Rudinger M, Takenaka M, Mireau H, Ostersetzer-Biran O (2020) Plant organellar RNA editing: what 30 years of research has revealed. Pl J 101:1040–1056. https://doi.org/10.1111/tpj.14578
Smith DR (2020) Unparalleled variation in RNA editing among Selaginella Plastomes. Pl Physiol 182:12–14. https://doi.org/10.1104/pp.19.00904
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Sun Y, Li X, Wu D, Pan Q, Ji Y, Ren H, Ding K (2016) RED: a Java-MySQL software for identifying and visualizing RNA editing sites using rule-based and statistical filters. PLoS ONE 11:e0150465. https://doi.org/10.1371/journal.pone.0150465
Takenaka M, Zehrmann A, Verbitskiy D, Hartel B, Brennicke A (2013) RNA editing in plants and its evolution. Annual Rev Genet 47:335–352. https://doi.org/10.1146/annurev-genet-111212-133519
Takenaka M, Zehrmann A, Verbitskiy D, Glass F, Brehme N, Brennicke A (2014) The world of RNA editing in mitochondria and chloroplasts in plants. Biocell 38:58–59
Tang W, Luo C (2018) Molecular and functional diversity of RNA editing in plant mitochondria. Molec Biotechnol 60:935–945. https://doi.org/10.1007/s12033-018-0126-z
Walkley CR, Li JB (2017) Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol 18:205. https://doi.org/10.1186/s13059-017-1347-3
Wang Z, Lian J, Li Q, Zhang P, Zhou Y, Zhan X, Zhang G (2016) RES-Scanner: a software package for genome-wide identification of RNA-editing sites. Gigascience 5:37. https://doi.org/10.1186/s13742-016-0143-4
Wu S, Liu W, Aljohi HA, Alromaih SA, Alanazi IO, Lin Q, Yu J, Hu S (2018) REDO: rna editing detection in plant organelles based on variant calling results. J Comput Biol 25:509–516. https://doi.org/10.1089/cmb.2017.0214
Xiong J, Tao T, Luo Z, Yan S, Liu Y, Yu X, Liu G, Xia H, Luo L (2017) RNA editing responses to oxidative stress between a wild abortive type male-sterile line and its maintainer line. Frontiers Pl Sci 8:2023. https://doi.org/10.3389/fpls.2017.02023
Xiong Y, Fang J, Jiang X, Wang T, Liu K, Peng H, Zhang X, Zhang A (2022) Genome-wide analysis of multiple Organellar RNA editing factor (MORF) Family in Kiwifruit (Actinidia chinensis) reveals its roles in chloroplast rna editing and pathogens stress. Plants (Basel) 11:146. https://doi.org/10.3390/plants11020146
Yagi Y, Tachikawa M, Noguchi H, Satoh S, Obokata J, Nakamura T (2013) Pentatricopeptide repeat proteins involved in plant organellar RNA editing. RNA Biol 10:1419–1425. https://doi.org/10.4161/rna.24908
Yan J, Zhang Q, Yin P (2018) RNA editing machinery in plant organelles. Sci China Life Sci 61:162–169. https://doi.org/10.1007/s11427-017-9170-3
Zahn LM (2017) The evolution of edited RNA transcripts. Science 355:1278–1279. https://doi.org/10.1126/science.355.6331.1278-d
Zhang F, Lu Y, Yan S, Xing Q, Tian W (2017) SPRINT: an SNP-free toolkit for identifying RNA editing sites. Bioinformatics 33:3538–3548. https://doi.org/10.1093/bioinformatics/btx473
Zhang A, Jiang X, Zhang F, Wang T, Zhang X (2020) Dynamic response of RNA editing to temperature in grape by RNA deep sequencing. Funct Integr Genomics 20:421–432. https://doi.org/10.1007/s10142-019-00727-7
Acknowledgements
We would like to thank the members of Bioinformatics Group of Wuhan Botanical Garden, Chinese Academy of Sciences, China, for the discussion and suggestion to improve the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, Grant Numbers 32070682, the National Science & Technology Innovation Zone Project, grant numbers 1716315XJ00200303 and 1816315XJ00100216, CAS Pioneer Hundred Talents Program, and the Key Research and Development Program of Hubei Province (2022BBA0076).
Author information
Authors and Affiliations
Contributions
ADZ and XJZ designed the experiments. ADZ and JF performed statistical analyses of gene expression data. ADZ performed statistical analyses. ADZ drafted the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Consent for publication
All authors have consented to publishing this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Handling Editor: Julien Boutte.
Supplementary Information
Below is the link to the electronic supplementary material.
Information on Electronic Supplementary Material
Information on Electronic Supplementary Material
Online Resource 1. SRA accessions and chloroplast genome for each plant used in the study.
Online Resource 2. The statistical result of RNA editing sites in 21 species.
Online Resource 3. Information of identified RNA editing sites by REDO.
Online Resource 4. Editing sites in 30 genes with most editing sites across 21 species.
Online Resource 5. Cytosines content of shared 50 genes across 21 plants.
Online Resource 6. a Average read depth of RNA-seq data across 21 species used in our study. b The attributes of RNA editing sites in chloroplast illustrated by samples of Adiantum aleuticum. c Hierarchical cluster analysis of numbers of RNA editing sites in chloroplast across 21 species. d Hierarchical cluster analysis of numbers of RNA editing sites in chloroplast across 11 angiosperms. e Hierarchical cluster analysis of numbers of RNA editing sites in chloroplast across 6 fern plants. f Hierarchical cluster analysis of numbers of RNA editing sites in chloroplast across 4 gymnosperm plants.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, A., Fang, J. & Zhang, X. Diversity of RNA editing in chloroplast transcripts across three main plant clades. Plant Syst Evol 309, 12 (2023). https://doi.org/10.1007/s00606-023-01849-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00606-023-01849-z