
Abstract
β-Nickel hydroxide nanoplatelets (NPs) were synthesized and used as a modifier on a carbon paste electrode (CPE) for the detection of hydrazine. Synthesis is based on a one-step hydrothermal method using L-arginine that acts as an agent to adjust the pH value and to shape the NPs. They were characterized by field emission scanning electron microscopy, X-ray diffraction and energy-dispersive X-ray spectroscopy. The composition of the modified electrode was optimized by changing the amount of NPs. Best results were achieved using a 10:70:20 weight ratio for nanoparticles, graphite and mineral oil (the binder). The electrochemical properties of the modified CPE were studied by cyclic voltammetry. The surface coverage of the NPs, the electron transfer coefficient and the charge transfer rate constant were calculated. The diffusion coefficient of hydrazine is 1.56 × 10−5 cm2 s−1. An amperometric method was worked out that has the following figures of merit: (a) A working applied potential of 500 mV (vs. Ag/AgCl reference electrode), (b) a linear range that extends from 1 to 1300 μmol L−1, (c) a 0.28 μmol L−1 detection limit, (d) a relative standard deviation of 1.3% (for n = 3 at a level of 5 μmol L−1), and (d) a sensitivity of 1.33 μA μmol−1 L cm−2. The performance of the sensor is compared to other nickel-based sensors for hydrazine. The sensor was successfully used to quantify hydrazine in spiked tap water samples, and the recoveries were 97 ± 2.3% (n = 3).

Schematic presentation of hydrazine electrocatalytic oxidation on NP.CPE*. *NP-CPE (β-nickel hydroxide nanoplatelet modified carbon paste electrode)
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrazine has gained wide industrial applications. During 1960s, hydrazine has been used mainly as propellant for rockets and spacecraft’s. But nowadays hydrazine alone has gained several roles in industries. It is used as corrosion inhibitor, reducing agent, catalyst, blowing agents and etc. Its derivatives also found wide applications in agriculture as an insecticide, and several hydrazine based medicines are present in pharmaceutical Industries.
Due to its toxicological effects and wide industrial applications, the demand for determination of hydrazine in trace level is always felt. Several methods for determination of hydrazine have been described in literature, but the most common analytical methods were spectrophotometry [1], GC [2], GC-MS [3], liquid chromatography [4] and electrochemical sensors [5]. Direct chromatographic techniques for hydrazine suffer from strong peak tailing [3]; therefore the most chromatographic methods for hydrazine are based on pre-column derivatization. However, in most cases, these derivatization techniques are time consuming and labor intensive procedures and suffer from low reproducibility. In addition, chromatographic methods need more expensive instrumentation compared to electrochemistry or spectrophotometry. In spectrophotometric techniques, due to absence of chromophore functional groups on hydrazine, a pre-derivatization reaction is needed. But, due to electroactive nature of hydrazine, electroanalytical techniques, having the advantages of low cost, fast response and portable nature remain unique for it and several electrochemical sensors have been developed for hydrazine [5].
Electrochemical studies of hydrazine are also important from another point of view. Hydrazine is a promising fuel for alkaline fuel cells and studies in this field has been dates back to 1960s [6]. Hydrazine as a fuel has several advantages. It is a well-behaved electroactive species with high theoretical electromotive force. During the electrocatalytic oxidation of hydrazine no carbon dioxide has been produced and therefore reduces emission of greenhouse gases. In addition, lack of carbon atom in electrooxidation products leads to no poisoning of electrocatalysts or anodes. Consequently, electrocatalytic oxidation of this compound has gained much attention. The developments in the electrooxidation of hydrazine on different catalysts have been reviewed [7] by Serov et al.
Nickel-based materials in different forms and compositions, such as alloys, oxides, oxyhydroxides, hydroxides, complexes, etc. are considered as important electrode materials in electrochemistry. This is due to their electrocatalytic behaviors toward many small organic molecules [8]. In comparison to other transition metals with electrocatalytic abilities, such as gold, platinum, copper or cobalt, nickel and its compounds are much more abundant and less toxic. Among the different nickel-based compounds, nickel hydroxide is a better candidate toward electrocatalytic activity due to its relative stability in alkaline medium. Therefore, it’s not strange to observe that several research studies have been focused on preparation of nickel hydroxide nanostructures in different shapes and on different substrates. Nickel hydroxide nanomaterials have found wide applications in different branches of electrochemistry. They are very important active electrode materials for alkaline rechargeable batteries [9] and high-performance supercapacitors [10]. Improved electrolysis of urea has been achieved on anode with nano-nickel hydroxide layers [11]. Several electrochemical sensors based on nickel hydroxide nanoparticles have been reported in literature for a variety of important molecules such as glucose [12], insulin [13], l-histidine [14], dopamine [15], hydrogen peroxide [16] and some drugs [17, 18].
