
Abstract
This paper reports on the development of an amperometric method for the determination of myo-inositol. The method involves coating of a glassy carbon electrode (GCE) with a molecularly imprinted polymer (MIP) and reduced graphene oxide (RGO) that was modified with nickel nanoparticles (NiNPs). The MIP was prepared by electropolymerization of pyrrole on the surface of the GCE in the presence of myo-inositol molecules. The construction steps of the modified electrode were monitored via cyclic voltammetry, atomic force microscopy, scanning electron microscopy and X-ray Photoelectron Spectroscopy. The results were evaluated using differential pulse voltammetry, in which hexacyanoferrate was used as an electrochemically active probe. Under optimized experimental conditions, the imprint-modified GCE has a linear response in the 1.0 × 10−10 mol L−1 to 1.0 × 10−8 mol L−1 concentration range, with a 7.6 × 10−11 mol L−1 detection limit and an electrochemical sensitivity of 4.5 μA·cm-2 μmol−1. The method showed improved selectivity even in the presence of molecules with similar chemical structure. The GCE modified was successfully applied to the determination of myo-inositol in sugarcane vinasse where it yielded recoveries that ranged from 95 to 102%.

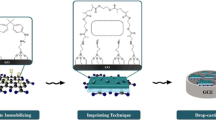
Schematic presentation of molecularly imprinted polymer (MIP) on a glassy carbon electrode (GCE) modified with nickel nanoparticles (NiNP) anchored in reduced graphene oxide (RGO). The resulting MIP/NiNP/RGO-GCE was used for indirect determination of myo-inositol.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Myo-inositol is a cyclic polyalcohol found in animals, plants, seeds, fungi and some bacteria [1]. Its benefits to human health include treatment and prevention of anxiety [2, 3], dietary supplement for the promotion of female fertility [4], besides playing an important role in monitoring the treatment of cardiovascular diseases [5], Alzheimer [6], diabetes and renal disease [7].
Several methods have been used in the determination of myo-inositol, including chromatographic methods such as gas chromatography/mass spectrometry (GC-MS) [8], high performance liquid chromatography (HPLC) [9], liquid chromatography tandem-mass spectrometry (LC-MS/MS) [10] high-performance anion-exchange chromatography with pulsed amperometric detection [11, 12]. As can be noted, no reports have been published in the literature regarding the use of electrochemical methods for direct determination of myo-inositol.
While the methods reported in the literature offer relatively good selectivity and low detection limit for myo-inositol, they present some disadvantages such as high cost of equipment and time-consuming sample pretreatment apart from requiring the use of well-trained professionals to operate them. By contrast, electrochemical assays involve a fast, reliable, simple and low-cost determination mechanism with high sensitivity and selectivity. In addition, this method is widely applied in medical, biological and environmental analyses [13,14,15,16].
Although myo-inositol is known to be electroactive, the direct oxidation of myo-inositol does not exhibit significant current at a conventional glassy carbon electrode (GCE). On the other hand, for electrodes modified with metal nanoparticles (such as Ni, Cu and Au), oxidation occurs in alkaline medium [13]. These conditions do not guarantee selectivity of the electrode, since other molecules such as amino sugars, alditols, and acidic sugars tend to cause interference in the process.
To tackle this problem, researchers have developed a combination of nanomaterials with molecular imprinting technology, thus incorporating the selectivity feature into the construction of nanomaterial-based electrodes [14]. Usually, for the formation of molecular imprinting polymer (MIP), a monomer is polymerized in the presence of the molecule of interest. Subsequently, this molecule is removed from the polymer matrix leaving cavities tailored for further recognition.
Many studies show that the combination of MIP with nanomaterials can increase the electrochemical signal, as it is the case of reduced graphene oxide (RGO) and nickel nanoparticles (NiNP) [15, 16]. Nickel nanoparticles have high conductivity, fast electron transfer and good stability and low cost [17]. RGO is a derivative of graphene that possesses excellent electrical conductivity and high surface area, apart from having several functional groups found to be essentially important for its functionalization [18]. The introduction of nickel nanoparticles onto RGO sheets helps to prevent the nanoparticles from undergoing agglomeration, due to the peculiar structure of RGO. In addition, the use of RGO provides a greater number of sites for the deposition of nanoparticles. One will note, however, that the high surface area exhibited by these modified electrodes contributes to a better electropolymerization, since more sites to form the complementary cavities of the molecule of interest are formed.
Among the wide range of methods employed for the preparation of molecularly imprinted polymer (MIP), one can mention free radical polymerization, photopolymerization, and electropolymerization [18]. Electropolymerization has advantages over other methods in that it allows one the possibility of controlling the thickness of the electrodeposited film and its morphology, this can be achieved through controllable parameters, such as applied potential difference and cyclic scan. In view of that, we chose the mechanism involving electropolymerization of polypyrrole (PPy) for the development of our method as a result of its stability, excellent biocompatibility, ease of preparation and good conductivity [19]. A further advantage associated with the use of PPy is its ability to incorporate anions into its polymer structure during electropolymerization, rendering myo-inositol easily imprinted for subsequent molecular recognition [20].
