
Abstract
Purpose
Hydrogen gas (H2) inhalation improved the survival rate of hemorrhagic shock. However, its mechanisms are unknown. We hypothesized that H2 protected the endothelial glycocalyx during hemorrhagic shock and prolonged survival time.
Methods
83 Sprague–Dawley rats were anesthetized with isoflurane. The animals were randomly assigned to 5 groups: room air with no shock, 1.2% H2 with no shock, room air with shock (Control-S), 1.2% H2 with shock (H21.2%-S), and 3.0% H2 with shock (H23.0%-S). Shock groups were bled to a mean arterial pressure of 30–35 mmHg and held for 60 min, then resuscitated with normal saline at fourfold the amount of the shed blood volume.
Results
The syndecan-1 level was significantly lower in the H21.2%-S [8.3 ± 6.6 ng/ml; P = 0.01; 95% confidence interval (CI), 3.2–35.8] than in the Control-S (27.9 ± 17.0 ng/ml). The endothelial glycocalyx was significantly thicker in the H21.2%-S (0.15 ± 0.02 µm; P = 0.007; 95% CI, 0.02–0.2) than in the Control-S (0.06 ± 0.02 µm). The survival time was longer in the H21.2%-S (327 ± 67 min, P = 0.0160) than in the Control-S (246 ± 69 min). The hemoglobin level was significantly lower in the H21.2%-S (9.4 ± 0.5 g/dl; P = 0.0034; 95% CI, 0.6–2.9) than in the Control-S (11.1 ± 0.8 g/dl). However, the H23.0%-S was not significant.
Conclusions
Inhalation of 1.2% H2 gas protected the endothelial glycocalyx and prolonged survival time during hemorrhagic shock. Therapeutic efficacy might vary depending on the concentration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hemorrhagic shock accounts for 50.7% of unexpected perioperative death in Japan [1]. Therefore, overcoming hemorrhagic shock is considered as a major issue in perioperative medicine.
The endothelial glycocalyx (EGCX) is located in the vascular endothelium and is responsible for vascular permeability. During hemorrhagic shock with fluid resuscitation, the EGCX is damaged [2]. Shedding of the EGCX increases vascular integrity, resulting in multiple organ failure and increased mortality [3]. The EGCX is also shed by ischemia–reperfusion, reactive oxygen species (ROS), inflammation, sepsis, hyperglycemia, and other conditions [4,5,6,7,8]. Protection of the EGCX might help to prevent the progression of multiple organ failure and/or decrease mortality.
Hydrogen gas (H2 gas) is an antioxidative and anti-inflammatory substance [9]. H2 gas is not explosive and can be safely used at a concentration of < 4%. H2 gas selectively reduces hydroxyl radical and peroxynitrite, which are ROS with particularly high levels in oxidation-induced damage; however, it does not reduce hydrogen peroxide, which acts as a gas mediator. Based on these characteristics, H2 gas is considered to be an ideal antioxidant that retains its property as a necessary gas mediator, while alleviating ROS-induced damage. H2 gas has a protective effect on cells and organs (brain, intestine, liver, kidney, lung, and heart) in several pathological conditions [10,11,12,13,14,15], especially ischemia–reperfusion [16]. Matsuoka et al. [17] demonstrated that inhalation of H2 gas improved the survival rate in a rat model of hemorrhagic shock and fluid resuscitation, although the underlying mechanism is unknown.
We hypothesized that H2 gas inhalation protects the EGCX and prolongs the survival time during hemorrhagic shock. We conducted the present study using a rat model of hemorrhagic shock to investigate the effects of H2 gas inhalation on the syndecan-1 level (a marker of EGCX shedding), EGCX thickness, markers of organ failure, and survival time.
Materials and methods
Animal preparation
This study was approved by the Ethical Committee for Animal Experiments and the Laboratory Animal Facility of Hamamatsu University School of Medicine (2018049). In total, 83 male Sprague–Dawley rats (10–11 weeks old; mean body weight, 355 ± 30 g) were purchased from Japan SLC, Inc. (Shizuoka, Japan). All rats were acclimatized to a 12-/12-hour light/dark cycle at a room temperature of 20 °C. The rats had free access to food and water before the experiment.
