
Abstract
A UV-vis, CD, and differential pulse voltammetric study was performed on the deactivation of the activity of parallel G-quadruplex/hemin DNAzymes (G4 DNAzymes) by Pb(II). The G4 DNAzyme carries a d[TC] sequence at its 3′ end and is stabilized by potassium(I). On addition of Pb(II), the K(I) ions in the parallel G4 are replaced by Pb(II) to keep the parallel topology. Intruded Pb(II) decrease the affinity between the topology and hemin, this leads to a decrease of DNAzyme activity for catalyzing the oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) by hydrogen peroxide to form a green dye with an absorption maximum at 420 nm. The assay does not use any amplification, and has a linear response in the 0.01 to 10 μM Pb(II) concentration range and a 7.1 nM limit of detection. The method was successfully applied to the analysis of spiked water samples.

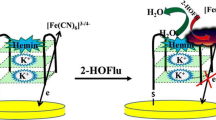
Schematic diagram of the colorimetric lead(II) assay based on the competition between K+ and Pb2+ stabilized G-quadruplex/hemin DNAzymes (G4 DNAzymes).
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pb2+ pollution has irreversible effects on humans, such as anemia, cardiovascular dysfunction, and irritability [1]. The safety limit of total lead in drinking water established by WHO is 10 ppb (48.3 nM) [2]. To monitor the Pb2+ pollution, traditional and reliable methods have been developed, such as atomic fluorescence spectroscopy (AFS), atomic absorption spectroscopy (AAS), anodic stripping voltammetry (ASV) and inductively coupled plasma-mass spectrometry (ICP-MS) [3]. These techniques are accurate but require sophisticated equipment or complicated sample preparations [4], most of them are costly and cannot be used on-site. Thus, it is highly desirable to design a sensitive method based on simple sample preparation, selective and inexpensive strategies for the detection and quantification of Pb2+.
Peroxidases has received intensive interests because of its functional diversity [5,6,7], and also exploited to catalyze a wide variety of chemical reaction, especially for analytical applications [8]. However, most natural peroxidases are proteins, which are often difficult and/or expensive to synthesize and sensitive to the environment. To overcome these limitations, a series of artificial enzymes have been designed to imitate the biological function of peroxidases [9]. These artificial enzymes can be organic compounds, metal and metal oxide-based nanomaterials, carbon-based nanomaterials, metal-organic frameworks, or catalytic nucleic acids (known as DNAzymes or ribozymes) [10]. In particular, DNA-based artificial enzymes have attracted a lot of attention due to their advantages of high selectivity, biocompatibility and sensitivity. The first DNAzyme for Pb2+ detection is 8–17 DNAzyme [11], which is the main detection element of nucleic acid-based Pb2+ probe [12, 13]. Even though this DNAzyme is highly sensitive and selective, its low robustness limits its application in complex environments, and sequences optimization is onerous to ensure high-efficiency.
G-quadruplexes (G4s) are highly ordered DNA structures, which involve four guanines arranged in a square-planar array via Hoogsteen hydrogen-bonding [14]. G4s can be stabilized by a number of metal ions, such as K+, Na+, and Pb2+. The binding ability of these cations to G4s generally depends on several factors, such as ionic radius, hydration effect, and coordination behavior [15]. In the meanwhile, G4s can effectively bind with hemin and possess catalytic properties as horseradish peroxidases (named as G4 DNAzymes) [16]. Additionally, the activities of G4 DNAzymes adopting a parallel conformation are higher than other topologies (antiparallel or hybrid) [17,18,19]. Compared with monovalent ions, Pb2+ has a greater ability to stabilize G4 [20, 21]. Li and coworkers discovered that Pb2+ can compete with K+ to induce a conformational change, and decrease the activity of G4 DNAzyme [22]. These G4 DNAzymes have been successfully used for colorimetric detection of Pb2+ [23,24,25,26]. In addition, G4 DNAzymes are also work with some nanomaterials to detect Pb2+, including graphene oxide and gold nanoparticles [27,28,29]. However, it should be noted that most G4 DNAzymes are limited by their low catalytic proficiency. In order to increase the detection sensitivity, several amplification strategies and nanomaterials are introduced, which make the detection systems too complicated, sample preparations too complex, and cannot be used to on-site detection. Therefore, the one-step method without any amplification assay deserves to be developed.
