
Abstract
Background
Hypoxic microenvironment is a common feature of most solid tumors including hepatocellular carcinoma (HCC). Vasculogenic mimicry (VM) formation by tumor cells could provide blood supply to tumor cells under hypoxia. NFE2 like basic leucine zipper (bZIP) transcription factor 2 (Nrf2), a regulator of cellular homeostasis, may promote tumor progression in the hypoxic conditions. However, the role and regulatory mechanisms of Nrf2 in HCC are not fully elucidated.
Methods
Nrf2 and assembly factor for spindle microtubules (ASPM) expression modulations were conducted by lentiviral transfections. Western blot, immunofluorescence, ChIP-qPCR, dual-luciferase reporter gene assay, flow cytometry, RNA sequencing, multiple bioinformatics databases analysis, cell function assays in vitro, mouse model in vivo and human HCC tissues were employed to assess the effect of Nrf2/ASPM axis on HCC progression under hypoxia.
Results
Nrf2 and ASPM expression facilitated epithelial–mesenchymal transition (EMT), cancer stem cells (CSCs) feature, and VM formation of HCC cells under hypoxia. Furthermore, Nrf2-regulated ASPM expression, via binding directly to the promoter region of ASPM and transcriptionally promoting ASPM expression. ASPM re-expression in Nrf2 knockdown cells or ASPM knockdown in Nrf2 overexpression cells reversed the cellular function caused by Nrf2. Meantime, retinol metabolism pathway was disrupted following abnormal ASPM expression. Nrf2/ASPM axis in murine models accelerated tumor growth and VM, corroborating in vitro findings. All-trans retinoic acid treatment reversed stemness and VM of HCC cells in vitro and in vivo. Clinically, Nrf2 and ASPM expressions were related to poor prognosis of HCC patients.
Conclusions
Nrf2 drives EMT, CSCs characteristics and VM in HCC under hypoxia through the modulation of ASPM. Retinol metabolism pathway was dysregulated in HCC cells with ASPM overexpression. Nrf2/ASPM axis and related pathway provided potential therapeutic target for HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) imposes a significant global burden, ranking sixth in global cancer incidence and being the second leading cause of premature cancer-related mortality in the 2020 global cancer statistics [1,2,3]. Despite several decades of medical advancements, HCC’s survival rates have remained largely stagnant, primarily due to its marked heterogeneity and high recurrence rates.
Throughout the course of HCC development, the presence of a hypoxic microenvironment has emerged as a pivotal factor at all stages [4]. Hypoxia, indeed, exerts cytotoxic effects on both cancer and normal cells. However, in response to the adverse conditions, tumor cells undergo adaptive changes to meet their survival and proliferative demands. In addition to the conventional angiogenesis, vasculogenic mimicry (VM), a process wherein tumor cells, as opposed to endothelial cells, form vascular-like structures to secure nutrients and oxygen perfusion, was found to be instrumental in cancer cell survival under hypoxia [5]. VM has been reported in various malignancies including HCC [6,7,8,9,10]. Patients exhibiting VM-positive status tend to have a poorer prognosis, rendering VM a promising novel target in anti-cancer therapies.
Epithelial–mesenchymal transition (EMT) plays a critical role in VM formation. During this process, certain epithelial cell markers like E-cadherin, zonula occludins-1, and α-catenin are downregulated, while mesenchymal cell markers, including VE-cadherin, fibronectin, cadherin-2, and vimentin, are upregulated. These changes occur through various mechanisms, thereby promoting tumor invasion and metastasis [11]. When EMT is aberrantly activated in cancer, cells gain attributes of stem cells capable of self-renewal and multipotent differentiation so called as cancer stem cells (CSCs) [12]. The resulting CSCs attain cellular plasticity and are involved in the formation of VM [8]. They possess the capability to differentiate to vascular like cells and contribute to VM formation, aligning themselves to form branching structures that provide nourishment to tumor masses. Furthermore, hypoxia also plays a role in maintaining the cancer stem cell-like phenotype, vital for VM formation, by activating stem cell-related factors [13].
NFE2 like basic leucine zipper (bZIP) transcription factor 2 (Nrf2) is ubiquitously expressed in all cell types. It regulates gene expression associated with oxidative stress responses and detoxification, enhancing cellular defenses against toxicity and oxidative damage [14]. However, Nrf2 does not consistently play the role of a guardian within the organism. Over the past decade, several studies have suggested that Nrf2 can promote cancer metastasis and enhance resistance to chemotherapy [15, 16]. DeNicola and colleagues have discovered that Nrf2 activates ATF4 to regulate the expression of genes involved in serine and glycine biosynthesis pathways, thereby supporting cancer cell proliferation and growth [17]. Additionally, Nrf2 could regulate stemness-related NOTCH signaling, while NOTCH activation further stimulates Nrf2, creating a positive feedback loop to maintain a stem cell-like state in liver development [18]. Moreover, Nrf2 expression correlates closely with venous hypertension induced angiogenesis in cerebral arteriovenous malformations [19].
Assembly factor for spindle microtubules (ASPM) was originally identified as a central spindle protein regulating neural development and brain size [20]. Recent studies implicate an oncogenic role of ASPM as its expression positively correlates with poor survival in patients with various types of cancer. ASPM was overexpressed in approximately 66% of HCC patients [21]. ASPM silencing would lead to reduced proliferation, migration, and invasion of cancer cells in various cancers by in vitro and in vivo experiments [22]. Data accumulated over recent years indicate that ASPM is preferentially expressed by CSCs in various solid tumors. ASPM has been indentified as a regulatory hub of stemness-associated signaling pathways, such as the Wnt, Hedgehog, and Notch pathways in cancer cells, which drives tumor stemness and progression in multiple cancers [22].