Hydrazine and nickel-based electrodes have had long common electrochemical history. In 1966, Nesterov and Korovin investigated anodic oxidation of hydrazine on nickel [19]. Nickel alloys, as anode materials and catalysts, are widely used in direct hydrazine fuel cells [20] and several nickel-based sensors for hydrazine have been reported in literature [21,23,24,25,26,27,28,29,29]. Despite these facts, our literature survey shows that less attention has been paid to the electrooxidation and detection of hydrazine with nickel hydroxide nanostructures. Thus in this work, a simple hydrothermal method has been used for preparation of NPs. Carbon paste electrodes modified with these NPs (NP-CPE) were used for electrocatalytic studies of hydrazine. Then, a simple amperometric sensor for hydrazine detection has been developed. Finally, figures of merit of NP-CPE are compared with several other nickel-based hydrazine sensors.
Experimental section
Materials
For synthesis of NPs, L-arginine and NiCl2.6H2O were purchased form Scharlau (www.scharlab.com), (http://www.scharlab.com/) and Rankem (www.rankem.in) chemical companies respectively. Mineral oil was obtained from Acros organic (www.acros.com). All other chemicals including hydrazine hydrate, KCl, NaOH and graphite fine powder (particle size ≤50 μm) were obtained from Merck (www.merck.com) and used without further purification. Nafion was obtained from Aldrich (www.sigmaaldrich.com) as a 5 wt% solution. In order to gain more stable and smooth baseline, graphite powder is refluxed in 1.0 mol L−1 HCl solution for one hour and then washed with distilled water and dried at 105 °C for two hours. All solutions were prepared in double-distilled water with conductivity less than 2 μS cm−1.
Synthesis of the nanoplatelets (NPs)
NPs were synthesized according to the reported simple one-pot hydrothermal method [17] with some modification. Synthesis procedure S1 in details was provided in Electronic Supplementary material (ESM).
Preparation of working electrodes
NP-CPE was prepared by careful hand-mixing of graphite powder, mineral oil and nickel hydroxide with 70/20/10% weight ratio in a small mortar for 15 min. This paste was packed firmly into a cylindrical cavity (3.0 mm diameter and 5.0 mm depth) at the end of Teflon body with stainless steel rod for electrical contact. The surface of resulting CPEs was polished on a white paper. Unmodified carbon paste electrode (U-CPE) was prepared in the same way with 80/20% weight ratio for graphite powder and mineral oil.
Electrochemical pretreatment of electrodes
An important final step for preparation of NP-CPE was electrochemical pretreatment of the electrode surface in order to observe reproducible and smooth nickel hydroxide anodic and cathodic peaks. For this purpose, the NP-CPE was transferred into the electrochemical cell filled with a 0.1 mol L−1 NaOH solution (the supporting electrolyte) and cyclic voltammetry in the potential range of 0.2–0.6 V at a scan rate of 50 mV s−1 for 50 cycles was applied. After about 50 cycles, stable anodic and cathodic peaks were attained. This electrochemical procedure was applied for regeneration of either used or new NP-CPE. It should be noted that time required for regeneration of an used modified electrode or preparation of a new electrode is about 20 min. This time include packing, polishing and electrochemical pretreatment steps.
For repeatability and stability test, after drawing a calibration plot by amperometric method the used electrode was stored for one month. After that period, it was transferred to a supporting electrolyte and only the electrochemical pretreatment procedure was applied on it. In other cases, the surface of CPE was scratched by a needle and new fresh paste was added to the surface and all the steps including packing, polishing and pretreatment were applied again.
Instruments and methods
μ-AutoLab type (III) potentiostat (The Netherlands) with GPES software (Version 4.9.007) and a conventional three-electrode system was used in all electrochemical experiments. An Ag/AgCl electrode 3.0 mol L−1 (Metrohm, 6.0733.100) and a Metrohm platinum rod electrode were used as the reference and counter electrodes respectively. All potentials in this study were reported with respect to the Ag/AgCl reference electrode. In all electrochemical studies a 0.1 mol L−1 NaOH solution was used as a supporting electrolyte in room temperature without oxygen purging.
Surface morphology studies of NPs were characterized by a Zeiss EM10C 80 Field emission scanning electron microscopy (FE-SEM). A Siemens D-500 X-ray diffractometer with a Cu Kα (λ = 1.5416 Ȧ) source was used for recording X-ray diffractogram in a 2θ ranges of 3 to 70°.
FESEM images were analyzed by Digimizer (Version 4.3.5) image analysis software.