In 2018, we performed a precise analysis and assessment of the position-dependent catalytic enhancement in the G4 DNAzymes model reactions [30]. The results show a highly enhanced activity of G4 DNAzyme when hemin binds to parallel G4 with a d[TC] flanking sequence at the 3′ end (PG4TC). So, we chose the constructed PG4TC as a probe to improve the sensitivity for Pb2+ detection. The experimental results demonstrate that the binding of Pb2+ into G4 does not induce a structural change, but decrease the binding affinity of hemin with G4. To the best of our knowledge, this is the first successful use of highly active G4 DNAzyme for the sensitive and selective monitoring of Pb2+ without introducing other signal amplification methods. Finally, this method was applied to the determination of Pb2+ in spiked water samples.
Experimental section
Materials and reagents
HPLC-purified DNA oligonucleotides listed in Table 1 were obtained from Sangon (Shanghai, China, https://www.sangon.com). Their concentrations were determined by measuring the absorbance at 260 nm using the extinction coefficients given by OligoAnalyzer 3.1 (http://sg.idtdna.com/calc/analyzer). Hemin, Triton X-100, DMSO, tris-(hydroxymethyl)-amino-methane (Tris), acetic acid (HAc), citric acid, tris(2-carboxyethyl) phosphine hydrochloride (TCEP), 6-mercapto-1-hexanol (MCH), KCl, Pb(NO3)2, Zn(NO3)2·6H2O, FeCl3·6H2O, CuSO4·5H2O, Hg(NO3)2, MnCl2, CaCl2, CdCl2·2.5H2O, BaCl2·2H2O, KSCN, SrCl2·6H2O, MgCl2·6H2O, 2,2′-azino-bis (3-ethylben-zothiazoline-6-sulfonic acid) (ABTS) and NaClO4 were purchased from Sigma-Aldrich (St. Louis, MO, USA, https://www.sigmaaldrich.com). H2SO4 and H2O2 were obtained from Lingfengshiji (Shanghai, China, http://lingfengshiji.foodmate.net).
Hemin was dissolved in DMSO and diluted to 50 μM, and then stored in the dark at 4 °C. Freshly prepared ABTS was dissolved in ultrapure water to 50 mM. DNA samples were prepared in 10 mM Tris-HAc buffer (pH titrated with acetic acid, HAc, to 6.5) with different concentrations of K+ or Pb2+. They were heated to 95 °C for 5 min, then cooled slowly to room temperature and stored at room temperature for 2 h to thoroughly fold into K+ or Pb2+ stabilized G4s.
Apparatus
UV absorbance spectra were measured with a Cary 100 UV-vis spectrophotometer (Agilent Technologies, USA, https://www.agilent.com) in a sealed tandem cuvette with a path length of 10 mm. Circular dichroism (CD) spectra were recorded on a Chirascan equipped with a Peltier temperature control accessory (Applied Photophysics, UK, https://www.photophysics.com). Electrochemical measurements were performed on a CHI 660D electrochemical workstation (CH Instruments Inc., USA, https://www.chinstr.com) at room temperature with a conventional three-electrode configuration composed of a platinum wire as counter, Ag/AgCl as reference, and a gold disk electrode (AuE) with 2 mm diameter as working electrodes.
Typical oxidation procedure
The K+ or Pb2+ stabilized G4s were incubated with hemin (at a molar ratio of 1:1) at 25 °C for 30 min in 10 mM Tris-HAc buffer (pH 6.5) containing 0.05% (v/v) Triton X-100. Then, 6 μL 50 mM ABTS was added. The reaction was started with the addition of 6 μL 50 mM H2O2. The generation of oxidation product ABTS·+ was estimated by monitoring the absorbance at 420 nm. The initial rate (V0, nM/s) of the oxidation reaction was obtained from the slope of the initial linear portion (the first 10 s) of the plot of absorbance versus reaction time. All kinetic measurements were repeated three times.
UV–vis absorption titration experiments
To evaluate the binding ratio between PG4TC and Pb2+, UV absorbance spectra were collected from 240 nm to 350 nm at 25 °C in a cuvette with a path length of 10 mm. The spectrophotometric titration of PG4TC with increasing concentrations of Pb2+ was performed by incubating Pb2+ (0 to 50 μM) with PG4TC (5.0 μM) in 10 mM Tris-HAc buffer at 25 °C for 2 h. The difference spectra ∆Aλ=\( {\mathrm{A}}_{\mathrm{F}}^{\uplambda}\hbox{--} {\mathrm{A}}_{\mathrm{U}}^{\uplambda} \) were obtained by subtracting the absorbance spectra of the unfolded G4 from the folded form. The resulting ∆A at 303 nm were then plotted as a function of the [Pb2+]/[PG4TC] molar ratio.