However, the mechanisms through which Nrf2 influences cancer cell progression and invasion in the hypoxic microenvironment of liver cancer, particularly in inducing stemness and VM formation, remain largely unknown so far. In this study, we aim to elucidate the potential role of the Nrf2/ASPM axis in shaping vascular mimicry formation in tumor cells of HCC. This research seeks to unveil potential therapeutic targets for disrupting VM formation as part of therapeutic strategies in HCC.
Methods
Cell culture, CoCl2 treatment and lentiviral transduction
HCCLM3 cells, sourced from the Peking Union Basic Medical Cell Center, and Huh7 cells, obtained from Pricela (China), were propagated in DMEM (KeyGEN Bio TECH, China), supplemented with 10% fetal bovine serum (FBS) (Gibco, USA) and 1% penicillin–streptomycin (P/S) (Gibco, USA). Cells were confirmed mycoplasma-free and authenticated via STR profiling. Cultivation was conducted at 37 °C and 5% CO2. Cells were pre-cultured for 24 h, and then subjected to 150 μM CoCl2 for another 24 h to simulate hypoxia. CoCl2 is a widely-used hypoxia-mimetic agent in research involving cellular invasion, differentiation, and various hypoxic responses [23].
Plasmids were sourced from FulenGen (China). The pEZ-Lv201 or pEZX-MR03 vectors and pcDNA3 vector were used for the overexpression of Nrf2, ASPM, and HIF-1α, respectively. The psi-LVRH1GP, psi-LVRU6GP, and psi-LVRHIMH vectors were used for the silencing of Nrf2, ASPM, and HIF-1α. The shRNA target sequences for Nrf2, ASPM, and HIF-1α are as follows: 5′-gatgcctgtagcactagaaat-3′, 5′-uguaaacauccuugacuggaag-3′, 5'-gccgaggaagaactatgaaca-3′. Transfected cells were treated with 2 μg/ml puromycin (Gibco, USA) for 2 weeks to obtain stably-transfected cell lines.
Western blot assays
Protein extracts were separated using SDS-PAGE gel electrophoresis and then transferred onto PVDF membranes. After blocking with 5% non-fat milk for 1 h, membranes were incubated with primary antibodies overnight at 4 °C, followed by 1 h incubation with secondary antibodies. Detection was performed using an ECL reagent (R-03031-D2; Advansta, USA) and imaged by a C-DiGit Blot Scanner (LI-COR Biosciences, USA).
The Western blot antibodies used were as follows: anti-HIF-1 alpha (ab279654, 1:1000; Abcam), anti-Nrf2 (ab62352, 1:1000; Abcam), anti-ASPM (NBP2-92395, 1:500; Novus), anti-VE-Cadherin (ab33168, 1:500; Abcam), anti-E-Cadherin (#3195, 1:1000; Cell Signaling), anti-N-Cadherin (ab245117, 1:1000; Abcam), anti-MMP2 (10373-2-AP, 1:500; Proteintech), anti-CD133 (18470-1-AP, 1:500; Proteintech) and anti-beta Actin (81115-1-RR, 1:2000; Proteintech).
Quantitative real-time PCR (qRT‒PCR)
Total RNA was extracted using an RNA extraction kit (DP451, TIANGEN). Subsequently, cDNA was synthesized using the PrimeScript™ RT Master Mix (TaKaRa). QRT‒PCR was conducted with TB Green Premix EX TaqTM II (Takara) in 7500HT Real-Time PCR System (Applied Biosystems, Foster City, CA). Gene expression levels were quantified using the 2−ΔΔCt method, with primer details provided in the supplementary materials.
Cell invasion assays
The assays were performed by using Transwell (Invitrogen) cell culture inserts. In the upper chamber coated with Matrigel, 5 × 104 cells were cultured in serum-free medium, while the lower chamber of 24-well plates contained DMEM medium with 20% FBS. After 36-h incubation, cells were fixed in methanol and stained with 0.5% crystal violet for 30 min. Cell counting under microscope was conducted.
Chromatin immunoprecipitation (ChIP) assays
EX-Nrf2 HCCLM3 cells were cultured, cross-linked with formaldehyde, quenched with glycine, and then lysed. DNA was fragmented using sonication, followed by chromatin immunoprecipitation with either Nrf2 antibody (Abcam) or IgG (Santa Cruz), and overnight incubation at 4 °C. Antibody-protein-DNA complexes were isolated by immunoprecipitation with protein A agarose beads. Following extensive washing, bound DNA fragments were eluted and analyzed by subsequent qRT-PCR. Primer sequences are listed in the supplementary materials.
Immunohistochemistry (IHC) assays and periodic acid-schiff stain (PAS) staining
This experiment was performed following standard procedures. The primary antibodies Nrf2 (16396-1-AP, 1:600; Proteintech), ASPM (26223-1-AP, 1:800; Proteintech), HIF-1alpha (20960-1-AP, 1:100; Proteintech), VE-cadherin (ab33168, 1:400; Abcam), E-cadherin (sc-7870, 1:50; SantaCruz), MMP-2 (10373-2-AP, 1:250; Proteintech), N-Cadherin (ab245117, 1:500; Abcam), CD133 (18470-1-AP, 1:200; Proteintech) and endomucin (ab106100, 1:1000, Abcam) were used. Finally, the sections were counterstained with Hematoxylin and Eosin (H&E) or PAS staining.
The percentage of the staining cells (P) was scored as follows: 0 (none), 1 (< 25% positive cells), 2 (25%–50% positive cells), 3 (> 50% positive cells). Staining intensity (I) was graded as follows: 0 (negative), 1 (weak yellow), 2 (medium yellow), 3 (deep yellow). Samples were evaluated for both factors, i.e. P multiplied by I. The samples with scoring > 3 were considered as positive.
Soft agar colony formation assays
In a 24-well plate pre-coated with 0.5% alkaline agar, cells were suspended in the top soft agar (0.35%) layer at approximately 5 × 102/ml. Allow colonies to grow in incubators at 37 °C with 5% CO2 for 3 weeks. Colony forming was quantified using the inverted microscope (Nikon, Japan).