The calibration plot of hydrazine was obtained by hydrodynamic amperometry. In the amperometric and chronoamperometric experiments the applied potential was selected as 500 mV. While the solution was heavily stirred by a magnet, the standard solutions were injected by a calibrated micro-pipette. Standard stock solutions of hydrazine were freshly prepared by diluting a stock solution of hydrazine hydrate (80%) with distilled water and kept in a dark and cool place.
The step by step procedure S2 for preparation and analysis of water sample spiked with hydrazine was explained in ESM.
Results and discussion
Characterization of the nanoplatelets (NPs)
The FESEM images of NPs with different magnification are shown in Fig. 1. The images show that by this one-pot simple synthesis method, it is possible to produce homogenous and reproducible disk-shape nickel hydroxide nanostructures. Using image analysis software, the smallest and largest dimensions of forty selected NPs were determined as 34 ± 9 and 306 ± 46 nm respectively. Therefore, these NPs have a 1D-nanostructure.

FE-SEM image of NPs with different magnifications
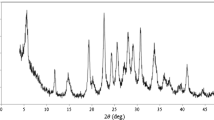
Figure 2b illustrates the XRD pattern of the NPs powder. All the main diffraction peaks at 19.10°, 32.97°, 38.40°, 51.85°, 58.89°, 62.57° and 70.00° are respectively indexed to (001), (100), (101), (102), (110), (111) and (103) standard reflections, and they are in agreement with the standard reflection values of JCPDS 14–0117. In this diffractogram, not only peak positions but also relative peak intensities have good compatibility with reported ones in handbook of mineralogy for nickel hydroxide [30]. The results indicate pure β-type nickel hydroxide phase with hexagonal structure was achieved.

a EDX spectrogram of NPs b XRD powder pattern of NPs
In order to determine elemental composition of NPs, Energy dispersive X-ray (EDX) technique was used, and the spectrogram is shown in Fig. 2a. The presence of nickel and oxygen peaks and absence of any other peaks in the spectrogram confirms high purity of the NPs.
Electrochemical characterization of NP-CPE
Optimization the amount of modifier in NP-CPE
Fig. S1 (ESM) shows cyclic voltammograms of NP-CPEs prepared with varying percentages of NPs in supporting electrolyte at a potential sweep rate of 50 mV s−1. To select the optimum percentage of the modifier, although the criteria of minimum differences between anodic-to-cathodic peak potential was committed [17] and the similar approach can be followed in the present study (Fig. S1 inset A), however, due to better baseline stability, larger anodic and cathodic peaks currents and more symmetric shapes of the peaks, 10% weight ratio of the modifier was selected for preparation of NP-CPE. A comparison between two similar works with different approaches in optimization of amount of modifier in carbon paste has been made in Table S1. As it is seen in Table S1, present approach led to better surface coverage on the surface of electrode. Obviously, due to presence of more electroactive species in surface of electrode, the kinetic of electron transfer reaction was also improved. In electrodes prepared with modifier of more than 10%, smaller and deformed anodic and cathodic peaks were observed.
Effect of potential sweep rate
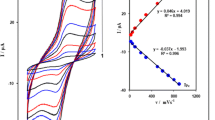
Figure 3a depicts cyclic voltammograms of NP-CPE in the supporting electrolyte recorded at different potential sweep rates of 1–600 mV s−1. The voltammograms are similar in pattern to those reported elsewhere for nickel-based electrodes [17]. In nickel-based electrodes, during positive potential sweep in alkaline solution a thin layer of nickel hydroxide is created on the surface of electrodes [8], and therefore voltammograms are more or less similar to voltammogram recorded for nickel hydroxide here or elsewhere. Nickel hydroxide voltammograms are extensively described in the literature [8] on the basis of the following half reaction.

a Cyclic voltammograms of NP-CPE recorded at different potential sweep rates of 1–600 mV s−1. b Linear dependency of anodic and cathodic peak currents on the potential sweep rates at values lower than 20 mV s−1. c Dependency of anodic and cathodic peak currents on the square root of the potential sweep rate at high values of ≥30 mV s−1 d Variation of the anodic and cathodic peak potentials with the natural logarithm of the potential sweep rate
The formal potential of this redox transition was calculated to be 422 mV (at potential sweep rates of <20 mV s−1). In the voltammograms, the anodic and cathodic current increased as the potential sweep rates increase, while, the ratio of Ipa/Ipc was almost equal to unity at all of the potential sweep rates. This confirmed a high degree of kinetic reversibility of the redox transition of electrode.