Circular dichroism (CD) experiments
CD spectra were collected from 220 nm to 350 nm at 25 °C. 5 μM DNA samples were prepared in 10 mM Tris-HAc buffer (pH 6.5) containing 2 mM KCl or 100 μM Pb(NO3)2, or 2 mM KCl in the presence of 1 to 100 μM Pb(NO3)2. The lamp was kept under a stable stream of dry purified nitrogen (99.99%) during experiments, and the measurements were repeated three times.
Electrochemical analysis
The AuE was firstly immersed into freshly prepared piranha solution (H2SO4/H2O2, 3:1) for 1 h and polished with 0.05 μm Al2O3 slurry. Then, the electrode was electrochemically activated by scanning the potential from −0.2 to +1.6 V in 0.5 M H2SO4 at a scan rate of 0.1 V s−1 for 80 cycles. The annealed SH-PG4TC (1 μM) and TCEP (500 μM) were mixed for 1 h to reduce disulfide bonds. An amount of 10 μL of PG4TC (1 μM) was dropped on the electrode surface to incubate at 37 °C for 2 h. After rinsing with 10 mM Tris-HAc buffer (pH 7.0) and drying with nitrogen, 10 μL of MCH (1 mM) was dropped on the electrode and incubated for 1 h to block the unmodified DNA. The MCH/SH-PG4TC/AuE was incubated in 10 mM Tris-HAc containing 2 mM KCl or 100 μM Pb(NO3)2 for 2 h at room temperature to allow the appropriate structure to fold. After thorough washing with 10 mM Tris-HAc buffer (pH 7.0), the electrode was immersed in 10 mM Tris-HAc buffer (pH 7.0) containing 0.1 M NaClO4 for differential pulse voltammetry (DPV) detection from – 0.1 V to – 0.6 V, using pulse amplitude of 50 mV and width of 50 ms.
Measurement procedure of Pb2+
1.25 μL, 100 μM DNA samples were prepared with 2 μL, 500 mM KSCN and different concentrations of Pb(NO3)2 in buffer (10 mM Tris-HAc, 1 mM citric acid, pH 6.5). Thereafter, the samples were heated to 95 °C for 5 min, slowly cooled to room temperature and stored 2 h at room temperature to thoroughly fold into K+ or Pb2+ stabilized G4s. After that, samples were incubated with 2.5 μL, 50 μM hemin in 40 μL buffer (100 mM Tris-HAc, 10 mM citric acid, pH 6.5), containing 0.05% (v/v) Triton X-100 and 1% DMSO for 30 min at 25 °C. Finally, 6 μL ABTS (50 mM) and 6 μL H2O2 (50 mM) were added. The detection procedures were the same as above mentioned.
Regarding to the environmental water sample detection (water from Xuanwu Lake in Nanjing, P. R. China). Firstly, 50 μL filtered environmental water sample (centrifuged at 10000 rpm for 15 min, and then filtered through a 0.45 μm membrane (www.tansoole.com)), mixed with 1.25 μL, 100 μM G-quadruplex, 2 μL, 500 mM KSCN, and 10 μL buffer (containing 100 mM Tris and 10 mM citric acid, pH 6.5), 36.8 μL ultrapure water added to make sure the total volume into 100 μL. Then the sample annealed at 95 °C for 5 min and naturally annealed to room temperature, kept for 2 h. Secondly, 2.5 μL, 50 μM Hemin, 40 μL buffer (containing 100 mM Tris, 10 mM citric acid and 0.5% v/v Triton X-100) and 345.5 μL ultrapure water were added into the sample. Finally, 6 μL 50 mM ABTS and 6 μL 50 mM H2O2 were added into this system to start the reaction, then collected the absorbance at 420 nm for 2 min.
Results and discussion
Influence of Pb2+ on catalytic ability of G4 DNAzyme
In our previous studies, dozens of G4s were designed to decipher the mechanism of G4 DNAzymes, and the result indicated that the d[TC] flanking sequence at the 3′ end of parallel G4s (PG4TC) presented the highest catalytic efficiency [19, 30]. Therefore, we chose PG4TC to investigate the effect of Pb2+ on K+-stabilized G4 DNAzyme. PS2.M, a well-known G4 sequence GTGGGTAGGGCGGGTTGG (Table 1), was exploited as a reference sequence because it was previously described as G4 DNAzyme-based assay for Pb2+ detection [23].