After its construction, the modified electrode was applied for the determination of myo-inositol in sugarcane vinasse sample. Sugarcane vinasse was chosen because it is a highly complex matrix that contains several molecules which are similar to myo-inositol. Some molecules found in sugarcane vinasse that deserve mentioning include D-mannitol, glycerol, L-arabitol and erythritol. Clearly, the presence of these molecules makes sugarcane vinasse interesting for the evaluation of selectivity Moreover, the concentration of these compounds in the matrix can reach up to 0.115 g L−1 [21]. Biorefineries make the reutilization of these compounds possible in addition to contributing towards aggregating value to them. The aforementioned benefits of myo-inositol makes it suitable for application in the pharmaceutical industry.
The present work reports the development of an electrochemical method for the determination of myo-inositol. To this end, nickel nanoparticles anchored on reduced graphene oxide were electrodeposited on GCE. After that, the electropolymerization of PPy and myo-inositol was performed on the modified NiNP/RGO-GCE for the formation of molecularly imprinted polymer. All the steps involving the development of the electrode were carried out electrochemically and under optimized conditions The method showed good stability, reproducibility, rapid construction and selectivity for the determination of myo-inositol in sugarcane vinasse sample.
Experimental
Materials and instruments
Pyrrole (purity: ≥ 98%), myo-inositol (purity: ≥ 98%), potassium ferricyanide (K3[Fe(CN)6], purity: 99%), nickel sulfate and graphene oxide (GO, purity: > 95%) were purchased from Sigma–Aldrich (http://www.sigmaaldrich.com). Potassium chloride, potassium dihydrogen phosphate, dipotassium hydrogen phosphate, lithium perchlorate and sodium sulfate were employed as supporting electrolyte and were also purchased from Sigma–Aldrich. The redox solution applied as probe was 5.0 × 10−3 mol L−1 K3Fe(CN)6 in 0.10 mol L−1 KCl. Phosphate buffer (PB, 0.10 mol L−1) of pH 6 was used as the rebinding solution to myo-inositol.
Electrochemical voltammetric measurements were performed using a potentiostat Autolab PGSTAT30 coupled to a microcomputer that records and stores data using the control software Nova 1.11. A conventional electrochemical cell with three electrodes was used, Ag/AgCl (KCl 3.0 mol L−1) being the reference electrode, platinum wire as the auxiliary electrode, and a galssy carbon electrode (GCE), with diameter of 3.0 mm, as working electrode. To carry out the measurements via differential pulse voltammetry (DPV), the following conditions were applied: pulse amplitude of 50 mV, pulse time of 100 ms, potential step of 2 mV and scan rate of 10 mVs−1. A magnetic stirrer was used for convective transport when necessary. All experiments were carried out at room temperature.
Cleaning procedure for GCE
The GCE electrode was polished with 0.30 μm alumina powder on a felt and electrochemically polished by successive scans at potential range of −0.50 to +1.5 V in 0.50 mol L−1 H2SO4 at 50 mVs−1 until voltammogram cyclic characteristics of a cleaned GCE were obtained.
Preparation of RGO-GCE
The RGO was eletrodeposited on GCE as reported in previous works by the authors [22]. Briefly, 0.50 mg mL−1 of graphene oxide suspension (acquired from Sigma Aldrich) was dispersed in 0.10 mol L−1 Na2SO4, used as supporting electrolyte. For the electrodeposition process, the potential applied was −1.5 V for 600 s. The electrode was subsequently dried at room temperature.
Preparation of NiNP on RGO-GCE
Following the drying of the RGO, NiNP was prepared as previously reported [23]. The NiNP was electrodeposited onto the RGO in solution containing 5.0 × 10−3 mol L−1 NiSO4 in 0.10 mol L−1 Na2SO4. The film was deposited under a constant potential of −1.3 V, using chronoamperometry with charge controller, the process continued until the charge of 5 mC was reached as demonstrated in SI, Fig. S1.
Preparation of imprinted myo-inositol
The MIP was prepared by cyclic voltammetry (CV) in the range of 0.0 V to 1.0 V at 50 mV s−1 for 10 cycles in 0.10 mol L−1 LiClO4 solution containing 7.0 × 10−3 mol L−1 myo-inositol and 2.5 × 10−2 mol L−1 pyrrole. The MIP modified electrode was denoted as MIP/NiNP/RGO-GCE.
For the formation of imprinted cavities, the MIP/NiNP/RGO-GCE was overoxidized in solution of 0.10 mol L−1 NaOH using CV. The CV employed here ranged from 0.0 V to 1.8 V at 50 mV s−1, the process lasted until the stabilization of the current, which occurred in the 8th cycle. This mechanism enables the removal of myo-inositol trapped in the polymer matrix, thus forming cavities. These cavities exhibit the same size and shape, and are capable of interacting with the same positioning of the functional groups of the template molecule. Non-imprinted polymer modified electrode (NIP/NiNP/RGO-GCE) used as control was prepared under the same aforementioned experimental conditions without adding myo-inositol.