After induction of anesthesia with 5% isoflurane (Mylan, Tokyo, Japan), the rats underwent tracheostomy and intubation using a 19-gauge fluororesin catheter (Hakko Medical Device Division, Nagano, Japan). The rats were artificially ventilated (rate, 50/min; tidal volume, 1.0 ml/100 g; FiO2, 21%; Shinano Seisakusho, Tokyo, Japan) with 2% isoflurane during the experiments. The core body temperature of the rats was measured using a rectal probe and maintained at 37 °C with a heating light. A 20-gauge catheter (B Braun, Melsungen, Germany) was placed into the right carotid artery to measure the arterial pressure, withdraw blood, and infuse normal saline. The electrocardiogram, heart rate, and arterial pressure were continuously recorded.
Hemorrhagic shock model
After stabilization, the rats were randomly divided into 5 groups: no shock with room air (Room-NS), no shock with 1.2% H2 gas (H21.2%-NS), hemorrhagic shock with room air (Control-S), hemorrhagic shock with 1.2% H2 gas (H21.2%-S), and hemorrhagic shock with 3% H2 gas (H23.0%-S). Hemorrhagic shock was induced by withdrawing blood until the mean arterial pressure (MAP) decreased to 30–35 mmHg in 5 min and maintained for 60 min at this value by further blood withdrawal. After the shock phase, the rats were fluid-resuscitated by normal saline at fourfold the amount of the shed blood volume at 2.5 ml/min. Rats that survived the experiment were killed by withdrawal of blood under isoflurane anesthesia.
H 2 gas inhalation
H2 gas was delivered by mixing with room air using an H2 gas supply device (Nihon Kohden, Tokyo, Japan). This device allows for the delivery of several concentrations of H2 gas with room air. We chose 1.2% and 3.0% H2 gas concentrations in this study. The concentration of 1.2% was chosen based on our pilot study with reference to Matsuoka et al. [17], and 3.0% was applied as the maximum concentration that our H2 gas supply device could deliver. H2 gas inhalation began at the time of shock induction and continued for 3 h.
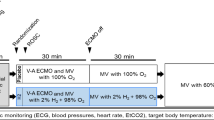
Experimental protocol (Fig. 1).

Experimental protocol of hemorrhagic shock and fluid resuscitation. H2 gas was inhaled by rats in three study groups: no shock with 1.2% H2 gas, hemorrhagic shock with 1.2% H2 gas, and hemorrhagic shock with 3.0% H2 gas. H2 gas inhalation began from initiation of hemorrhagic shock and continued for 180 min.H2 gas, hydrogen gas; MAP, mean arterial pressure
We performed three sets of experiments to investigate whether H2 gas inhalation protects the EGCX against hemorrhagic shock and improves the survival time, as described below.
Experiment 1: Glycocalyx analysis (n = 6 per group)
Two hours after the beginning of fluid resuscitation, we collected blood samples from the catheter of the right carotid artery. Serum was used to measure syndecan-1 using an enzyme-linked immunosorbent assay kit (Cloud-Clone Corp., Katy, TX, USA), creatinine using a clinical chemistry analyzer (JCA-BM8060; JEOL Ltd., Tokyo, Japan), and blood gas analysis (ABL90 FLEX; Radiometer Medical ApS, Brønshøj, Denmark). We then performed a thoracotomy and inferior vena cava incision, and lactated Ringer’s solution was administered into the left ventricle for 2 min for removal of blood. A fixing/staining solution (2% glutaraldehyde, 30-mM HEPES buffer, and 2% lanthanum nitrate) was then used at a dose of 8 ml/min for 5 min for perfusion fixation. The heart was removed and cut up for immersion fixation with a fixing/staining solution for 24 h at 4 °C. A transmission electron microscope (JEM-1400 Plus; JEOL, Ltd.) was used to observe the EGCX with a 10-µm capillary endothelium [18, 19]. Five perfused vessels from different perspectives were chosen. For measurement, the shortest distance between the lumen and the vascular endothelium were measured using ImageJ (US National Institutes of Health, Bethesda, MD, USA) as our previous study [19].