Dual-luciferase reporter gene experiment
A promoter clone for ASPM (HPRM38862-PG04) was obtained from FulenGen (China). The aforementioned plasmids were co-transfected with varying concentrations of EX-Nrf2 or control plasmids into 293 T cells, and with varying concentrations of SH-Nrf2 or control plasmids into Huh7 cells. After 48 h, culture supernatant was collected and luciferase activities were measured according to the manufacturer’s instructions (Secrete-Pair TM Dual Luminescence Assay Kit, GeneCopoeia, LF031).
Flow cytometry
Cells were collected, 75% ethanol-fixed, and incubated in the dark with APC-conjugated anti-human CD133 antibody (397,905, BioLegend). Flow cytometry was conducted using Accuri C6 laser flow cytometer (BD, USA), and data analysis was performed with FlowJo 10.0 software, with gating based on an unstained control reference.
Immunofluorescence (IF) assays
Cells were permeabilized with 0.1% Triton X-100, blocked with 5% FBS, and incubated overnight at 4 °C with anti-Nrf2 (1:100) and anti-ASPM (1:100) antibodies, followed by secondary antibodies conjugated with Alexa 488 or Alexa 568 (Molecular Probes, Eugene, OR, USA). Cell nuclei were stained with DAPI (Sigma) for 10 min and analyzed using a Leica laser scanning confocal microscope.
3D matrigel culture
The bottom of 24-well plate was coated with DMEM: Matrigel (BD Biosciences) (4:1) mixture. Tumor cells were seeded and incubated for 36 h. VM tubes were photographed using a phase contrast microscope.
RNA sequencing
Total RNA was extracted from HCCLM3 cells transfected with EX-ASPM and EX-Control using TRIzol reagent, and was sent to LC-Bio Technologies (Zhejiang, China) for RNA sequencing.
Xenograft animal models
The animal experiment was approved by the Ethics Committee of Tianjin Medical University. Four-week-old nude mice were randomly divided into nine groups, subcutaneously inoculated with 1 × 107 cells. Tumor volume was monitored weekly using digital calipers. The tumor volume (TV) was calculated by the following formula: TV = 1/2 × a × b2 (in which a was the length and b was the width of tumor). When the tumor volume reached 50–100 mm3, ATRA was administered intraperitoneally to the mice in the ATRA treatment group daily (10 mg/kg). After 4 weeks mice were sacrificed and xenograft tumors were processed for histology and IHC analyses.
Bioinformatics technology
Cistrome DB, MotifMap and CHEA were used for the prediction of Nrf2 target genes. RNA-sequencing expression (level 3) profiles and corresponding clinical information were downloaded from the TCGA dataset (https://portal.gdc.com). R software GSVA package was used to analyze, choosing parameter as method = ‘sgsea’.
Patient samples
HCC tissues and paired normal liver tissues were collected at the Tianjin Cancer Hospital of Tianjin Medical University. The written informed consents from all the patients were obtained. The study was conducted in accordance with Declaration of Helsinki and was approved by the review board of Tianjin Medical University, China.
Statistical analysis
Experimental data were analyzed using GraphPad Prism, presented as mean ± standard deviation. Two-group differences were assessed with T-tests, and one-way ANOVA was used for more than two groups. A significance level of p < 0.05 was applied (*p < 0.05, **p < 0.01, ***p < 0.001), and all tests were two-tailed.
Results
Under hypoxic environment, Nrf2 can promote EMT, VM, stemness and invasion ability of HCC cells
We first observed the Nrf2 protein expression level in four HCC cell lines HCCLM3, Huh7, MHCC-97H and MHCC-97L under normoxic and hypoxic conditions. The Nrf2 protein expression level was the lowest in HCCLM3 and was the highest in Huh7 under normoxia. By using CoCl2 to mimic the hypoxic environment, the expression of HIF-1α protein of the four HCC cell lines significantly increased following treatment with CoCl2 (Fig. 1a). Importantly, Nrf2 protein expression level of the four HCC cell lines was also increased under hypoxia (Fig. 1a).

Nrf2 can promote EMT, VM, stemness and invasion ability of HCC cells under hypoxic environment. a Expression of HIF-1α and Nrf2 in four HCC cells was increased under hypoxia than in under normoxic condition. b Nrf2 overexpression inhibited E-cadherin expression, promoted N-cadherin, VE-cadherin, MMP2 and CD133 expression in HCCLM3 cells, especially under hypoxia. Meantime, Nrf2 knockdown in Huh7 cells induced increased E-cadherin expression, and decreased N-cadherin, VE-cadherin, MMP2 and CD133 expression. Although CoCl2 treatment led to moderate increase of Nrf2 expression in SH-Nrf2 Huh7 cells, SH-Nrf2 Huh7 cells showed increased E-cadherin expression and decreased N-Cadherin, VE-Cadherin, MMP2 and CD133 expression compared with SH-Control Huh7 cells under hypoxia. c, d Nrf2 overexpression promoted the invasion (c) and VM formation (d) of HCCLM3 cells, especially under hypoxia. Meantime, Nrf2 knockdown in Huh7 significantly reduced the invasion capability (c) and VM formation (d), while CoCl2 treatment slightly restored these abilities of SH-Nrf2 Huh7 cells when Nrf2 expression was silenced. e Flow cytometry analysis of CD133+ cells in EX-control and EX-Nrf2 HCCLM3 cells, and in SH-Control and SH-Nrf2 Huh7 cells (with blue and red denoting negative controls and samples, respectively). f Soft agar colony formation assay showed that HCCLM3 cells overexpressing Nrf2 exhibited a higher capability to form colonies while Nrf2 silencing in Huh7 cells inhibited the formation of cell colonies
Subsequently, we performed lentiviral transfection to induce Nrf2 silencing in Huh7 cells and Nrf2 overexpression in HCCLM3 cells (Fig. 1b). Western Blot demonstrated that overexpression of Nrf2 in HCCLM3 cells resulted in a significant reduction in the protein levels of the epithelial marker, E-Cadherin, while concurrently elevating the protein levels of the mesenchymal marker, N-Cadherin. Furthermore, Nrf2 upregulation was associated with an upregulation of the VM marker VE-cadherin and MMP2. Notably, the protein levels of CSCs marker CD133 was elevated by the upregulation of Nrf2. Meantime, silencing Nrf2 in Huh7 cells led to the suppression of protein expression of N-Cadherin, VE-Cadherin, MMP2 and CD133, concomitant with an increase in E-Cadherin protein levels (Fig. 1b).