At low values of the potential sweep rates of ≤20 mV s−1, the anodic and cathodic currents depend linearly on the potential sweep rate which indicative of a surface-confined redox process. Surface coverage (Γ*) can be calculated from the slopes of two straight lines for anodic and cathodic currents (Fig. 3b) and following equation [31].
where Ip, n, F, υ, A and Γ* are the peak current, number of transferred electron in the redox transition, Faraday constant, potential sweep rate, geometric surface area and surface coverage of the redox species, respectively. Γ* was obtained as 2.15 × 10−8 mol cm−2 from average value of the slopes of the lines in Fig. 3b. This concentration of nickel species on the NP-CPE surface is higher than other nickel hydroxide based modified electrodes [17] and was compared in Table S1.
On the other hand, at higher values of the potential sweep rates of >30 mV s−1, a good linear relationship between the anodic and cathodic peak currents versus the square root of the potential sweep rate was attained (Fig. 3c). This behavior shows a diffusion control overall electrode reaction for the NP-CPE and, it was reported in several other nickel-based sensors [17, 32]. Because redox species have stuck to the electrode surface, a diffusion-control behavior was explained due to slow diffusion of hydroxide ions toward electrode [28]. This diffusion process of hydroxide ions was rate limiting step and obviously slower than the electron transfer reaction.
Kinetic parameters
Laviron-derived equations [33] were used for calculation of the kinetic parameters in redox reaction. This method is based on Butler-Volmer approach and is widely used for redox couples when they were adsorbed on the surface of electrode. This approach only needs the overpotentials in different scan rates for calculation of the kinetic parameters. According to Laviron, when the peak-to-peak potential deference (ΔE) exceed the limit of 200/n (n: number of exchanged electron in the redox transition), a plot of anodic and cathodic peak potentials versus natural logarithm of sweep rate (lnυ) should be linear with a slope equal to \( \left(\frac{\mathrm{RT}}{\left(1-{\upalpha}_{\mathrm{s}}\right)\mathrm{nF}}\right) \) for anodic peak and \( \left(\frac{\mathrm{RT}}{\upalpha_{\mathrm{s}}\mathrm{nF}}\right) \) for the cathodic peak. The general equations for these two lines for anodic (Eq. 3) and cathodic (Eq. 4) branches are as follow:
where Ep,a, Ep,c, E0,υ, αs, ks and ΔEp are the anodic and cathodic peak potentials, formal potential, potential sweep scan rate, the electron transfer coefficient for the redox transition, the apparent rate constant for the electron transfer across the NP-CPE/solution interface, and the peak potential separation, respectively.
Plots of anodic and cathodic peak potentials versus natural logarithm of the potential sweep rate were shown in Fig. 3d. It is important to note that the Laviron predicted linear pattern was observed at potential sweep rates higher than 50 mV s−1 and it was reported in several others Ni-based sensors previously [17, 32]. By using the linear regression analysis electron transfer coefficients αs were calculated from slopes of both lines and average value was obtained as 0.59. The x-intercepts of the lines for the anodic and the cathodic branches provide υ values that are used in Eq. 5 to calculate ks as 1.8 s−1. The calculated ks showes a faster kinetic for electron exchange reaction on the surface of electrode and is compared with a previously published similar work [17] in Table S1.
Electrocatalytic oxidation of hydrazine on NP-CPE
Figure 4 shows cyclic voltammograms of unmodified U-CPE and NP-CPE in supporting electrolyte in the absence and presence of 1.0 mmol L−1 hydrazine. In the voltamograms of U-CPE, there was a negligible difference in anodic current in absence (curve a) and presence (curve b) of hydrazine. This indicates a slow rate of hydrazine electrooxidation on U-CPE in mentioned potential range. Voltammogram (c) in Fig. 4 is a typical voltammogram of NP-CPE with two well-behaved anodic and cathodic peaks belongs to nickel, which explained precisely in literature [8]. In the presence of hydrazine, as we see in voltammogram (d), an increase in anodic current and decrease in cathodic current of NP-CPE were observed. This behavior is explained due to electrocatalytic oxidation of hydrazine on surface of NP-CPE. Several mechanisms have been proposed for explanation of electrocatalytic behavior of nickel hydroxide, but the most acceptable one [8] is based on following two steps Ni(OH)2/NiOOH redox reactions. At the first step, NiOOH the main oxidizing agent is produced. Then, NiOOH oxidizes the hydrazine to its oxidation products. The increase of anodic peak current is explained by the production of more nickel hydroxide through the reaction of NiOOH with hydrazine in second reaction.

Cyclic voltammograms of U-CPE (a, b) and NP-CPE (c, d) in 0.1 mol L−1 NaOH electrolyte in the absence (a, c) and presence (b, d) of 1.0 mmol L−1 hydrazine
It is important to note that, the increased anodic current was proportional to hydrazine concentration, and it has been used as analytical signal for determination of hydrazine on NP-CPE.