To investigate the catalytic activities of the resulting DNAzymes formed by the PG4TC-hemin (PG4TC DNAzyme) or PS2.M-hemin (PS2.M DNAzyme) complexes, the oxidation substrate ABTS was used. The green-color oxidized product ABTS•+ can be conveniently monitored by absorbance around 420 nm, so the catalytic efficiency of the DNAzyme was assessed by the measurement of absorbance at this wavelength as a function of time (Fig. 1a). Strikingly, both PG4TC and PS2.M DNAzymes exhibit a remarkable decrease in catalytic efficiency when 50 μM Pb2+ were added. We quantified the catalytic activity of DNAzymes via the initial velocity V0, defined as the concentration of ABTS•+ generated as a function of time (V0 expressed in nM/s). The V0 values of PG4TC and PS2.M DNAzymes are 275.8 nM/s and 42.0 nM/s, which are dropped to 70.5 nM/s and 10.5 nM/s in the presence of 50 μM Pb2+, respectively (Fig. 1b). Both DNAzymes show a similar decrease rate (4-fold), but the PG4TC DNAzyme system exhibit a greater extent signal change than PS2.M, implying that G4TC DNAzyme has potential in further visual detection (Fig. 1b).

a Kinetic curves of ABTS-H2O2 reaction catalyzed by hemin (blank), PG4TC DNAzyme and PS2.M DNAzyme with 2 mM K+ in the absence or presence of 50 μM Pb2+. b DNAzyme activity of two G4 DNAzymes under 2 mM K+ in the absence (1, 3) or presence (2, 4) of 50 μM Pb2+. Inset: Solutions catalyzed by two G4 DNAzymes in the absence or presence of 50 μM Pb2+
The Pb2+-induced decrease in PS2.M DNAzyme catalytic efficiency has been attributed to a Pb2+-dependent conformational change, further hindering hemin from stacking on G4 [22]. To verify the interaction between PG4TC and hemin, UV-vis absorption spectroscopy was conducted to study their interaction. As shown in Fig. S1, the absorption spectra of PG4TC/hemin complex in the absence of metal ion feature an absorption peak around 400 nm. Upon addition of 2 mM K+, a concomitant hyperchromism at 404 nm is observed, indicating the formation of PG4TC DNAzyme. Subsequently, the Soret band intensity at 404 nm diminish with increasing concentration of Pb2+ ([Pb2+]/[PG4TC] = 1, 2, 4, 8, 12, 16, 20) (Fig. S1). This hypochromism indicate that Pb2+ will introduce the dissociation between PG4TC and hemin. To assess the role of Pb2+ on DNAzymes, we systematically study the difference between Pb2+ and K+-stabilized G4 in all subsequent experiments.
Binding mechanism of PG4TC with Pb2+
To understand the effect of Pb2+ on the PG4TC DNAzyme, the binding mode of Pb2+ to PG4TC was investigated by UV–vis absorbance titration experiments. The difference spectrum obtained by subtracting the spectrum of the fully unfolded oligomer from the absorption spectrum of PG4TC in the presence of Pb2+. As shown in Fig. S2a, the difference absorbance spectra at 303 nm increase, but at 245 and 275 nm decrease with increasing the concentration of Pb2+. Different from unfolded oligonucleotide, the formation of Pb2+-driven G4 is accompanied by a hyperchromic effect at 303 nm [31]. Therefore, we monitor the absorbance change at 303 nm for PG4TC, with different molar ratios of [Pb2+]/[PG4TC] (Fig. S2b). We get 2:1 stoichiometric, suggesting that two Pb2+ cations are sufficient to induce one PG4TC molecule to form G4, suggesting that each of the two quartets coordinate with one Pb2+ ion.
Comparation of thermal properties of K+ or Pb2+ stabilized G4
To elucidate which cation is embedded in PG4TC when K+ and Pb2+ coexist, it is necessary to compare their stability in the presence of these two metal ions. The Tm (melting temperature) of K+ and Pb2+-PG4TC are 73.5 and 83.1 °C in 2 mM K+ or 10 μM Pb2+, respectively, with 9.6 °C difference (Fig. S3). Furthermore, the ΔGo (298 K) of PG4TC, calculated by Van’t Hoff analysis [32], transforming into K+-PG4TC was – 31.2 kJ/mol, which is less favorable (by 7.3 kJ/mol) than that of Pb2+-PG4TC (−38.5 kJ/mol) (Table S1). These results illustrate that the Pb2+ stabilized PG4TC is more stable than that of K+ one. Therefore, we hypothesize that Pb2+ may replace the position of K+ in G4 and decrease the catalytic activity of PG4TC DNAzyme.