Particularly under hypoxia, the protein levels of Nrf2 in HCCLM3 EX-Nrf2 cells were further increased compared to EX-Control HCCLM3 cells under hypoxia and EX-Nrf2 HCCLM3 cells under normoxia (Fig. 1b). In addition, protein levels of N-Cadherin, VE-Cadherin, MMP2 and CD133 were all significantly higher and E-Cadherin protein levels were notably lower in HCCLM3 EX-Nrf2 cells under hypoxia compared to EX-Control HCCLM3 cells under hypoxia and EX-Nrf2 HCCLM3 cells under normoxia (Fig. 1b), suggesting EMT phenotype, VM and stemness was promoted by Nrf2 overexpression under hypoxia. Importantly, although hypoxia condition restored moderate Nrf2 expression of SH-Nrf2 Huh7 cells, these cells showed increased E-cadherin expression and decreased N-Cadherin, VE-Cadherin, MMP2 and CD133 expression compared with SH-Control Huh7 cells under hypoxia (Fig. 1b).
Through transwell experiments (Fig. 1c) and matrix gel-based tube formation assay (Fig. 1d), we observed that Nrf2 promoted the invasion and VM formation of HCCLM3 cells, especially under hypoxia. Conversely, inhibiting Nrf2 significantly reduced the invasion capability and VM formation of Huh7 cells, while hypoxia slightly restored these abilities of SH-Nrf2 Huh7 cells when Nrf2 expression was silenced (Fig. 1c, d).
To further explore the influence of Nrf2 on HCC cell stemness, flow cytometry analysis were performed to assess the expression of CD133. The results revealed that in the EX-Nrf2 HCCLM3 cells, the proportion of CD133-positive cells was 52%, as opposed to 23% in the control HCCLM3 cells. In addition, in the SH-Nrf2 Huh7 cells, the CD133-positive cell population was 24%, in contrast to 45% in the control Huh7 cells (Fig. 1e). Subsequently, soft agar colony formation experiments demonstrated that HCCLM3 cells overexpressing Nrf2 exhibited a higher capability to form colonies and generated a greater number of colonies. Conversely, Nrf2 expression silencing in Huh7 cells inhibited the formation of cell colonies (Fig. 1f).
Nrf2 directly binds to the ASPM promoter to promote ASPM expression
To identify the downstream target genes regulated by Nrf2, cistrome DB, MotifMap, and CHEA databases were used and a Venn diagram revealed the identification of four differentially expressed genes including GAPDH, RXRA, IKBKG and SQSTM1 as genes regulated by Nrf2 (Fig. 2a). Meantime, given that Nrf2 was a transcription factor which bound to the antioxidant response element (ARE) (TGACnnnGC) and promoted stemness of HCC cells in the above experiment, we also analyzed cancer stemness related genes which contain ARE elements in the promoter region. Emerging evidence suggests that ASPM plays a crucial role in the regulation of cancer stemness across various malignancies [20, 24,25,26]. Interestingly, we found ARE was rich in the ASPM promoter region, indicating a potential Nrf2 binding site in the ASPM promoter. Further JASPAR analysis was instrumental in revealing Nrf2 binding sites within the ASPM promoter region, with predicted binding scores consistently surpassing 80% (Fig. 2b, supplementary Table 1). Subsequently, we selected five candidate genes, including GAPDH, RXRA, IKBKG, SQSTM1 and ASPM, to perform real-time quantitative PCR analysis employing gene-specific primers. ASPM showed significant expression increase in EX-Nrf2 HCCLM3 cells than in EX-Control HCCLM3 cells (Fig. 2c). These results suggested that ASPM may play a relatively independent role in the signal pathway regulated by Nrf2.

Nrf2 directly binds to the ASPM promoter to promote ASPM expression. a Venn diagram of three databases that predicted downstream target of Nrf2. b Predicted Nrf2 binding site sequence within the ASPM promoter region from JASPAR. c Relative expression levels of Nrf2 downstream target genes were quantified by real-time PCR. ASPM showed significant expression increase in EX-Nrf2 HCCLM3 cells than in EX-Control HCCLM3 cells. d ChIP-qPCR showed Nrf2 could bind to the promoter of ASPM. e Dual-luciferase reporter assay in 293 T cells co-transfected with the ASPM promoter reporter gene and EX-Nrf2. f Dual-luciferase reporter assay in HCC cells co-transfected with the ASPM promoter reporter gene and SH-Nrf2. g Immunofluorescent character of Nrf2 and ASPM protein expression and localization dynamics in HCCLM3 and Huh7 cell lines under normoxic and hypoxic conditions. CoCl2 treatment of EX-Nrf2 HCCLM3 cells or SH-Control Huh7 cells led to Nrf2 protein translocation to the cell nucleus from the cytoplasm, and a concurrent increase in ASPM expression
To investigate whether Nrf2 can directly bind to the ASPM promoter region to modulate ASPM expression, we detected the direct binding of Nrf2 to the ASPM promoter through ChIP experiments (Fig. 2d). Nrf2 bound to the ASPM promoter within the region spanning from − 1457 bp to − 1660 bp. To observe the impact of Nrf2 on ASPM promoter activity, we constructed luciferase reporter gene vector containing a segment of the ASPM promoter. In 293 T cells, Nrf2 overexpression significantly increased the luciferase activity of the ASPM promoter in a dose-dependent manner compared to control (Fig. 2e), additionally, in Huh7 cells, SH-Nrf2 inhibited ASPM promoter reporter gene activity, indicating that Nrf2 directly promotes ASPM expression (Fig. 2f).