Experiment 2: survival time analysis (n = 7 per group)
Two hours after the beginning of fluid resuscitation, we stopped the H2 gas and changed to room air as described by Matsuoka et al. [17]. We then continued our observation of the survival time under general anesthesia. Death was defined as a decrease in the MAP to < 10 mmHg.
Experiment 3: serum hemoglobin and TNF-α analysis (n = 6 per group)
We performed an additional experiment to investigate serum hemoglobin and tumor necrosis factor-α (TNF-α) levels. We investigated blood hemoglobin to evaluate hemodilution after fluid resuscitation. H2 gas may contribute to EGCX protection via multiple pathways, including an anti-inflammatory effect; we, therefore, evaluated serum TNF-α in the three shock groups (Control-S, H21.2%-S, and H23.0%-S; n = 6 per group). Animals were prepared in the same way as for the experiment 1. Two hours after initiating fluid resuscitation, we collected blood samples for the measurement of hemoglobin and TNF-α level using an enzyme-linked immunosorbent assay kit (R&D Systems, Inc., Minneapolis, MN, USA).
Statistical analysis
Data are expressed as mean ± standard deviation. The means of each group were compared using one-way analysis of variance followed by the Turkey–Kramer post hoc test. All data with a P value of < 0.05 were considered significant except survival times. Survival times were evaluated using the Kaplan–Meier method and a log rank test. These data with a P value of < 0.166 were considered significant by Bonferroni correction. All statistical analyses were performed using JMP 14.3 for Windows (SAS Institute Inc., Cary, NC, USA).
Results
No significant differences except hemoglobin were found in blood gas analysis (Table 1) and the amount of bleeding among the Control-S, H21.2%-S, and H23.0%-S groups. Similarly, no significant differences were found in the heart rate, MAP and systolic arterial pressure among the groups (Fig. 2a–c). However in diastolic arterial pressure, H21.2%-S (41.7 ± 10.4 mmHg) was higher than Control-S group [20.7 ± 13.2 mmHg; P = 0.0096; 95% confidence interval (CI), 4.4–37.5] (Fig. 2d).

Hemodynamic parameters. a Heart rate. b Mean arterial pressure. c Systolic arterial pressure. d Diastolic arterial pressure. The error bars represent the standard deviation of the mean
The level of serum syndecan-1 was significantly lower in the H21.2%-S group (8.3 ± 6.6 ng/ml; P = 0.01; 95% CI 3.2–35.8) than in the Control-S group (27.9 ± 16.9 ng/ml), but it was not significantly lower in the H23.0%-S group (18.6 ± 11.2 ng/ml; P = 0.4645; 95% CI − 6.9 to 25.5) (Fig. 3a). The glycocalyx as assessed by electron microscopy was significantly thicker in the H21.2%-S group (0.15 ± 0.02 µm; P = 0.007; 95% CI 0.02–0.2) than in the Control-S group (0.06 ± 0.02 µm). The glycocalyx in the H23.0%-S group (0.13 ± 0.05 µm) also tended to be thicker, but not significantly (P = 0.06; 95% CI − 0.003 to 0.1) (Fig. 3b, c).

Evaluations of glycocalyx (n = 6 per group). a Serum concentration of syndecan-1. b Endothelial glycocalyx of myocardium under transmission electron microscopy. c Glycocalyx thickness of myocardial capillaries. d Serum concentration of creatinine. Control-S hemorrhagic shock with room air, H21.2%-S hemorrhagic shock with 1.2% H2 gas, H23.0%-S hemorrhagic shock with 3.0% H2 gas, Room-NS no shock with room air, H21.2%-NS no shock with 1.2% H2 gas, EGCX endothelial glycocalyx
The level of serum creatinine was significantly lower in the H21.2%-S group (0.43 ± 0.10 mg/ml; P = 0.008; 95% CI 0.05–0.4) than in the Control-S group (0.64 ± 0.12 mg/ml), but it was not significantly lower in the H23.0%-S group (0.55 ± 0.14 mg/ml; P = 0.52; 95% CI − 0.08 to 0.3) (Fig. 3d).