Studies of competing process between Pb2+ and K+-induced PG4TC
To prove the hypothesis, we measured CD spectra of PG4TC in the presence of both K+ and Pb2+ firstly. CD spectra for K+- and Pb2+-induced PG4TC both show a negative peak around 240 nm and a positive peak around 260 nm (Fig. S4a), indicating that both of them promote parallel conformation PG4TC [33]. However, subtle differences are found upon addition of 100 μM Pb2+. Interestingly, the negative peak at 295 nm will gradually increase with lead ion addition (Fig. S4b). We think the negative peak at 295 nm maybe attributed to the interaction between Pb2+ and dT in loops and flanking regions due to single thymine (dT) has a small negative CD signal at 295 nm [34]. Therefore, all dT bases were replaced by dA bases in the PG4AC sequence, but displays the same CD spectra (Fig. S4c). These results suggest that the negative peak around 295 nm can be used as a useful character of Pb2+-induced G4.
Then, differential pulse voltammetry (DPV) method was introduced to check whether Pb2+ replace K+ to get a new G-quadruplex. From Fig. S5, PG4TC system does not show distinctive reduction peak ranging from 0 to – 0.6 V, while a strong negative reduction peak at −0.3 V (vs. Ag/AgCl) shown in the presence of 100 μM Pb2+ (Fig. S5). These results indicate the direct reduction of Pb2+ in PG4TC [35]. When 100 μM Pb2+ coexist with 2 mM K+ (blue curve in Fig. S5), strong negative reduction peak at −0.3 V still exist, with a bit lower reduction current than Pb2+-stabilized PG4TC. From these results, we propose that Pb2+ replaces K+ within the G4 instead of inducing a conformational switch.
Highly active DNAzyme for Pb2+ detection
The results above illustrate the influence of Pb2+ on the structure of PG4TC and the activity of PG4TC DNAzyme (Scheme 1). In order to assess the potential applications of the highly active DNAzyme on Pb2+ detection, the effects of various experimental parameters such as K+ pH, and DNAzyme concentration were investigated.

Schematic diagram of the colorimetric assay of Pb2+ based on the competition between K+ and Pb2+ stabilized G4
Since the low activity of Pb2+- PG4TC DNAzyme results from Pb2+ competing and replacing the position of K+, the minimum concentration of K+ to stabilize and keep the high activity of PG4TC DNAzyme was determined. As shown in Fig. 2a and Fig. S6a, the CD spectra of PG4TC at 265 nm reach a maximum value at 2 mM K+, consistent with the activity analysis of PG4TC DNAzyme under different concentrations of K+ (maximum activity was observed at 2 mM K+, Fig. 2b and Fig. S6b). These results illustrate that 2 mM K+ is sufficient for PG4TC to fold into a parallel structure and simultaneously binds to hemin to form the PG4TC DNAzyme. Furthermore, the optimal pH value is demonstrated to be 6.5 (Fig. 2c and Fig. S6c). The optimal amount of PG4TC is also optimized to 0.25 μM, which was chosen for colorimetric assay, based on the signal-to-noise ratio value before and after the addition of 50 μM Pb2+ (Fig. 2d and Fig. S6d). To mask Hg2+, which may interact with thymine residues of PG4TC to form T-Hg2+-T, and other potential interference ions, KSCN was used to provide the coordination cation K+ and introduce the chelator SCN− [23]. Furthermore, citric acid was introduced to chelate Fe3+ [36]. Both additives (KSCN and citric acid) proved to have no influence on the catalytic activity of the DNAzyme (Fig. S7).

a CD intensities of 5 μM PG4TC at 265 nm with different K+ concentration. b Absorbance at 420 nm of 0.4 μM PG4TC DNAzyme with different K+ concentration. c Absorbance at 420 nm of 0.4 μM PG4TC DNAzyme with different pH. d A0/A1 with different PG4TC concentration. A0 and A1 represent the absorption at 420 nm for ABTS-H2O2 system in the absence or presence of 50 μM Pb2+
Under optimized conditions, various Pb2+ concentrations were added to 2 mM K+ stabilized PG4TC, subsequently incubated with hemin for 30 min and then corresponding kinetic curves were measured. As shown in Fig. 3a, the color change of the solution (from green to colorless) can be used to estimate the Pb2+ concentration visually. The kinetic curves indicate that increasing concentrations of Pb2+ ions induce a decrease of the generation velocity of ABTS.+ (Fig. 3b). Notably, we find a linear relationship between the absorbance at 420 nm and the logarithm value of the lead ion concentration ranging from 0.01 to 10 μM with a correlation coefficient of 0.9948 (Fig. 3c). The regression equation is expressed as follow:
where A0 and A1 are the absorbance at 420 nm in the absence and presence of corresponding Pb2+, [Pb2+] is the concentration of Pb2+ in solution.