Experimental measurements
To rebind the target molecule prior to performing electrochemical measurements, the MIP/NiNP/RGO-GCE was immersed in a PB (pH 6.0) containing 5.0 × 10−8 mol L−1 of myo-inositol under mildly magnetic stirring at 500 rpm for 20 min. The electrode was then washed carefully with distilled water to remove the physisorbed compounds. After that, the electrode MIP/NiNP/RGO-GCE was used as working electrode in a three-electrode conventional cell. Solution of 5.0 × 10−3 mol L−1 [Fe(CN)6]3−/4- in 0.10 mol L−1 KCl was used as an electrochemical active probe to evaluate the performance of the electrode. This probe was chosen due to the poor electroactivity of myo-inositol under neutral pH. When the imprinted cavities are formed, they can provide pathway for the diffusion of the probe. When myo-inositol are rebound in these complementary cavities, they block the transfer of electrons from the [Fe(CN)6]3−/4− probe to the electrode surface. In this sense, the electrochemical signal intensity is found to be related to myo-inositol concentration. As a result, all the electrochemical experiments were carried out in 0.1 mol L−1 KCl containing 5.0 × 10−3 mol L−1 [Fe(CN)6]3−/4− using different electrochemical techniques, including CV and DPV.
The analytical data were obtained based on the variation of the current. The current shift (ΔI) was calculated taking into account the oxidation peak currents before and after the combination of the cavities formed with myo-inositol. After each experimental run, the MIP/NiNP/RGO-GCE was cleaned by immersing the electrode in 0.10 mol L−1 NaOH with CV scanning between 0.0 and 1.8 V until stabilization of cycles. The electrode was reused after this cleaning process.
The figures of merit including limit of detection (LOD), limit of quantification (LOQ) and amperometric sensitivity (As) were calculated. LOD and LOQ were calculated according to the equation LOD = 3.3 SD/S and LOQ = 10 SD/S, where SD is the standard deviation of the intercept, and S is the slope of the calibration curve.
To study the morphology of the original surface, scanning electron microscope with field emission gun (SEM-FEG) of Jeol, model JSM 7500F was used. Furthermore, atomic force microscopy (AFM) of Bruker nanoscope V, operating in tapping mode with 256 × 256 pixels resolution and scan rate was 2.0 μm s−1 was also used to characterize the surface. For the monitoring of chemical interactions of the surface, X-ray Photoelectron Spectroscopy, XPS, model commercial spectrometer (UNI-SPECS UHV) were used. The analysis was performed at a pressure of less than 10−7 Pa. Here MgKα line was employed (hν = 1253.6 eV) and the analyzer pass energy was set to 10 eV. Shirley’s method was used to substract the core-level spectra of the inelastic background of Ni1s, N1 s, O1s and C1s electron. The composition (in %) was determined by Scofield’s sensitivity factors of the elements with an accuracy of ±10% based on the ratio of the corrected relative peak areas. Aiming at correcting the binding energy scale of the spectra, the value of 285 eV was set for C1s hydrocarbon component. Multiple Voigt profiles were used without placing constraints to fit the spectra. The width at half maximum (FWHM) varied between 1.2 and 2.1 eV while accuracy of the peak positions was ±0.10 eV.
Detection of myo-inositol in sugarcane vinasse sample
The vinasse samples were centrifuged at 4000 rpm for 20 min aiming at removing the solid particles. The supernatant was collected and filtered with filters 0.47 and 0.22 μm of porosity. Subsequently, 5.0 μL of the filtered vinasse were diluted in 10 μL of PB under pH 6. For the analyte determination myo-inositol concentrations ranging from 2.0 × 10−10 mol L−1 to 6.0 × 10−10 mol L−1 were added to the sample by the standard addition method. Recovery tests were done by adding a defined concentration of myo-inositol to the samples.
Results and discussion
Preparation of the GCE modified with MIP/NiNP/RGO
In order to obtain a good molecular imprint, the substrate needs to guarantee a good adhesion to the monomer to be electropolymerized. In view of that, nanomaterials are regarded interesting structures used as substrates for the formation of MIPs. This class of materials is endowed with specific characteristics, such as good electrical properties, strong adsorption ability, high surface reaction activity, small particle size and good surface properties. In this work, reduced graphene oxide with nickel nanoparticles was employed with the aim of increasing the surface area and conductivity, thus allowing us to obtain a highly sensitive electrode. The anode peak current of NiNP/RGO-GCE is found to be about 3.2 times greater than that of NiNP/GCE, as it has already been demonstrated by our research group in a recently published work [24]. This can be attributed to the larger surface and the better conductivity of the surface of the modified GCE.