There were no survival rats in Control-S, H21.2%-S, and H23.0%-S groups. The survival time in the H21.2%-S group (327 ± 67 min, P = 0.0160) was significantly longer than that in the Control-S group (246 ± 69 min), but it was not significantly longer in the H23.0%-S group (242 ± 79 min, P = 0.67) (Fig. 4).

Survival time (n = 7 per group). *P = 0.0160 compared with the Control-S group. Control-S hemorrhagic shock with room air, H21.2%-S hemorrhagic shock with 1.2% H2 gas, H23.0%-S hemorrhagic shock with 3.0% H2 gas
The hemoglobin level was significantly lower in the H21.2%-S group (9.4 ± 0.5 g/dl; P = 0.003; 95% CI 0.6–2.9) than in the Control-S group (11.1 ± 0.8 g/dl), but was not significantly different in the H23.0%-S group (10.1 ± 0.9 mg/ml; P = 0.07; 95% CI − 0.1 to 2.2). The amount of bleedings were not significant between Control-S, H21.2%-S, and H23.0%-S groups. The serum TNF-α level was not significantly different in the H21.2%-S group (144.7 ± 103.8 pg/ml; P = 0.68; 95% CI − 105.7 to 206.2) or the H23.0%-S group (147.0 ± 77.5 pg/ml; P = 0.66; 95% CI − 103.4 to 208.5) compared with the Control-S group (94.4 ± 152.2 pg/ml).
Discussion
The present study demonstrated that 1.2% H2 gas inhibited the increase in the serum syndecan-1 and creatinine levels, preserved the EGCX layers after hemorrhagic shock and fluid resuscitation, and further prolonged survival time. These effects were reduced under 3.0% H2 gas inhalation. These findings suggest that H2 gas inhalation may have a protective effect during hemorrhagic shock and fluid resuscitation, although an optimal inhalational concentration might exist. We evaluated the blood vessels in the myocardium to analyze the EGCX, similar to our previous study [19] and other previous studies [20,21,22] because the heart has continuous capillaries [4] that can easily be observed by electron microscopy. To our knowledge, this is the first study to show that H2 gas protects the glycocalyx as shown by electron microscopy images.
Several studies have demonstrated that H2 gas has antioxidant and anti-inflammatory effects [10, 23], and these effects have been suggested during hemorrhagic shock in rat models [17, 24,25,26]. Matsuoka et al. indicated that 1.3% H2 gas inhalation prolonged survival time during hemorrhagic shock and fluid resuscitation [17]. However, they did not explore the mechanism underlying why H2 gas was effective for hemorrhagic shock. The present study supports their findings, and we think the protective effects of H2 gas on the EGCX could be one of the mechanisms. The EGCX is a polysaccharide layer located at the vascular endothelium. The EGCX controls vascular permeability and tonus [27]. When the EGCX is shed, vascular permeability is enhanced [20]. In our experiments, the Control-S group showed greater EGCX injury and higher hemoglobin level compared with the H21.2%-S group. We propose that, in the Control-S group, infused fluid likely extravasated to interstitium and it must be difficult to maintain blood volume because of injury to the EGCX. The EGCX also exerts anti-inflammatory and anti-coagulant effects [27]. The EGCX covers the surface of vascular endothelial cells with various receptors (selectin, integrin, and toll-like receptors). In normal condition, these receptors cannot combine leucocyte or ligands because of covered by EGCX [28]. When the glycocalyx is shed, vascular permeability is enhanced and the pathological condition is aggravated. Integrin helps to combine leukocytes with endothelial cells, and Toll-like receptors easily combine with ligands. They will progress inflammatory. Osuka et al. [29] reported that glycocalyx damage was correlated with a deterioration in the condition of patients with burn injuries. Thus, protection of the glycocalyx might be important to prevent the progression of multiple organ failure and/or decrease the mortality.
H2 gas selectively and directly reduces hydroxyl radical and peroxynitrite [9], which are detrimental ROS that induce ischemia–reperfusion injury and, thus, cause EGCX shedding. H2 gas combines hydroxyl radical and peroxynitrite, forming water. The other mechanism is gene expression and anti-inflammatory effect. H2 gas decreases the expression of certain signal transduction pathways via oxidized phospholipid species, including HMOX1 [heme oxygenase 1, hypoxia-inducible factor 1 (HIF-1) signaling pathway], tumor necrosis factor, interleukin-8 (IL-8), and nuclear factor of activated T cells [30]. TNF-α is also a cause of inflammatory and sheds EGCX [31]. However, in our additional experiment, no significant difference in TNF-α was observed between the shock groups.