Chronoamperometric studies
Chronoamperometry was used to calculate the average diffusion coefficient (D) of hydrazine upon electrooxidation on NP-CPE surface. The first chronoamperogram was recorded in the absence of hydrazine and the rest of them were recorded in different concentrations of hydrazine, which are shown in Fig. 5. By increasing hydrazine concentration, transient current was increased by a Cottrelian pattern. The experimental plots of I vs. t-1/2 were best fitted by a linear model. These linear behaviors show the electrooxidation process of hydrazine on NP-CPE was controlled by diffusion. Slopes of these linear plots (I vs. t-1/2) and Cottrell equation [31] were used to calculate diffusion coefficients of hydrazine.

Chronoamperograms recorded at 500 mV applied potential and in different concentrations of: (a) 0, (b) 125, (c) 275, (d) 750 μmol L−1 hydrazine using NP-CPE. Inset (A) shows Cottrell plots of hydrazine Chronoamperograms
Where D is the diffusion coefficient (cm2 s−1), n is the hydrazine electrooxidation electron number (equal to 4), F is the Faraday constant (96,485 C), A is the electrode surface area (0.072 cm2), C is hydrazine concentration (mol cm−3). The average value of D was obtained as 1.56 × 10−5 cm2 s−1. This value is very close to estimated diffusion coefficient for hydrazine [34].
Amperometric determination of hydrazine using NP-CPE
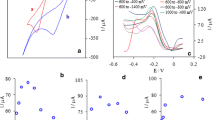
Hydrodynamic amperometry method was optimized for determination of hydrazine. Figure 6a shows a typical recorded amperogram when microliter amounts of hydrazine from standard solution were injected to supporting electrolyte.

a Typical amperometric signals of NP-CPE obtained during successive increments of hydrazine in working potential of 500 mV; b Related calibration plot of amperometric signal in Fig. 6(a), c Long-time amperometric response and response time of the NP-CPE toward a supporting electrolyte with 20 μmol L−1 hydrazine, d Interference effects on the hydrazine amperometric response with successive addition of a (10 μmol L−1 Hydrazine), b (10 mmol L−1 NaCl), c (6.8 mmol L−1 NaF), d (4.0 mmol L−1 Na3PO4), e (10 mmol L−1 Na2CO3), f (1.0 mmol L−1 Na2C2O4) g (10 mmol L−1 Na2SO4)
NP-CPE shows a fast and linear response to hydrazine in a wide concentration range that extends from 1 to 1300 μmol L−1. Related calibration plot has been shown in Fig. 6b. In the optimum condition, following linear Eq. (9) for calibration plot was achieved:
Detection limit
Detection limit (DL) of the amperometric signals were calculated by Eq. 10.
Where m is the slope of calibration plot, and sb is the standard deviation of blank signals respectively. In the amperometric method, unlike other analytical methods, there are a lot of data for the blank signal. Therefore it is possible to calculate DL more accurately. DL was obtained as 0.28 μmol L−1. The reported detection limit has good compatibility with the first point of calibration plot (1 μmol L−1).
Signal stability
In order to show the signal stability, two longtime amperograms in 500 s were recorded. One of them is electrolyte solution without hydrazine and the other is the same electrolyte containing 20 μmol L−1 hydrazine. These two amperograms have been subtracted from each other, and resultant curve was shown in Fig. 6c. As can be seen, in the first 200 s, sensor response was very stable and only 4% decrease in current compared to blank was observed. The signal was reduced to 90% of its original value after 400 s. By a simple coulometric calculation can be shown that the reduction of the signal is not due to the decrease in hydrazine concentration. This stable signal shows that, hydrazine oxidation products do not contaminate the surface of sensor. For an electrochemical sensor, with a fast response time (< 2 s), a stable signal for 200 s is acceptable and more than enough.
Response time
Another attractive feature of the NP-CPE is its fast amperometric response towards the oxidation of hydrazine. The electrode responds rapidly to the concentration change of hydrazine producing steady-state signal in less than 2 s (inset of Fig. 6c).
Precision
In order to show precision of the method, a single electrode was optimized and three consecutive calibration plots were drawn. A relative standard deviation of 1.3% (n = 3) for 5 μmol L−1 hydrazine concentration was achieved.
The storage and regeneration
The ability of a sensor to be stored and reused in a cyclic manner is important for practical applications. Thus the storage ability and reusability of NP-CPE was studied by storing it in air for three months. In a similar study, Heli et al. [17] store their nickel hydroxide based CPE in 0.1 mol L−1 NaOH electrolyte. After each month the electrode surface only regenerated by electrochemical pretreatment method explained in experimental section. The procedure took less than 20 min to prepare electrode. Then, the electrode was used to draw amperometric calibration plot and stored again for another month. After three months these recorded calibration plots were compared with each other. The recorded calibration plot had excellent similarity with only less than ±5% differences in slopes. It was concluded that, due to stability of electrode signal for several months, it is possible to prepare electrode several months forward and use it in a cyclic manner.