The electropolymerization process on the NiNP/RGO-GCE using CV is shown in Fig. S2. The formation of the PPy film can be easily seen by increasing the intensity of the peaks as the film grows. A large oxidation peak was observed at the peak potential of +0.15 V. During this process, myo-inositol diffuses towards the electrode surface and is trapped in the polymer matrix; this can be explained by the ability of these molecules to interact with the pyrrole units. Theoretically, this interaction is considered possible due to the interactions of hydrogen between the hygrogen of the hydroxyl group of the template molecule and the nitrogen atom of the N-H group of the pyrrole units.
The stepwise preparation of the modified electrodes was characterized by CV, using 0.10 mol L−1 KCl containing 5.0 × 10−3 mol L−1 [Fe(CN)6]3−/4− at 50 mV s−1 in the range of −0.20 to 0.60 V as shown in Fig. 1. In curve c, well defined reversible redox peaks can be observed on the bare GCE. When the GCE is modified with NiNP and RGO, an increase in the peak current is observed. This result suggests that the modification of the GCE results in a larger electrochemical surface area while contributing towards accelerating electron transfer of [Fe(CN)6]3−/4- probe (curve e). The modified surface provides more room for the formation of the MIP, thus increasing its capacity by up to 15 times [25]. However, when the NiNP/RGO-GCE is electropolymerized with PPy and myo-inositol to form MIP/NiNP/RGO-GCE, the pair of the redox peaks undergoes a noticeable decline as can be seen in curve a. This is attributed to the fact that the [Fe(CN)6]3−/4- molecules are unable to penetrate through the polymer layer and reach the surface of the electrode. The same behavior was observed for NIP/NiNP/RGO-GCE after electropolymerization, as demonstrated in Fig. S3.

Cyclic voltammograms of (a) MIP/NiNP/RGO-GCE after electropolimerization (b) bare GCE, (c) MIP/NiNP/RGO-GCE after 20 min rebinding in 5.0 × 10−9 mol L−1 myo-inositol solution (d) MIP/NiNP/RGO-GCE sensor after removal of myo-inositol (e) NiNP/RGO-GCE. All measurements were performed in solution containing 5.0 × 10−3 mol L−1 [Fe(CN)6]3−/4- in 0.10 mol L−1 KCl, scan rate 50 mV s−1
When myo-inositol is removed from the polymer matrix, the current response increases, as one can observe in curve d. It seems that cavities are formed when the template is removed from the polymeric matrix. The formation of these cavities enables the electronic transfer, which in turn allows the reversible redox peak to be observed again. When the MIP/NiNP/RGO-GCE is placed in a solution containing 5.0 × 10−8 mol L−1 of myo-inositol for 20 min, a decline is observed in the electrode response - see curve b. This result shows that some of the cavities can be rebinding to the myo-inositol, resulting in the availability of less cavities for the transfer of electrons from the redox probe to the electrode surface.
The removal of myo-inostol templates from the polymeric matrix was necessary for the creation of complementary imprinted sites for subsequent rebinding. To this end, the electrochemical overoxidation process of PPy was employed. The irreversible oxidation is referred to as overoxidation of the PPy, as reported by Beck et al. [26]. Overoxidation is found to occur relatively more easily in the presence of strong nucleophiles, such as OH−, at potential around 1.0 V [27]. In alkaline solutions, myo-inositol undergoes oxidation on the surface modified with NiNP/RGO-GCE at the potential of 0.70 V, as can be seen in Fig. 2. Nonetheless, the oxidation is not observed in neutral and acid solutions [13]. Figure 2b shows the first and the last cycle of overoxidation of PPy by scanning potential in the range of 0.0 V to +1.8 V in 0.10 mol L−1 NaOH at a rate of 50 mV s−1 in the presence and absence of myo-inositol. During the elution scan, an irreversible voltammetric peak is observed at 0.70 V when myo-inositol is present. Another wide peak was also found at 1.3 V, this is attributed to PPy overoxidation. Essentially, electrochemical overoxidation leads to the removal of myo-inositol from the polymer matrix while carboxylic groups are inserted. As such, subsequent scan cycles exhibit a decrease in peak current as a result of the conductivity loss of PPy during this process [19].

Cyclic voltammogram for (a) NiNP/RGO-GCE in (blue line) 0.1 mol L−1 NaOH and (black line) 1.0 × 10−4 mol L−1 of myo-inositol in 0.1 mol L−1 Na2SO4 and (red line) 0.1 mol L−1 NaOH. In (b) the first and the last cycle of overoxidation for PPy in 0.1 mol L−1 NaOH in the presence (red line) and absence (black line) of myo-inositol
Characterization of MIP/NiNP/RGO-GCE
The surface morphological analysis was conducted by SEM and AFM. Fig. S4 shows surface morphologies of NiNP/RGO-GCE and MIP/NiNP/RGO-GCE using SEM. Fig. S4a and Fig. 4Sb shows the image of a uniform electrodeposition of NiNP on the graphene nanosheets with a homogeneous distribution on the GCE. This indicates the success of the deposition of nickel nanoparticles onto the RGO-GCE surface, with dimensions of 50 nm in diameter. As can be seen in Fig. S4c, MIP/NiNP/RGO-GCE was successfully constructed. Fig. S4d shows MIP/NiNP/RGO-GCE following the overoxidation of PPy for myo-inositol removal, exhibiting a smoother and less wrinkled surface. The overoxidation process leads to some rearrangement of the polymer chain packing. By analyzing the images, it appears that the overoxidized structure has more pores compared to the PPy film. In both cases, it is possible to see the typical ‘cauliflower’ morphology.