We evaluated creatinine as organ dysfunction. Kidney is weaker than heart and intestine for oxygen debt [32], and acute kidney injury is independent risk of adverse outcomes in critically ill patients [33]. In this study, 1.2% H2 gas inhalation reduces creatinine and it might protect kidney.
We examined the effects of H2 gas under inhalational concentrations of 1.2% and 3.0%. Protective effects were shown with 1.2% H2 gas inhalation, but these effects were reduced at 3.0% inhalation. Ohsawa et al. [9] demonstrated that 2% H2 gas was more effective than edaravone but that 4% was not significantly effective in a rat model of brain ischemia–reperfusion. Similarly, Hayashida et al. [16] showed that 2% H2 gas was more effective than control but that 4% gas was not significantly effective in a rat model of myocardial ischemia–reperfusion. These findings suggest that a high concentration of H2 gas inhalation attenuates the protective effects of H2 gas, similar to our results, although previous authors did not discuss why 4% reduced the effects. We speculate that a preconditioning effect of isoflurane is involved. In the present study, we used isoflurane, which has a preconditioning effect of ischemia–reperfusion. Although isoflurane activates HIF-1, H2 gas decreases the expression of HMOX1. Further studies are required to investigate the relationship between H2 gas inhalational concentrations and optimal protective effects.
Our study has some limitations. We used the serum concentration of syndecan-1 as an indicator of glycocalyx damage [34], and the syndecan-1 level might depend on the intravascular volume such as hemodilution by fluid resuscitation. However, the blood loss volumes were similar among the groups, and the trend of syndecan-1 levels actually reflected the glycocalyx thickness as shown by electron microscopy; these findings indicate that the syndecan-1 level was suitable as an index of glycocalyx damage in this study. Next, we could not measure ROS directly. H2 gas can have multiple mechanism of prolong survival time and EGCX protection. These markers would help to understand which mechanism is most contributing. Another point is type of fluid. Although we used normal saline for resuscitation, EGCX damage depends on the type of fluid. If we use fresh frozen plasma or another type of fluid for resuscitation, damage level or EGCX may be different. In addition, our hemorrhagic shock model simulated compensated shock because blood was withdrawn, but not reinfused, to maintain an MAP of 30–35 mmHg. This is less severe than a decompensating model, which requires reinfusion of blood [35]. Our results might not be applicable to severe hemorrhagic shock. Finally, as mentioned above, we used isoflurane, which has a protective effect against ischemia–reperfusion injury [36]. Although all groups of rats were anesthetized by isoflurane and significant differences were observed in the glycocalyx thickness and survival rate, the results (including the appropriate concentration of H2 gas) might be different when using other anesthetics.
In conclusion, inhalation of 1.2% H2 gas protected the glycocalyx, reduced the creatinine level, and prolonged the survival time during hemorrhage shock and fluid resuscitation in rats, although inhalation of 3.0% attenuated these effects. Further studies are needed to identify the optimal concentration that shows maximal protective effects.
Part of this article was presented at The 66th Annual Meeting of the Japanese Society of Anesthesiologists. May 30th–June 1st, 2019, Kobe, Japan.
References
Kawashima Y, Irita K, Morita K, Tuzaki K, Sawa T. Preoperative hemorrhagic shock and intraoperative bleeding: two main causes of surgical deaths in Japan. J Jpn Soc Blood Trans. 2005;51(1):23–31.
Kozar RA, Peng Z, Zhang R, Holcomb JB, Pati S, Park P, Ko TC, Paredes A. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth Analg. 2011;112(6):1289–95.
Chappell D, Westphal M, Jacob M. The impact of the glycocalyx on microcirculatory oxygen distribution in critical illness. Curr Opin Anaesthesiol. 2009;22(2):155–62.