To further investigate the relationship between Nrf2 and ASPM, as well as their protein expression locations, we conducted immunofluorescence experiments. The results revealed that under normoxic conditions, both Nrf2 and ASPM exhibited low expression levels in HCCLM3 and were located in the cytoplasm. However, after treatment with CoCl2, their expression levels increased. Interestingly, under hypoxic conditions, Nrf2 overexpression in HCCLM3 cells led to its translocation to the cell nucleus from the cytoplasm, and a concurrent increase in ASPM expression. Furthermore, under normoxic conditions in Huh7 cells, both Nrf2 and ASPM exhibited high expression levels, but after Nrf2 knockdown, their expression levels significantly decreased. Similarly, hypoxia led to an increase in Nrf2 expression, its translocation to the nucleus, and an increase in ASPM expression in control Huh7 cells. However, similar phenomena were not observed in SH-Nrf2 Huh7 cells (Fig. 2g).
Under hypoxic condition, ASPM can promote EMT, VM, stemness and invasion ability of HCC cells
We assessed the protein expression levels of ASPM in different HCC cell lines and observed that under normoxic conditions, ASPM was highly expressed in Huh7 cells, while its expression was relatively lower in HCCLM3 cells. Importantly, hypoxic conditions significantly elevated the protein levels of ASPM following HIF-1α expression increase (Fig. 3a). Furthermore, when HIF-1α was upregulated in HCCLM3 cells, the expression levels of Nrf2 and ASPM proteins were significantly increased. Conversely, under hypoxic conditions, downregulation of HIF-1α resulted in a marked decrease in Nrf2 and ASPM expression levels (Fig. S1).

ASPM can promote EMT, VM, stemness and invasion ability of HCC cells under hypoxic condition. a Expression of HIF-1α and ASPM in four HCC cells was increased under hypoxia than in under normoxic condition. b Western blot analysis of EMT markers (E-cadherin, N-cadherin), VM markers (VE-cadherin, MMP2), and CSC marker (CD133) in HCCLM3 and Huh7 cell lines with regulated ASPM expression under normoxic and hypoxic conditions. c Immunofluorescence showed that ASPM expression was further augmented when EX-ASPM HCCLM3 cells were exposed to CoCl2 treatment. Meantime, the expression levels of ASPM in SH-Control Huh7 cells were further enhanced simultaneously accompanying Nrf2 translocating to the nucleus following CoCl2 treatment. However, in SH-ASPM Huh7 cells, CoCl2 treatment couldn’t induce significant ASPM expression increase although there was a slight Nrf2 expression increase. d, e ASPM overexpression promoted the invasion (d) and VM formation (e) of HCCLM3 cells, especially under hypoxia. Meantime, ASPM silencing in Huh7 significantly reduced the invasion capability (d) and VM formation (e), while CoCl2 treatment slightly restored these abilities of SH-ASPM Huh7 cells when ASPM expression was silenced. f Flow cytometry analysis of CD133+ cells in EX-control and EX-ASPM HCCLM3 cells, and in SH-Control and SH-ASPM Huh7 cells (with blue and red denoting negative controls and samples, respectively). g Soft agar colony formation assay showed that HCCLM3 cells overexpressing ASPM exhibited a higher capability to form colonies while ASPM knockdown in Huh7 cells inhibited the formation of cell colonies
To demonstrate the direct role of ASPM in HCC cells, HCCLM3 cells was transfected with ASPM overexpression plasmids and Huh7 cells was transfected with ASPM shRNA plasmids. Western blot (Fig. 3b) and immunofluorescence (Fig. 3c) revealed that ASPM expression in EX-ASPM HCCLM3 cells was significantly higher in comparison to control cells transfected with empty vectors, and this was notably augmented when EX-ASPM HCCLM3 cells were exposed to CoCl2 treatment. In contrast, in Huh7 cells under normoxic conditions, ASPM expression significantly decreased upon ASPM silencing (Fig. 3b, c). Furthermore, immunofluorescence showed hypoxia further enhanced the expression levels of ASPM in SH-Control Huh7 cells (Fig. 3c) simultaneously accompanying Nrf2 translocating to the nucleus. However, in SH-ASPM Huh7 cells, hypoxia could not induce significant ASPM expression increase although there was a slight Nrf2 expression increase (Fig. 3c).
The upregulation of ASPM in HCCLM3 cells led to a decrease in E-Cadherin expression, and this decrease was further pronounced in the CoCl2-treated condition, compared to the control cells. Meantime, the expression of N-Cadherin, VE-Cadherin, MMP2, and CD133 was elevated, with further enhancement following CoCl2 treatment (Fig. 3b). Correspondingly, the downregulated ASPM expression in Huh7 cells led to decreased levels of N-Cadherin, VE-Cadherin, MMP2, and CD133, and increased E-Cadherin expression. Importantly, even under hypoxic conditions, ASPM knockdown in Huh7 cells still led to decreased N-Cadherin, VE-Cadherin, MMP2 and CD133 expression and increased E-cadherin expression, compared to control cells under hypoxia (Fig. 3b).
In HCCLM3 cells, the upregulation of ASPM, or combined with hypoxia, significantly increased VM formation and enhanced cell invasion capabilities (Fig. 3d, e). In contrast, the silencing of ASPM in Huh7 cells suppressed VM formation and invasion. Importantly, the VM tube formation and invasive cell number were fewer in SH-ASPM Huh7 cells than in SH-Control Huh7 cells under hypoxia (Fig. 3d, e). Flow cytometry results demonstrated that in HCCLM3 cells overexpressing ASPM, the proportion of CD133-positive cells increased from 16 to 54%, while in Huh7 cells with ASPM knockdown, this proportion decreased from 59 to 31%, indicating a positive correlation between ASPM and CD133 expression (Fig. 3f). Additionally, soft agar colony formation experiments revealed that HCCLM3 cells overexpressing ASPM exhibited a significantly higher capacity to form colonies compared to the control group, while the knockdown of ASPM in Huh7 cells inhibited colony formation (Fig. 3g).