Selectivity studies
For an electrochemical sensor, selectivity is the one of most important significant figures. Therefore interfering effect of some common anions and cations on amperometric signal of hydrazine have been studied. Among the studied anions, only iodide and bromide at applied potential were electroactive and drastically increase amperometric signal. Other anions and cations, even at high concentration, hadn’t any interference effect. Figure 6d shows effects of some common anions on amperometric signal of hydrazine.
Interferences of some small organic molecules, such as ethylenediaminetetraacetic acid (EDTA), aniline and hydroquinone on amperometric signals of hydrazine were studied. Among these molecules, aniline was the only interferences. It produced a signal equal to hydrazine signal in similar concentration. Upon addition of hydroquinone and EDTA to supporting electrolyte, they didn’t produce any amperometric signals.
Humic substances were present in surface waters and groundwaters in concentrations ranging from less than 1 ppm to 100 ppm. Therefore, Interference effects of humic and fulvic acids on amperometric signal of hydrazine were studied in concentration range extended from 10 to 150 ppm. The NP-CPE, armed with a nafion polymeric layer, brilliantly responded to hydrazine concentration when the humic and fulvic acids were present in concentration above 100 ppm. Fig. S2 (ESM) shows amperometric response of the NP-CPE to hydrazine when different concentration of humic and fulvic acids were added to supporting electrolyte.
The results have been collected in Table S.2, where tolerance limit was defined as the maximum concentration ratio of interfering species to hydrazine, which cause an error less than 5%. According to results, we can conclude that this sensor able to show good performance in aqueous media in the presence of humic substances, inorganic cations and anions.
Accuracy of method
In order to study the accuracy of method, three tap water samples were spiked with hydrazine. In all samples, the final concentrations of hydrazine were 200 μmol L−1. The three determinations were performed in different days and with separate calibration plots. The accuracy of method was 97 ± 2.3%.
Significant figures of this method along with some other important parameters have been presented in Table 1 where they are compared with some Ni-based hydrazine sensors. As it is seen from Table 1, NP-CPE is competitive to other sensors and in some cases, its response time, linear range, detection limit and sensitivity is better. Most of the hydrazine electrochemical sensors which presented in literature were made from complicated composites with several components. Most of the times, their components had unspecified role in electrochemistry. The results show that NP-CPE has several advantages compared to other sensors. The simplicity of components, ease of preparation, low cost, fast response time, competitive detection limit and sensitivity are its advantages.
It is important to note, in a similar work Ji et al. [28] were grown β-nickel hydroxide nanoplates on the surface of a copper rod as a hydrazine sensor. It is interesting to compare NP-CPE with that one. Because in NP-CPE β-nickel hydroxide was in the bulk and Ji et al. used it on surface of a copper rod. When nanoparticles have been grown on surface, it is anticipated to observe better performance compared to the same nanoparticles in the bulk. But at least in this case, there is no superiority in significant figures. For example NP-CPE response time (< 2 s) is better than mentioned Cu rod electrode (3 s). When comparing surface coverages of these two sensors, despite the fact that surface coverage of Cu rod sensor was five times more, but there was no significant difference in sensitivity. In addition, Cu rod sensor had very complicated cyclic voltammograms due to its copper base but, in NP-CPE only two well-defined Ni(OH)2/NiOOH redox peaks were present. This simplicity made electrochemical studies much easier.
Conclusions
Uniform nanoplatelets were synthesized by a simple hydrothermal method. Electrochemical behaviors were studied in carbon paste media. An amperometric sensor was worked out and optimized for quantification of hydrazine. The simplicity, sensitivity and fast response time were main advantages of NP-CPE. Electrocatalytic oxidation of hydrazine on NP-CPE is possibly attractive for researchers who are involved in direct hydrazine fuel cell studies.
Change history
09 July 2019
The authors of “Electrocatalytic oxidation and amperometric determination of hydrazine using a carbon paste electrode modified with β-nickel hydroxide nanoplatelets (Microchimica Acta (2019) 186:441)” wish to update the incorrect panel/labels of Figure 3.