Application of K+ stabilized PG4TC for colorimetric analysis of Pb2+ in ABTS-H2O2 system: a Solutions with different concentrations of Pb2+. b Kinetic absorbance curves for 0.25 μM PG4TC in the presence of 0, 0.01, 0.05, 0.1, 0.5, 1, 5, 10, 30, 50, 100 μM Pb2+, respectively (from top to bottom). c Plot of absorbance at 420 nm vs logarithm of target Pb2+ concentration. Inset: Calibration plot
In consequence, the detection limit is 7.1 nM at a signal-to-noise ratio of 3, which is much lower than the concentration of 48.3 nM in drinking water permitted by the WHO. Relative to previous colorimetric strategies based on G4 DNAzymes and G4s, this high catalytic DNAzyme has a lower detection limit in the meanwhile (Table 2) [23,24,25, 27, 28].
Selectivity was evaluated by adding 5 or 15-folds common metal ions of Pb2+ concentration to the system respectively, such as Ba2+, Ca2+, Cd2+, Hg2+, Cu2+, Mg2+, Sr2+, Zn2+, Fe2+, Fe3+. As shown in Fig. 4, the A0-A1 signal is about 5-fold more selective for Pb2+ than other metal ions, indicating its good selectivity against other environmentally relevant metal ions.

Selectivity of PG4TC DNAzyme toward Pb2+ ion by comparing it to the other metal ions: 50 μM Ba2+, Cd2+, Hg2+, Sr2+, Fe2+, Fe3+, 150 μM Ca2+, Cu2+, Mg2+, Zn2+, and 10 μM Pb2+. A0 and A1 are the absorbance at 420 nm in the absence and presence of corresponding metal ions, respectively
To further validate the performance of the PG4TC DNAzyme for Pb2+ ion in practical applications, spiked-recovery experiments of Pb2+ in environmental water samples were performed. The water samples were spiked with 10 nM, 500 nM and 10 μM for recovery experiments, then quantitatively determined via the regression equation acquired above. The recovery is from 95.90 to 103.27%, and the RSD is from 2.55 to 4.84% for environmental water (Table 3), demonstrate that this assay has a prospect application for real samples.
Conclusions
This work bases on high activity G-quadruplex/hemin DNAzyme, PG4TC DNAzyme. Owing to the higher affinity of Pb2+ to PG4TC, Pb2+ can replace K+ inside of G4, then a non-amplification and cost-efficient colorimetric strategy is built to detect Pb2+. This method has good sensitivity and detection range, and also show high tolerance to other metal ions. Our study not only introduces a new method for the sensitive detection of Pb2+, but also provides new insight into the competition of K+ and Pb2+ in parallel G4 structure. The use of high activity G4 DNAzyme makes it hopeful to be applied to other analytical applications related to peroxidases. However, the process is still not fast enough, even though our probe can be prepared quickly. Therefore, future researches in our group will focus on simplify this method and verify the feasibility of this method for on-site application.