Nrf2 affects EMT, VM, stemness and invasion ability of HCC cells via ASPM expression
Next we conducted rescue experiments to assess whether the impact of Nrf2 on HCC cells was mediated through ASPM expression. Co-transfection of HCCLM3 cells with overexpressing Nrf2 plasmids and shRNA ASPM plasmids confirmed that ASPM knockdown could reverse the elevation of ASPM protein levels induced by hypoxia and Nrf2 overexpression (Fig. 4a). Similarly, ASPM silencing also counteracted the decrease in E-Cadherin protein expression and the increase in N-Cadherin, VE-Cadherin, MMP2, and CD133 expression induced by hypoxia and Nrf2 overexpression (Fig. 4a). As expected, ASPM knockdown counteracted the promoting effects of hypoxia and Nrf2 expression on HCCLM3 cell invasion and VM formation (Fig. 4b, c).

Rescue experiments show Nrf2 affects EMT, VM, stemness and invasion ability of HCC cells via ASPM expression. a–c ASPM knockdown in EX-Nrf2 HCCLM3 cells could reverse the elevation of ASPM protein levels, EMT, CSCs and VM markers expression (a), cell invasion ability (b) and VM formation (c) induced by hypoxia and Nrf2 overexpression. Meantime overexpressing ASPM in Huh7 SH-Nrf2 cells under hypoxia restored ASPM high expression and subsequent EMT phenotype, VM and CSCs marker expression (a), cell invasion ability (b) and VM formation (c)
In addition, Nrf2 knockdown in Huh7 cells could inhibit the elevation of ASPM protein levels induced by hypoxia, as well as inhibited the decrease in the E-Cadherin expression and the increase in N-Cadherin, VE-Cadherin, MMP2 and CD133 expression (Fig. 4a). Importantly, overexpressing ASPM in Huh7 SH-Nrf2 cells under hypoxia restored ASPM high expression and subsequent EMT phenotype, VM and CSCs marker expression (Fig. 4a). Meantime, Nrf2 knockdown suppressed the increased invasion and VM tube formation capabilities induced by hypoxia in Huh7 cells, and overexpression of ASPM in Huh7 SH-Nrf2 cells restored these capabilities under hypoxia (Fig. 4b, c).
Elevated ASPM in HCC cells impacts retinol metabolism, with all-trans retinoic acid (ATRA) treatment reversing stemness and VM
Further mRNA transcriptome sequencing was conducted on HCCLM3 cells overexpressing ASPM and control cells. Figure 5a and supplementary Table 2 illustrate all differentially expressed genes. Subsequently, enrichment analysis of differentially expressed genes was conducted using KEGG. Figure 5b displayed the top 20 pathways with the smallest p-values, and the distribution of upregulated and downregulated genes within these pathways is shown in Fig. 5c. We focused on the retinol metabolism pathway, in which the expression of 14 genes related to retinol metabolism was found to increase or decrease with ASPM upregulation (Fig. 5d). Furthermore, TCGA database analysis revealed a negative correlation between ASPM expression and retinol metabolism pathway in 371 liver cancer patients (Fig. 5e).

Elevated ASPM in HCC cells impacts retinol metabolism, with ATRA treatment reversing stemness and VM. a Heatmap of the differentially expressed genes between EX-ASPM and EX-Control HCCLM3 cells. b The top 20 enriched KEGG pathways of differentially expressed genes. c The distribution of upregulated and downregulated differentially expressed genes in the top 20 pathways. d The differential expression of retinol metabolism-related genes between EX-control and EX-ASPM HCCLM3 cells. e The Spearman correlation analysis of TCGA data between ASPM expression and retinol metabolism pathway. The X-axis in the graph represents gene expression, and the Y-axis denotes the pathway scoring. f ATRA treatment induced a significant reduction in CD133 expression in HCCLM3 cells overexpressing ASPM by flow cytometry. g ATRA treatment reversed EMT, CSCs and VM marker expression induced by ASPM overexpression by Western Bolt. f–h Inhibition of soft agar clonogenicity and invasion in ASPM overexpressing HCCLM3 cells by ATRA treatment
Recent research showed that CSCs could be eradicated by targeting the retinol metabolism pathway [27]. Dysregulation of cellular cytoplasmic aldehyde dehydrogenase, which is responsible for synthesizing ATRA, occurs in various human cancer types. Targeting ATRA synthesis and its receptors has provided promising approaches to eliminating CSCs. Given that our results showed HCC cells with ASPM overexpression displayed cancer stemness, we observed the effect of ATRA treatment on HCC cells. The results showed a significant reduction in CD133 expression in HCCLM3 cells overexpressing ASPM compared to untreated HCCLM3 cells overexpressing ASPM (Fig. 5f, g). Consistent with the flow cytometry and Western blot finding, soft agar colony formation experiment revealed that ATRA treatment counteracted the enhanced colony-forming capacity induced by ASPM overexpression (Fig. 5h).
In addition, ATRA treatment reversed EMT and VM phenotype induced by ASPM overexpression and led to E-Cadherin protein levels increase, VE-Cadherin and N-Cadherin protein levels decrease (Fig. 5g). VM tube formation experiments revealed that ATRA treatment counteracted the enhanced tube formation capacity induced by ASPM overexpression (Fig. 5i).