Okada H, Takemura G, Suzuki K, Oda K, Takada C, Hotta Y, Miyazaki N, Tsujimoto A, Muraki I, Ando Y, Zaikokuji R, Matsumoto A, Kitagaki H, Tamaoki Y, Usui T, Doi T, Yoshida T, Yoshida S, Ushikoshi H, Toyoda I, Ogura S. Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic conditions. Crit Care. 2017;21(1):261.
Rubio-Gayosso I, Platts SH, Duling BR. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;290(6):H2247–H2256256.
Iba T, Levy JH. Derangement of the endothelial glycocalyx in sepsis. J Thromb Haemost. 2019;17(2):283–94.
Diebel ME, Martin JV, Liberati DM, Diebel LN. The temporal response and mechanism of action of tranexamic acid in endothelial glycocalyx degradation. J Trauma Acute Care Surg. 2018;84(1):75–80.
Kazuma S, Tokinaga Y, Kimizuka M, Azumaguchi R, Hamada K, Yamakage M. Sevoflurane promotes regeneration of the endothelial glycocalyx by upregulating sialyltransferase. J Surg Res. 2019;241:40–7.
Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S, Ohta S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007;13(6):688–94.
Ichihara M, Sobue S, Ito M, Ito M, Hirayama M, Ohno K. Beneficial biological effects and the underlying mechanisms of molecular hydrogen—comprehensive review of 321 original articles. Med Gas Res. 2015;5:12.
Xie K, Liu L, Yu Y, Wang G. Hydrogen gas presents a promising therapeutic strategy for sepsis. Biomed Res Int. 2014;2014:807635.
Yu Y, Yang Y, Bian Y, Li Y, Liu L, Zhang H, Xie K, Wang G, Yu Y. Hydrogen gas protects against intestinal injury in wild type but not NRF2 knockout mice with severe sepsis by regulating HO-1 and HMGB1 release. Shock. 2017;48(3):364–70.
Yonamine R, Satoh Y, Kodama M, Araki Y, Kazama T. Coadministration of hydrogen gas as part of the carrier gas mixture suppresses neuronal apoptosis and subsequent behavioral deficits caused by neonatal exposure to sevoflurane in mice. Anesthesiology. 2013;118(1):105–13.
Shinbo T, Kokubo K, Sato Y, Hagiri S, Hataishi R, Hirose M, Kobayashi H. Breathing nitric oxide plus hydrogen gas reduces ischemia-reperfusion injury and nitrotyrosine production in murine heart. Am J Physiol Heart Circ Physiol. 2013;305(4):H542–H550550.
Hayashida K, Sano M, Kamimura N, Yokota T, Suzuki M, Ohta S, Fukuda K, Hori S. Hydrogen inhalation during normoxic resuscitation improves neurological outcome in a rat model of cardiac arrest independently of targeted temperature management. Circulation. 2014;130(24):2173–80.
Hayashida K, Sano M, Ohsawa I, Shinmura K, Tamaki K, Kimura K, Endo J, Katayama T, Kawamura A, Kohsaka S, Makino S, Ohta S, Ogawa S, Fukuda K. Inhalation of hydrogen gas reduces infarct size in the rat model of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun. 2008;373(1):30–5.
Matsuoka T, Suzuki M, Sano M, Hayashida K, Tamura T, Homma K, Fukuda K, Sasaki J. Hydrogen gas inhalation inhibits progression to the “irreversible” stage of shock after severe hemorrhage in rats. J Trauma Acute Care Surg. 2017;83(3):469–75.
Kataoka H, Ushiyama A, Akimoto Y, Matsubara S, Kawakami H, Iijima T. Structural behavior of the endothelial glycocalyx is associated with pathophysiologic status in septic mice: an integrated approach to analyzing the behavior and function of the glycocalyx using both electron and fluorescence intravital microscopy. Anesth Analg. 2017;125(3):874–83.
Kobayashi K, Mimuro S, Sato T, Kobayashi A, Kawashima S, Makino H, Doi M, Katoh T, Nakajima Y. Dexmedetomidine preserves the endothelial glycocalyx and improves survival in a rat heatstroke model. J Anesth. 2018;32(6):880–5.
van den Berg BM, Vink H, Spaan JA. The endothelial glycocalyx protects against myocardial edema. Circ Res. 2003;92(6):592–4.