References
Flegal AR, Smith DR (1992) Current needs for increased accuracy and precision in measurements of lead in blood. Environ Res 58(2):125–133. https://doi.org/10.1016/s0013-9351(05)80209-9
World Health Organization (2017) Guidelines for drinking water quality. WHO, Geneva
Liang G, Man Y, Li A, Jin X, Liu X, Pan L (2017) DNAzyme-based biosensor for detection of lead ion: a review. Microchem J 131:145–153. https://doi.org/10.1016/j.microc.2016.12.010
Ma Y, Yu C, Yu Y, Chen J, Gao R, He J (2019) DNAzyme assisted recycling amplification method for ultrasensitive amperometric determination of lead (II) based on the use of a hairpin assembly on a composite prepared from nitrogen doped graphene, perylenetetracarboxylic anhydride, thionine and gold nanoparticles. Microchim Acta 186:677. https://doi.org/10.1007/s00604-019-3790-1
Krieg R, Halbhuber KJ (2003) Recent advances in catalytic peroxidase histochemistry. Cell Mol Biol 49(4):547–563
Veitch NC (2004) Horseradish peroxidase: a modern view of a classic enzyme. Phytochemistry 65(3):249–259. https://doi.org/10.1016/j.phytochem.2003.10.022
Nagaraja P, Shivakumar A, Shrestha AK (2009) Peroxidase-catalyzed oxidative coupling of paraphenylenediamine with 3-dimethylaminobenzoic acid: application in crude plant extracts. J Agric Food Chem 57(12):5173–5177. https://doi.org/10.1021/jf900696p
Ryan O, Smyth MR, O’Fágáin CO (1994) Horseradish peroxidase: the analyst’s friend. Essays Biochem 28:129–146
Lin Y, Ren J, Qu X (2014) Nano-gold as artificial enzymes: hidden talents. Adv Mater 26(25):4200–4217. https://doi.org/10.1002/adma.201400238
Willner I, Shlyahovsky B, Zayats M, Willner B (2008) DNAzymes for sensing, nanobiotechnology and logic gate applications. Chem Soc Rev 37(6):1153–1165. https://doi.org/10.1039/b718428j
Li J, Lu Y (2000) A highly sensitive and selective catalytic DNA biosensor for lead ions. J Am Chem Soc 122(42):10466–10467. https://doi.org/10.1021/ja0021316
Liu J, Lu Y (2004) Accelerated color change of gold nanoparticles assembled by DNAzymes for simple and fast colorimetric Pb2+ detection. J Am Chem Soc 126(39):12298–12305. https://doi.org/10.1021/ja046628h
Zhao X, Kong R, Zhang X, Meng H, Liu W, Tan W, Shen G, Yu R (2011) Graphene-DNAzyme based biosensor for amplified fluorescence “turn-on” detection of Pb2+ with a high selectivity. Anal Chem 83(13):5062–5066. https://doi.org/10.1021/ac200843x
Gellert M, Lipsett MN, Davies DR (1962) Helix formation by guanylic acid. Proc Natl Acad Sci U S A 48(12):2013–2018. https://doi.org/10.1073/pnas.48.12.2013
Hardin CC, Perry AG, White K (2001) Thermodynamic and kinetic characterization of the dissociation and assembly of quadruplex nucleic acids. Biopolymers 56(3):147–194. https://doi.org/10.1002/1097-0282(2000/2001)56:3<147::aid-bip10011>3.0.co;2-n
Travascio P, Li Y, Sen D (1998) DNA-enhanced peroxidase activity of a DNA aptamer-hemin complex. Chem Biol 5(9):505–517. https://doi.org/10.1016/S1074-5521(98)90006-0
Zhu L, Li C, Zhu Z, Liu D, Zou Y, Wang C, Fu H, Yang CJ (2012) In vitro selection of highly efficient G-quadruplex-based DNAzymes. Anal Chem 84(19):8383–8390. https://doi.org/10.1021/ac301899h
Guo Y, Chen J, Cheng M, Monchaud D, Zhou J, Ju H (2017) A thermophilic tetramolecular G-quadruplex/hemin DNAzyme. Angew Chem Int Ed 129(52):16636–16640. https://doi.org/10.1002/anie.201708964
Chen J, Guo Y, Zhou J, Ju H (2017) The effect of adenine repeats on G-quadruplex/hemin peroxidase mimicking DNAzyme activity. Chem A Eur J 23(17):4210–4215. https://doi.org/10.1002/chem.201700040
Kotch FW, Fettinger JC, Davis JT (2000) A lead-filled G-quadruplex: insight into the G-quartet’s selectivity for Pb2+ over K+. Org Lett 2(21):3277–3280. https://doi.org/10.1021/ol0065120
Smirnov I, Shafer RH (2000) Lead is unusually effective in sequence-specific folding of DNA. J Mol Biol 296(1):1–5. https://doi.org/10.1006/jmbi.1999.3441
Li T, Wang E, Dong S (2009) Potassium-lead-switched G-quadruplexes: a new class of DNA logic gates. J Am Chem Soc 131(42):15082–15083. https://doi.org/10.1021/ja9051075