Nrf2/ASPM axis affects HCC cells growth, EMT, stemness and VM in vivo
To further elucidate the molecular mechanisms linking the Nrf2/ASPM axis to in vivo tumor progression, validation experiments were conducted in nude mice model. HCCLM3 cells overexpressing Nrf2 or ASPM displayed a higher tumor growth rate in xenografts compared to the control group, while Nrf2 or ASPM silencing in Huh7 cells significantly delayed tumor growth. Additionally, ATRA treatment effectively inhibited tumor growth in ASPM-overexpressing tumors (Figs. 6a, b, 7a, d).

Nrf2 affects HCC cells growth, EMT, stemness and VM in vivo. a, b Enhanced tumor growth in Nrf2-overexpressing HCCLM3 xenografts compared to controls; Nrf2 knockdown in Huh7 xenografts significantly inhibits tumor growth. c VM channels were indicated by red arrows, and endomucin-positive genuine blood vessels were indicated by black arrows. High Nrf2 expression xenografts exhibited increased VM formation rate. d Tumor sections with H&E staining showed necrosis in EX-Nrf2 and control HCCLM3, SH-Nrf2 and control Huh7 xenografts. The expression levels of HIF-1α, Nrf2, ASPM, VE-Cadherin, N-Cadherin, E-Cadherin, and CD133 were evaluated in EX-Nrf2 and control HCCLM3, SH-Nrf2 and control Huh7 xenografts by IHC staining

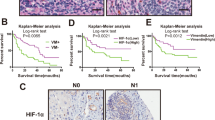
ASPM affects HCC cell growth, stemness, and VM in vivo, and these effects can be reversed by ATRA treatment, and the expression of Nrf2/ASPM is associated with poor prognosis in HCC patients. a Tumor growth in ASPM-knockdown Huh7 xenografts is inhibited compared to the control group. b Endomucin/PAS staining shows VM channels indicated by red arrows and true blood vessels positive for endomucin indicated by black arrows. The rate of VM formation is reduced in ASPM-knockdown xenografts. c IHC staining for HIF-1α, ASPM, VE-Cadherin, and CD133 expression levels in control Huh7 and SH-ASPM xenografts. d Tumor growth in ASPM-overexpressing HCCLM3 xenografts is enhanced compared to the control group, while ATRA treatment inhibits tumor growth. e Endomucin/PAS staining shows VM channels indicated by red arrows and true blood vessels positive for endomucin indicated by black arrows. The rate of VM formation is increased in ASPM-overexpressing xenografts compared to the HCCLM3 control group, while the VM formation rate is lower in the ATRA treatment group than in the EX-ASPM group. f IHC staining for HIF-1α, ASPM, VE-Cadherin, and CD133 expression levels in control HCCLM3, EX-ASPM, and EX-ASPM ATRA treatment group xenografts. g qRT-PCR analysis of Nrf2 and ASPM mRNA in HCC and adjacent normal liver tissues (n = 104). h Correlation analysis of Nrf2 and ASPM mRNA levels in HCC samples (n = 104). i Representative immunohistochemical staining of Nrf2 and ASPM protein in HCC tissues. j Correlation analysis of Nrf2 and ASPM protein expression levels by IHC in HCC samples (n = 88). k Kaplan–Meier survival curves compared the survival rates of patients with Nrf2 and ASPM positive (IHC score ≥ 4) versus negative (IHC score ≤ 3) expression in 88 HCC patients
Next, endomucin/PAS dual staining was employed to identify VM channels (Fig. 6c) (red arrows denoted endomucin-positive genuine vessels, black arrows indicated VM). VM channels were vascular-like patterns containing red blood cells composed of HCC cells and basement membrane stained positive with PAS in the absence of endothelial cells. VM structures were typically found adjacent to the normal vascular system, indicating coordination and blood supply between them. In HCCLM3 EX-Control xenografts, VM formation was observed in only one case (1/7, 14.3%). However, VM formation was significantly increased in the HCCLM3 EX-Nrf2 and EX-ASPM groups (4/7, 57% and 5/7, 71.4%, respectively). The EX-ASPM ATRA treatment group showed a reduction in VM cases (2/7, 28%). Additionally, VM formation was present in all xenografts derived from Huh7 control cells (7/7, 100%). In contrast, silencing Nrf2 and ASPM significantly decreased the percentage of VM formation (2/7, 28.6% and 3/7, 42.9%, respectively) (Figs. 6c, 7b, e).
In addition, the presence of tumor hypoxic microenvironment in this model was indicated by necrosis and HIF-1α activation (Fig. 6d). Interestingly, we observed more red blood cells (hemorrhaging) in the necrotic areas of Huh7 xenografts. This blood perfusion in the necrotic regions, observed in Huh7 but not in LM3 xenografts, may contribute to the faster tumor growth and larger tumor volume of Huh7 tumors. Consequently, tumor cells can mimic endothelial cells to form VM channels, further enhancing the blood supply for Huh7 xenografts. Compared to the control HCCLM3 group, increased expression of ASPM, CD133 and VM markers VE-Cadherin and MMP2 was observed in EX-Nrf2 HCCLM3 xenografts by immunohistochemistry (IHC). EMT-related markers IHC staining showed a decrease in E-cadherin, an increase in N-cadherin in EX-Nrf2 HCCLM3 xenografts (Fig. 6d). Conversely, when Nrf2 expression was reduced in Huh7 cells, ASPM and CD133 expression decreased, E-Cadherin expression increased, and N-Cadherin, VE-Cadherin and MMP2 expression decreased compared to the control Huh7 group (Fig. 6d). Similarly, IHC staining revealed that in xenografts of Huh7 cells with ASPM knockdown, the expression levels of ASPM, CD133, and VE-Cadherin were reduced compared to the control cells (Fig. 7c). In HCCLM3 cells overexpressing ASPM, levels of ASPM, CD133, and VE-Cadherin were higher than those in the control group. However, in the xenografts of EX-ASPM Huh7 cells treated with ATRA, the expression levels of ASPM, CD133, and VE-Cadherin were lower than in the EX-ASPM group (Fig. 7f).