Rehm M, Bruegger D, Christ F, Conzen P, Thiel M, Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Reichart B, Peter K, Becker BF. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation. 2007;116(17):1896–906.
Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res. 2010;87(2):300–10.
Tamura T, Hayashida K, Sano M, Onuki S, Suzuki M. Efficacy of inhaled HYDROGEN on neurological outcome following BRAIN ischemia during post-cardiac arrest care (HYBRID II trial): study protocol for a randomized controlled trial. Trials. 2017;18(1):488.
Kohama K, Yamashita H, Aoyama-Ishikawa M, Takahashi T, Billiar TR, Nishimura T, Kotani J, Nakao A. Hydrogen inhalation protects against acute lung injury induced by hemorrhagic shock and resuscitation. Surgery. 2015;158(2):399–407.
Du Z, Liu J, Jia H, Xu W, Zhao X. Three hydrogen-rich solutions protect against intestinal injury in uncontrolled hemorrhagic shock. Int J Clin Exp Med. 2015;8(5):7620–6.
Du Z, Jia H, Liu J, Zhao X, Xu W. Effects of three hydrogen-rich liquids on hemorrhagic shock in rats. J Surg Res. 2015;193(1):377–82.
Aguirre JA, Lucchinetti E, Clanachan AS, Plane F, Zaugg M. Unraveling interactions between anesthetics and the endothelium: update and novel insights. Anesth Analg. 2016;122(2):330–48.
Iba T. Glycocalyx regulates the intravascular hemostasis. Juntendo Med J. 2016;62(4):330–5.
Osuka A, Kusuki H, Yoneda K, Matsuura H, Matsumoto H, Ogura H, Ueyama M. Glycocalyx shedding is enhanced by age and correlates with increased fluid requirement in patients with major burns. Shock. 2018;50(1):60–5.
Iuchi K, Imoto A, Kamimura N, Nishimaki K, Ichimiya H, Yokota T, Ohta S. Molecular hydrogen regulates gene expression by modifying the free radical chain reaction-dependent generation of oxidized phospholipid mediators. Sci Rep. 2016;6:18971.
Chappell D, Hofmann-Kiefer K, Jacob M, Rehm M, Briegel J, Welsch U, Conzen P, Becker BF. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol. 2009;104(1):78–89.
van Bommel J, Siegemund M, Henny ChP, Ince C. Heart, kidney, and intestine have different tolerances for anemia. Transl Res. 2008;151(2):110–7.
Bihorac A, Delano MJ, Schold JD, Lopez MC, Nathens AB, Maier RV, Layon AJ, Baker HV, Moldawer LL. Incidence, clinical predictors, genomics, and outcome of acute kidney injury among trauma patients. Ann Surg. 2010;252(1):158–65.
Chelazzi C, Villa G, Mancinelli P, De Gaudio AR, Adembri C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care. 2015;19:26.
Kurita T, Morita K, Fukuda K, Uraoka M, Takata K, Sanjo Y, Sato S. Influence of hemorrhagic shock and subsequent fluid resuscitation on the electroencephalographic effect of isoflurane in a swine model. Anesthesiology. 2005;103(6):1189–94.
Nakajima Y, Moriwaki G, Ikeda K, Fujise Y. The effects of sevoflurane on recovery of brain energy metabolism after cerebral ischemia in the rat: a comparison with isoflurane and halothane. Anesth Analg. 1997;85(3):593–9.
Acknowledgements
The authors thank I. Ohta, Y. Kumakiri, and Y. Tokunaga (Hamamatsu University School of Medicine) for their technical assistance with the electron microscopy. We thank T. Ojima (Hamamatsu University School of Medicine) for his statistical assistance. We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript. This work was supported by JSPS KAKENHI (Grant Numbers JP19K09371 and JP18K08885).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by TS, SM, and KK. The first draft of the manuscript was written by TS and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Sato, T., Mimuro, S., Katoh, T. et al. 1.2% Hydrogen gas inhalation protects the endothelial glycocalyx during hemorrhagic shock: a prospective laboratory study in rats. J Anesth 34, 268–275 (2020). https://doi.org/10.1007/s00540-020-02737-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00540-020-02737-3