References
George M, Nagaraja KS, Balasubramanian N (2008) Spectrophotometric determination of hydrazine. Talanta 75(1):27–31. https://doi.org/10.1016/j.talanta.2007.09.002
Selim S, Warner CR (1978) Residue determination of hydrazine in water by derivatization and gas chromatography. J Chromatogr A 166(2):507–511. https://doi.org/10.1016/S0021-9673(00)95634-6
Oh J-A, Park J-H, Shin H-S (2013) Sensitive determination of hydrazine in water by gas chromatography–mass spectrometry after derivatization with ortho-phthalaldehyde. Anal Chim Acta 769:79–83. https://doi.org/10.1016/j.aca.2013.01.036
Smolenkov AD, Shpigun OA (2012) Direct liquid chromatographic determination of hydrazines: a review. Talanta 102:93–100
Hamidi H, Bozorgzadeh S, Haghighi B (2017) Amperometric hydrazine sensor using a glassy carbon electrode modified with gold nanoparticle-decorated multiwalled carbon nanotubes. Microchim Acta 184(11):4537–4543. https://doi.org/10.1007/s00604-017-2480-0
Rees NV, Compton RG (2011) Carbon-free energy: a review of ammonia-and hydrazine-based electrochemical fuel cells. Energy Environ Sci 4(4):1255–1260
Serov A, Kwak C (2010) Direct hydrazine fuel cells: a review. Appl Catal B 98:1):1–1):9. https://doi.org/10.1016/j.apcatb.2010.05.005
Miao Y, Ouyang L, Zhou S, Xu L, Yang Z, Xiao M, Ouyang R (2014) Electrocatalysis and electroanalysis of nickel, its oxides, hydroxides and oxyhydroxides toward small molecules. Biosens Bioelectron 53:428–439
Zhu X, Zhong Y, Zhai H, Yan Z, Li D (2014) Nanoflake nickel hydroxide and reduced graphene oxide composite as anode materials for high capacity lithium ion batteries. Electrochim Acta 132:364–369. https://doi.org/10.1016/j.electacta.2014.03.132
Lakshmi V, Ranjusha R, Vineeth S, Nair SV, Balakrishnan A (2014) Supercapacitors based on microporous β-Ni(OH)2 nanorods. Colloids Surf A Physicochem Eng Asp 457:462–468. https://doi.org/10.1016/j.colsurfa.2014.06.016
Wang D, Yan W, Vijapur SH, Botte GG (2012) Enhanced electrocatalytic oxidation of urea based on nickel hydroxide nanoribbons. J Power Sources 217:498–502. https://doi.org/10.1016/j.jpowsour.2012.06.029
Qiao NQ, Zheng JB (2012) Nonenzymatic glucose sensor based on glassy carbon electrode modified with a nanocomposite composed of nickel hydroxide and graphene. Microchim Acta 177(1–2):103–109. https://doi.org/10.1007/s00604-011-0756-3
Martínez-Periñán E, Revenga-Parra M, Gennari M, Pariente F, Mas-Ballesté R, Zamora F, Lorenzo E (2016) Insulin sensor based on nanoparticle-decorated multiwalled carbon nanotubes modified electrodes. Sens Actuators B 222:331–338. https://doi.org/10.1016/j.snb.2015.08.033
Chen Z, Nai J, Ma H, Li Z (2014) Nickel hydroxide nanocrystals-modified glassy carbon electrodes for sensitive l-histidine detection. Electrochim Acta 116:258–262. https://doi.org/10.1016/j.electacta.2013.10.153
Babaei A, Yousefi A, Afrasiabi M, Shabanian M (2015) A sensitive simultaneous determination of dopamine, acetaminophen and indomethacin on a glassy carbon electrode coated with a new composite of MCM-41 molecular sieve/nickel hydroxide nanoparticles/multiwalled carbon nanotubes. J Electroanal Chem 740:28–36. https://doi.org/10.1016/j.jelechem.2014.12.042
Zhang X, Huang Y, Gu A, Wang G, Fang B, Wu H (2012) Hydrogen peroxide sensor based on carbon nanotubes/β-Ni (OH) 2 nanocomposites. Chin J Chem 30(3):501–506
Heli H, Sattarahmady N, Vais RD, Karimian K (2014) Nickel hydroxide nanopetals: one-pot green synthesis, characterization and application for the electrocatalytic oxidation and sensitive detection of montelukast. Sensors Actuators B 196:631–639. https://doi.org/10.1016/j.snb.2014.02.057
Hajjizadeh M, Jabbari A, Heli H, Moosavi-Movahedi AA, Haghgoo S (2007) Electrocatalytic oxidation of some anti-inflammatory drugs on a nickel hydroxide-modified nickel electrode. Electrochim Acta 53(4):1766–1774. https://doi.org/10.1016/j.electacta.2007.08.026
Nesterov B, Korovin N (1966) Anodic oxidation of hydrazine on smooth nickel in alkaline solution. Elektrokhimiya 2(11):1296