Li T, Wang E, Dong S (2010) Lead(II)-induced allosteric G-quadruplex DNAzyme as a colorimetric and chemiluminescence sensor for highly sensitive and selective Pb2+ detection. Anal Chem 82(4):1515–1520. https://doi.org/10.1021/ac902638v
Zhu X, Gao X, Liu Q, Lin Z, Qiu B, Chen G (2011) Pb2+-introduced activation of horseradish peroxidase (HRP)-mimicking DNAzyme. Chem Commun 47(26):7437–7439. https://doi.org/10.1039/c1cc11349f
Elbaz J, Shlyahovsky B, Willner I (2008) A DNAzyme cascade for the amplified detection of Pb2+ ions or L-histidine. Chem Commun 13:1569–1571. https://doi.org/10.1039/b716774a
Tang D, Xia B, Tang Y, Zhang J, Zhou Q (2019) Metal-ion-induced DNAzyme on magnetic beads for detection of lead(II) by using rolling circle amplification, glucose oxidase, and readout of pH changes. Microchim Acta 186:318. https://doi.org/10.1007/s00604-019-3454-1
Chen X, Zhai N, Snyder JH, Chen Q, Liu P, Jin L, Zheng Q, Lin F, Hu J, Zhou H (2015) Colorimetric detection of Hg2+ and Pb2+ based on peroxidase-like activity of graphene oxide-gold nanohybrids. Anal Methods 7(5):1951–1957. https://doi.org/10.1039/c4ay02801e
Xu H, Liu B, Chen Y (2012) A colorimetric method for the determination of lead(II) ions using gold nanoparticles and a guanine-rich oligonucleotide. Microchim Acta 177(1):89–94. https://doi.org/10.1007/s00604-011-0744-7
Ravikumar A, Panneerselvam P, Radhakrishnan K (2018) Fluorometric determination of lead(II) and mercury(II) based on their interaction with a complex formed between graphene oxide and a DNAzyme. Microchim Acta 185:2–8. https://doi.org/10.1007/s00604-017-2585-5
Chen J, Zhang Y, Cheng M, Guo Y, Šponer J, Monchaud D, Mergny JL, Ju H, Zhou J (2018) How proximal nucleobases regulate the catalytic activity of G-quadruplex/hemin DNAzymes. ACS Catal 8(12):11352–11361. https://doi.org/10.1021/acscatal.8b03811
Hud NV, Plavec J (2006) The role of cations in determining quadruplex structure and stability. In: Neidle S, Balasubramanian S (eds) Quadruplex nucleic acids. Royal Society of Chemistry, London, pp 100–130
Hatzakis E, Keika O, Yang D (2010) Thermodynamic stability and folding kinetics of the major G-quadruplex and its loop isomers formed in the nuclease hypersensitive element in the human C-Myc promoter: effect of loops and flanking segments on the stability of parallel-stranded intramolecular G-quadruplexes. Biochemistry 49(43):9152–9160. https://doi.org/10.1021/bi100946g
Kypr J, Kejnovska I, Renciuk D, Vorlickova M (2009) Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res 37(6):1713–1725. https://doi.org/10.1093/nar/gkp026
Hazel P, Huppert J, Balasubramanian S, Neidle S (2004) Loop-length-dependent folding of G-quadruplexes. J Am Chem Soc 126(50):16405−16415. https://doi.org/10.1021/ja045154j
Zhai W, Du C, Li X (2014) A series of logic gates based on electrochemical reduction of Pb2+ in self-assembled G-quadruplex on the gold electrode. Chem Commun 50(17):2093−2095. https://doi.org/10.1039/C3CC47763K
Bobtelsky M, Jordan J (1947) The structure and behavior of ferric tartrate and citrate complexes in dilute solution. J Am Chem Soc 69(10):2286−2290. https://doi.org/10.1021/ja01202a014
Acknowledgements
We acknowledge the financial support of the National Natural Science Foundation of China (21503229, 21635005, and 21361162002), Fundamental Research Funds for the Central Universities (020514380070, 020514380085, 020514380105, 020514380144), the funds of Nanjing University (020514912216, 020514911200, 0205145051), and self-funding projects from State Key Laboratory of Analytical Chemistry for Life Science, Nanjing University (5431ZZXM1905). J. Chen acknowledges funding from the China Scholarship Council.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare they have no competing financial interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 789 kb)
Rights and permissions
About this article

Cite this article
Chen, J., Zhang, Y., Cheng, M. et al. Highly active G-quadruplex/hemin DNAzyme for sensitive colorimetric determination of lead(II). Microchim Acta 186, 786 (2019). https://doi.org/10.1007/s00604-019-3950-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-019-3950-3