The AFM 3D is shown in Fig. 3. In Fig. 3a, one can observe the homogeneous modification of the electrode by NiNP/RGO, with nanoparticles having an average of 62 nm of diameter and 28 nm of height. The value of roughness, expressed in terms of the root-mean-square (RMS) value (RMS is proportional to roughness), is 10 nm. A marked change in roughness is evident when the electrode is modified with the polymeric film during the molecular imprinting process. The RMS values obtained for the MIP was 88 nm (Fig. 3b) and for the NIP was 70 nm (Fig. 3d). The average height of the PPy film before the overoxidation process was 297 nm; this shows that the film was relatively flat and compact. The RMS value for MIP after removal of myo-inositol is 42 nm (Fig. 3c). When the PPy film is overoxidized it becomes more porous, and its original morphology is altered due to degradation. This degradation process is able to remove the imprinted molecule, generating complementary cavities.

AFM imagens, (a) NiNP/RGO-GCE electrode, (b) MIP before template removal, (c) MIP after template removal and (d) NIP
The surface chemistry of the MIP was also characterized using XPS (Fig. 4). This characterization was aimed at confirming the imprinting process in addition to investigating the oxidation level (verifying the effect of overoxidation conditions on the imprinting process) as well as the nature of the chemical bonding of the polymer. The high-resoluton C1s spectra of the MIP after electropolymerization (Fig. 4a) can be deconvoluted into five components. A component with BE, corresponding to peaks 285.0 (C-H), which is derived from the aromatic carbon of PPy. The remaining components corresponding to peaks 285.5 (C-H), 286.8 (C-O), 287.7 (C=O) and 289.2 (O-C=O) eV and are attributed to the groupings that occur during the formation of the polymer film. During the electropolymerization process, there is a possible occurrence of oxidation of polypyrrole carbons, generating C-OH group in the first step and C=O group in the subsequent step.

Deconvoluted C1s (a), O1s (b) and N1 s (c) XPS spectrum beforte overoxidation, and C1s (d) and O1s (e) XPS spectrum after overoxidation of the MIP/NiNP/RGO-GCE
Figure 4c shows the presence of nitrogen. In the polymer matrix PPy, N atoms can be present in the form of imine (=N- with binding energy, BE at 399.4 eV), amine (-NH; BE ∼ 399.5 eV) and electron deficient nitrogen (N+; BE ∼ 401.5 eV) [28].
The high-resolution O1s spectra of the PPy film can be deconvoluted into four components corresponding to oxygen atoms in different functional groups at 531.8 (O=C), 532.3 (C-O), 533.6 (O-C=O) and 535.0 (H2O) (Fig. 4b). The formation of these groups reinforces the possible occurrence of polypyrrole overoxidation during electropolymerization. For illustration purposes, the curves in Fig. 4b show the O1s spectra after the overoxidation of molecularly imprinted PPy film aimed at removing myo-inositol. The increase observed in the intensity of the O-C, O=C and O-C=O groups indicates the superoxidation of the film.
Optimization of experimental parameters
The main parameters that can crucially affect the formation of the MIP are: pyrrole concentration, myo-inositol concentration, number of cycles during electropolymerization, number of cycles of pyrrole overoxidation for the removal of the template molecule, incubation time and pH of the incubation solution, as shown in Fig. S5. These parameters were chosen because they are key factors that allow one to determine the thickness, physical stability of the polymer and quality of the cavities formed on the electrode. To evaluate the ideal conditions for the development of the MIP, the current change [Fe(CN)6]3- /4- (ΔI) was evaluated using DPV. In the SI, we discuss how each parameter affects the development of the MIP.
In short, the optimized experimental conditions included the following: electropolymerization carried out within the potential range of 0.0 to 1.0 V for 10 cycles in 0.10 mol L−1 LiClO4 solution containing 7.0 × 10−3 mol L−1 myo-inositol and 2.5 × 10−2 mol L−1 pyrrole. For the template removal, the polymer was overoxidized by scanning the potential in the range of 0.0 to 1.8 V for 10 cycles. Myo-inositol rebinding in the formed cavities occurred after 25 min of treatment in PB under pH 6.