Sakamoto T, Asazawa K, Sanabria-Chinchilla J, Martinez U, Halevi B, Atanassov P, Strasser P, Tanaka H (2014) Combinatorial discovery of Ni-based binary and ternary catalysts for hydrazine electrooxidation for use in anion exchange membrane fuel cells. J Power Sources 247:605–611. https://doi.org/10.1016/j.jpowsour.2013.08.107
Abbaspour A, Khajehzadeh A, Ghaffarinejad A (2009) Electrocatalytic oxidation and determination of hydrazine on nickel hexacyanoferrate nanoparticles-modified carbon ceramic electrode. J Electroanal Chem 631(1–2):52–57. https://doi.org/10.1016/j.jelechem.2009.03.011
Adekunle AS, Ozoemena KI (2010) Electron transport and electrocatalytic properties of MWCNT/nickel nanocomposites: hydrazine and diethylaminoethanethiol as analytical probes. J Electroanal Chem 645(1):41–49. https://doi.org/10.1016/j.jelechem.2010.04.010
Azad UP, Ganesan V (2011) Determination of hydrazine by polyNi(II) complex modified electrodes with a wide linear calibration range. Electrochim Acta 56(16):5766–5770. https://doi.org/10.1016/j.electacta.2011.04.051
Jeevagan AJ, John SA (2013) Synthesis of non-peripheral amine substituted nickel(ii) phthalocyanine capped gold nanoparticles and their immobilization on electrode for the electrocatalytic oxidation of hydrazine. RSC Adv 3(7):2256–2264. https://doi.org/10.1039/c2ra22895e
Kazemi SH, Hosseinzadeh B, Zakavi S (2015) Electrochemical fabrication of conducting polymer of Ni-porphyrin as nano-structured electrocatalyst for hydrazine oxidation. Sensors Actuators B Chem 210:343–348. https://doi.org/10.1016/j.snb.2014.12.131
Pandey PC, Panday D (2016) Novel synthesis of nickel–iron hexacyanoferrate nanoparticles and its application in electrochemical sensing. J Electroanal Chem 763:63–70. https://doi.org/10.1016/j.jelechem.2015.12.048
Zheng L, J-f S (2009) Ni(II)–baicalein complex modified multi-wall carbon nanotube paste electrode toward electrocatalytic oxidation of hydrazine. Talanta 79(2):319–326. https://doi.org/10.1016/j.talanta.2009.03.056
Ji R, Huang Y, Wang L, Yu L, Wang J, Wang G, Zhang X (2013) Synthesis of Ni(OH)2 nanoplates on cu rod and its applications for electrochemical sensors. Mater Res Bull 48(10):3729–3734. https://doi.org/10.1016/j.materresbull.2013.05.115
Anu Prathap MU, Anuraj V, Satpati B, Srivastava R (2013) Facile preparation of Ni(OH)2–MnO2 hybrid material and its application in the electrocatalytic oxidation of hydrazine. J Hazard Mater 262:766–774. https://doi.org/10.1016/j.jhazmat.2013.09.050
Anthony J, Bideaux R, Bladh K, Nichols M (1997) Handbook of mineralogy, halides, hydroxides, oxides, Vol. III. Mineral Data Pub
Bard AJ, Faulkner LR (eds) (2001) Electrochemical methods, fundamentals and applications, 6th edn. Wiley, New York
Sattarahmady N, Heli H, Faramarzi F (2010) Nickel oxide nanotubes-carbon microparticles/Nafion nanocomposite for the electrooxidation and sensitive detection of metformin. Talanta 82(4):1126–1135. https://doi.org/10.1016/j.talanta.2010.06.022
Laviron E (1979) General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J Electroanal Chem Interfacial Electrochem 101(1):19–28. https://doi.org/10.1016/S0022-0728(79)80075-3
Karp S, Meites L (1962) The Voltammetric characteristics and mechanism of Electrooxidation of hydrazine. J Am Chem Soc 84(6):906–912. https://doi.org/10.1021/ja00865a006
Acknowledgements
The authors wish to express their thanks to the research council of university of Maragheh for financial support of this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author(s) declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: Unfortunately in the online version of the article, Fig. 3 has four parts which should be labeled as a, b, c, and d; but incorrectly labeled as a, b, d, and d. There is two “d” in and no “c”. The correct image is given in this article.
Electronic supplementary material
ESM 1
(DOCX 260 kb)
Rights and permissions
About this article

Cite this article
Avanes, A., Hasanzadeh-Karamjavan, M. & Shokri-Jarcheloo, G. Electrocatalytic oxidation and amperometric determination of hydrazine using a carbon paste electrode modified with β-nickel hydroxide nanoplatelets. Microchim Acta 186, 441 (2019). https://doi.org/10.1007/s00604-019-3555-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-019-3555-x