Analytical performance
Sensitivity is one of the most important features of the imprinted electrode. In this study, both the sensitivity and linear range of the MIP/NiNP/RGO-GCE were evaluated by DPV under pH 6.0 using 5.0 × 10−3 mol L−1 [Fe(CN)6]3−/4− as probe. Figure 5 demonstrates the analytical and DPV curves of the MIP/NiNP/RGO-GCE after rebinding with a series of concentrations of myo-inositol solutions. The peak current was found to decrease as the myo-inositol concentration was increased. This can be said to be as a result of the occupation of the cavity sites in the MIP film by myo-inositol, which blocks the diffusion of the Fe(CN)63−/4- probe. The decreased peak current (ΔI) is related to the concentration of myo-inositol. Accordingly, the reduction in ΔI was proportional to the myo-inositol concentration in two linear ranges: 1.0 × 10−10 to 1.0 × 10−9 mol L−1 and 1.0 × 10−9 to 1.0 × 10−8 mol L−1, this is the first interval used to calculating the figures of merit. The linear regression equation was ΔI(μA) = 4.5 × 102 Cmyo-inostol + 3.24 × 10−6 with a correlation coefficient of 0.997, LOD of 7.6 × 10−11 mol L−1, LOQ of 2.0 × 10−10 mol L−1 and amperometric sensitivity (As) of 4.5 mA L mol−1, (n = 3).

Calibration curves for myo-inositol detection using MIP/NiNP/RGO-GCE in 5.0 × 10−3 mol L−1 [Fe(CN)6]3−/4- after 20 min of rebinding in phosphate buffer of pH 6.0. DPV in accordance with myo-inositol concentration in the range of 1.0 × 10−12 to 1.0 × 10−8 mol L−1 are in the bottom-left inset
The existence of two linear ranges is attributed to the affinity between the imprinted sites and myo-inositol. When the myo-inostol concentration is lower, the molecules tend to prefer the cavities with high affinity cavities located in the upper part of the film. Conversely, the lower affinity sites located more deeply are occupied when the myo-inositol concentration is higher, causing a decline in the linear slope.
The performance of the MIP/NiNP/RGO-GCE was compared with that of other methods reported in the literature for myo-inositol detection. The methods reported in the literature include the following: gas chromatography/mass spectrometry, liquid chromatography tandem mass spectrometry, inductively coupled plasma atomic emission spectrometry, high-performance liquid chromatography coupled with electrospray ionization mass spectrometry and high-performance anion-exchange chromatography with pulsed amperometric detection. Our method exhibits a relatively lower detection limit compared to those reported in the literature (Table 1).
The reproducibility of the MIP/NiNP/RGO-GCE was evaluated before and after the rebinding of 5.0 × 10−8 mol L−1 myo-inositol in five different electrodes, which have been independently prepared. Each electrode was constructed in three replications under the same conditons. The relative standard deviation (RSD) was 4.2%. The repeatability was assessed three consecutive times using the same electrode with RSD of 2.5%. Following the storage of the MIP/NiNP/RGO-GCE at room temperature (25 °C) for 30 days, the electrode still showed 87% of the initial current, proving to have good stability. It is worth pointing out that, the MIP/NiNP/RGO-GCE developed in this study can be used approximately 20 times. Essentially, these results demonstrate that our electrode presents excellent repeatability, reproducibility and stability. Such features make the method suitable for myo-inositol analysis.
Selectivity of the modified GCE
Fundamentally, the selectivity of the electrode is influenced by two factors: the positioning of functional groups and the size of the molecule. Molecules with different chemical structures are found to be relatively more difficult to cause interference in the analysis; this is largely as a result of steric hindrance [31]. As such, four molecules of similar weight, structure and position of the hydroxyl groups, which are also found in the sample in relevant quantities, were chosen aiming at evaluating the selectivity of the MIP. The molecules included L-arabitol, D-mannitol, erythritol and glucose. The changes in current response (ΔI) of [Fe(CN)6]3−/4- on the MIP/NiNP/RGO-GCE and NIP/NiNP/RGO-GCE, at the same concentration (5.0 × 10−8 mol L−1) of each molecule that can interfere in the analysis, were determined by DPV method, as shown in Fig. 6. The respective CVs are shown in Fig. S6.

Selectivity comparison between the MIP and NIP sensors. Concentration of 5.0 × 10−8 mol L−1 in PB of pH 6.0 for each molecule analyzed (rebinding time: 20 min). Response in DPV for solution contained 5.0 × 10−3 mol L−1 [Fe(CN)6]3−/4- in 0.1 mol L−1 KCl
As can be noted in Fig. 6, the response of the MIP and NIP to myo-inositol was found to be about 10 times higher for the MIP. This is, in effect, a clear proof of the formation of selective cavities. The values for L-arabitol, D-mannitol, erythritol and glucose were 2.5, 3, 1 and 4.5 respectively. These results indicate that the rebinding of myo-inositol to the MIP cavities was much more specific for myo-inositol than for other similar molecules, thus confirming the selectivity relative to this molecule.
Determination of myo-inositol in sugarcane vinasse samples
To evaluate the applicability of the developed MIP/NiNP/RGO-GCE, myo-inositol was determined in sugarcane vinasse samples. The samples were prepared as described in “Detection of myo-inositol in sugarcane vinasse sample” section. The standard addition method was used for determining the concentration of myo-inositol in the samples. The concentration of myo-inostol applied in this work ranged from 2.0 × 10−10 to 6.0 × 10−10 mol L−1. All measurements were conducted in triplicate using different electrodes. The DPV technique was used to perform the measurements. The results are presented in Table 2. The concentration of 91 ± 5.1 mg of myo-inositol was the concentration found per liter of vinasse. The production of ethanol for 2017/2018, in Brazil, is estimated to be around 24.70 billion liters. This will yield nearly 308.7 billion liters of vinasse [32]. Biorefinery can be used for myo-inositol recovery in the billion liters of vinasse that are often thrown away. When properly treated, approximately 2400 tons can be recovered and used by the pharmaceutical, food and chemical industries.
Conclusion
The MIP/NiNP/RGO-GCE developed in this work exhibited low limits of detection and quantification, wide linear concentration range, excellent selectivity, good repeatability and stability for up to 30 days upon storage. The MIP/NiNP/RGO-GCE can be reused approximately 20 times. The recovery value ranged between 95 and 102%, this demonstrates that the method presents excellent degree of accuracy. The MIP/NiNP/RGO-GCE was successfully applied toward the determination of myo-inositol in complex samples such as sugarcane vinasse. The applicability potential of the MIP for the selective recognition of myo-inositol renders it excellent as an alternative for the determination of myo-inositol. The MIP reported in this study is a fast method that is free of interferents and does not require pre-sample preparation.
References
Holub BJ (1982) The nutritional significance, metabolism, and function of myo-inositol and phosphatidylinositol in health and disease. In: Advances in Nutritional Research Springer, Boston, pp 107–141
Njau S, Joshi SH, Leaver AM, Vasavada M, Van Fleet J, Espinoza R, Narr KL (2016) Variations in myo-inositol in fronto-limbic regions and clinical response to electroconvulsive therapy in major depression. J Psychiatr Res 80:45–51
Strawn JR, Chu W-J, Whitsel RM, Weber WA, Norris MM, Adler CM, Eliassen JC, Phan KL, Strakowski SM, DelBello MP (2013) A pilot study of anterior cingulate cortex neurochemistry in adolescents with generalized anxiety disorder. Neuropsychobiology 67:224–229
Pande M, Seal A, Mishra S, Dasgupta A, Sengupta M, Dastider R (2017) The effects of combined therapy of myo-inositol and D-chiro inositol in reduction of the individual components of metabolic syndrome in overweight PCOS patients compared to myo-inositol supplementation alone: a prospective randomised controlled trial. Int J Reprod Contraception, Obstet Gynecol 6:2939–2943
D’Anna R, Santamaria A, Cannata ML, Interdonato GM, Giorgianni R, Granese F, Corrado A, Bitto A (2014) Effects of a new flavonoid and Myo-inositol supplement on some biomarkers of cardiovascular risk in postmenopausal women: a randomized trial. Int J Endocrinol 653561:1–7
Marjańska M, Weigand SD, Preboske G, Wengenack TM, Chamberlain R, Curran GL, Poduslo JF, Garwood M, Kobayashi D, Lin JC, Jack CR (2014) Treatment effects in a transgenic mouse model of Alzheimer’s disease: a magnetic resonance spectroscopy study after passive immunization. Neuroscience 259:94–100
Niwa H, Ojika M, Wakamatsu K, Yamada K, Hirono I (1983) Ptaquiloside, a novel norsesquiterpene glucoside from bracken, Pteridium aquilinum var. latiusculum. Tetrahedron Lett 24:4117–4120
Lee J, Chung BC (2006) Simultaneous measurement of urinary polyols using gas chromatography/mass spectrometry. J Chromatogr B 831:126–131
Duong QH, Clark KD, Lapsley KG, Pegg RB (2017) Determination of myo-inositol phosphates in tree nuts and grain fractions by HPLC–ESI–MS. J Food Compos Anal 59:74–82
Kindt E, Shum Y, Badura L, Snyder PJ, Brant A, Fountain S, Szekely-Klepser G (2004) Development and validation of an LC/MS/MS procedure for the quantification of endogenous myo-inositol concentrations in rat brain tissue homogenates. Anal Chem 76:4901–4908
Cataldi TRI, Campa C, De Benedetto GE (2000) Carbohydrate analysis by high-performance anion-exchange chromatography with pulsed amperometric detection: the potential is still growing. Fresenius J Anal Chem 368:739–758
Cataldi TRI, Centonze D, Casella IG, Desimoni E (1997) Anion-exchange chromatography with electrochemical detection of alditols and sugars at a Cu2O-carbon composite electrode. J Chromatogr A 773:115–121
Prabhu SV, Baldwin RP (1989) Electrocatalysis and detection of amino sugars, alditols, and acidic sugars at a copper-containing chemically modified electrode. Anal Chem 61:2258–2263
Roy E, Patra S, Madhuri R, Sharma PK, Tiwari A, Madhuri R (2017) Introduction of selectivity and specificity to graphene using an inimitable combination of molecular imprinting and nanotechnology. Biosens Bioelectron 89(1):234–248
Lim WQ, Gao Z (2015) Metal oxide nanoparticles in electroanalysis. Electroanalysis 27(9):2074–2090
Campbell FW, Compton RG (2010) The use of nanoparticles in electroanalysis: an updated review. Anal Bioanal Chem 396(1):241–259
Cheng Q, Wu C, Chen J, Zhou Y, Wu K (2011) Electrochemical tuning the activity of nickel nanoparticle and application in sensitive detection of chemical oxygen demand. J Phys Chem C 115(46):22845–22850
Hilder M, Winther-Jensen B, Li D, Forsyth M, MacFarlane DR (2011) Direct electro-deposition of graphene from aqueous suspensions. Phys Chem Chem Phys 13(20):9187–9193
Deore B, Chen Z, Nagaoka T (2000) Potential-induced enantioselective uptake of amino acid into molecularly imprinted overoxidized polypyrrole. Anal Chem 72(17):3989–3994
Tonelli D, Ballarin B, Guadagnini L, Mignani A, Scavetta E (2011) A novel potentiometric sensor for l-ascorbic acid based on molecularly imprinted polypyrrole. Electrochim Acta 56(20):7149–7154
Parnaudeau V, Condom N, Oliver R, Cazevieill P, Recous S (2008) Vinasse organic matter quality and mineralization potential, as influenced by raw material, fermentation and concentration processes. Bioresour Technol 99(6):1553–1562
Beluomini MA, da Silva JL, Sedenho GC, Stradiotto NR (2017) D-mannitol sensor based on molecularly imprinted polymer on electrode modified with reduced graphene oxide decorated with gold nanoparticles. Talanta 165(1):231–239
da Silva JL, Beluomini MA, Stradiotto NR (2017) Cathodic electrochemical determination of furfural in sugarcane bagasse using an electrode modified with nickel nanoparticles. Anal Methods 9(5):826–834
da Silva JL, Beluomini MA, Sedenho GC, Stradiotto NR (2017) Determination of amino acids in sugarcane vinasse by ion chromatographic using nickel nanoparticles on reduced graphene oxide modified electrode. Microchem J 134:374–382
Whitcombe MJ, Chianella I, Larcombe L, Piletsky SA, Noble J, Porter R, Horgan A (2011) The rational development of molecularly imprinted polymer-based sensors for protein detection. Chem Soc Rev 40(3):1547–1571
Beck F, Braun P, Oberst M (1987) Organic electrochemistry in the solid state-Overoxidation of Polypyrrole. Ber Bunsenges Phys Chem 91:967–974
Li Y, Qian R (2000) Electrochemical overoxidation of conducting polypyrrole nitrate film in aqueous solutions. Electrochim Acta 45(11):1727–1731
Renbi B, Zhang X (2001) Polypyrrole-coated granules for humic acid removal. J Colloid Infertace Sci 243(1):52–60
F. Grases, J. Perelló, B. Isern, R.M. Prieto, (2004) Determination of myo-inositol hexakisphosphate (phytate) in urine by inductively coupled plasma atomic emission spectrometry. Analytica Chimica Acta 510(1):41-43
Harvey E. Indyk, David C. Woollard, (1994) Determination of free myo-inositol in milk and infant formula by high-performance liquid chromatography. The Analyst 119(3):397
Kor K, Zarei K (2016) Development and characterization of an electrochemical sensor for furosemide detection based on electropolymerized molecularly imprinted polymer. Talanta 146(1):181–187
Portal Brasil (2017) Safra 2017/2018 de cana-de-açúcar deve ser de 647 milhões de toneladas. http://www.brasil.gov.br/economia-e-emprego/2017/04/safra-2017-2018-de-cana-de-acucar-deve-ser-de-647-milhoes-de-toneladas. Accessed 1 Aug 2017
Acknowledgements
Our sincerest gratitude to the Brazilian Research Funding Agencies – São Paulo Research Foundation (FAPESP) process n° 2014/23846-5 and Coordination for the Improvement of Higher Education Personnel (process n° 33004030072p8) for the financial support granted in the course of this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author(s) declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOCX 2851 kb)
Rights and permissions
About this article

Cite this article
Beluomini, M.A., da Silva, J.L. & Stradiotto, N.R. Amperometric determination of myo-inositol by using a glassy carbon electrode modified with molecularly imprinted polypyrrole, reduced graphene oxide and nickel nanoparticles. Microchim Acta 185, 170 (2018). https://doi.org/10.1007/s00604-018-2710-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-018-2710-0