Nrf2/ASPM expression was related to poor prognosis of HCC patients
To further validate the clinical relevance of Nrf2 and ASPM, we collected frozen tissue specimens from 104 HCC patients. The mRNA expression levels of Nrf2 and ASPM were determined in both HCC tissues and adjacent normal liver tissues using qRT-PCR. The results showed that the mRNA levels of Nrf2 and ASPM in HCC tissues were significantly higher than those in adjacent normal tissues (Fig. 7g). Additionally, in the 104 HCC cases, Nrf2 exhibited a positive correlation with ASPM (R = 0.201, p < 0.001) (Fig. 7h).
We further analyzed the expression of Nrf2 and ASPM protein in 88 HCC specimens by IHC. Consistent with mRNA analysis results (Fig. 7i), a statistically significant positive correlation was observed between Nrf2 and ASPM protein expression (R = 0.215, p < 0.001) (Fig. 7j). Importantly, the results revealed that patients with high Nrf2 expression (n = 61) exhibited significantly shorter overall survival compared to those with low Nrf2 expression (n = 27). Similarly, patients with high ASPM expression (n = 66) had a poorer prognosis than those with low ASPM expression (n = 22) (Fig. 7k). Additionally, further clinical feature analysis revealed that increased expression of Nrf2 and ASPM was significantly associated with pathological grade and metastasis, but not with age, gender, or tumor size (supplementary Table 3).
Discussion
Hypoxia has long been acknowledged as one of the fundamental characteristics of solid tumors, playing a pivotal role in tumor recurrence and metastasis. HCC, in particular, is recognized as one of the most hypoxic malignant tumors [28]. Nrf2, a redox-sensitive transcription factor that is also overexpressed in various tumors, could induce hypoxia-like metabolic changes and stabilize HIF-1α expression in HCC [29]. In addition, an accumulation of evidence supports the overexpression of Nrf2 in liver cancer, and high levels of Nrf2 are an independent factor indicating poor prognosis in patients with liver cancer. Moreover, the overexpression and nuclear localization of Nrf2 in HCC cells suggest its constitutive activation under hypoxia [30]. Our results showed that a significant upregulation of Nrf2 protein levels and its expression translocating from cytoplasm to nucleus in HCC cell lines under hypoxic conditions, demonstrating Nrf2 was activated under hypoxia in HCC.
It has been found that the increased expression of Nrf2 leads to morphological changes in tumor cells that facilitate the EMT, cancer stemness and promote tumor metastasis [31,32,33]. Coherently, our in vitro and in vivo experiments have also substantiated that Nrf2 within liver cancer cells inhibits E-cadherin expression while enhancing N-cadherin expression. Simultaneously, Nrf2 promoted CSCs marker CD133 expression and colony-forming capabilities of HCC cells. Furthermore, Nrf2 upregulated the expression of VM marker MMP2 and VE-cadherin, and promoted VM tubules formation. These outcomes indicated that the aberrant activation of Nrf2 facilitated EMT, CSCs population expansion and VM formation. Importantly, our results further displayed that under hypoxia Nrf2 upregulation could still further enhance EMT phenotype, stemness and VM formation. However, Nrf2 silencing under hypoxia reversed EMT, stemness and VM formation in HCC cells. These results collectively suggested the hypoxic microenvironment of liver cancer cells triggered the aberrant activation of Nrf2 and Nrf2 upregulation kept on promoting HCC progression under hypoxia.
Amidst deepening insights into the role of Nrf2 in HCC, our study sheds further light on the mechanisms by which Nrf2 drives HCC process. Utilizing bioinformatics tools, ChIP assays and dual-luciferase reporter gene experiments, we identified ASPM as a downstream target of Nrf2. There was a direct binding between Nrf2 protein and the promoter of ASPM, thus promoting ASPM expression. Our results have, for the first time, identified a potential key role for Nrf2 in regulating the expression of ASPM. We observed that under hypoxic conditions, the accumulation of Nrf2 in the cytoplasm markedly increased and subsequently translocated into the nucleus, while concurrently, the expression of ASPM in the cytoplasm was also elevated. Further human HCC tissue experiments showed that both Nrf2 and ASPM were highly expressed compared to normal live tissue, and there existed a positive correlation between them. In vivo mice experiments have demonstrated that the expression of Nrf2 can promote the expression of ASPM, impacting EMT and CSCs, and therefore, Nrf2 can facilitate the formation of VM and tumor growth in mice xenografts, thus providing a reasonable explanation for Nrf2 playing in the malignant progression of HCC via regulating ASPM expression.
In the symmetrical division of neural stem cells, a process critical to the renewal of stem cells, ASPM is a pivotal regulatory factor [34]. Previous research has demonstrated that ASPM expression is associated with poor prognosis in HCC [35]. Furthermore, interfering with ASPM expression has been shown to suppress HCC cell invasion and EMT [36]. Our data consistently underscores the high expression of ASPM in liver cancer cells, with notable effects extending beyond promoting EMT conversion to significantly enhancing CSCs characteristics. Earlier research has pointed out the enrichment of liver CSCs within the CD133-expressing cancer cell population [37]. In our study, ASPM overexpression promoted CD133 expression and colony-forming capabilities of HCC cells. Under hypoxic stimulation, EMT and CD133 expression were further strengthened in liver cancer cells overexpressing ASPM and these cells were organized into VM tubular structures. These findings suggested that ASPM activation might enhance liver cancer VM and invasive properties by bolstering both EMT and CSCs development under hypoxia.
In addition, rescue experiments in our study showed that ASPM overexpression could rescue the inhibitory effects of Nrf2 knockdown on EMT, cancer stemness, VM and invasive ability of HCC cells. Moreover, ASPM silencing in HCC cells overexpressing Nrf2 could mitigate the promotional effects on EMT, cancer stemness, VM and invasive ability of HCC cells induced by Nrf2 overexpression. Meantime, we further found Nrf2/ASPM expression was related to poor prognosis of HCC patients. Therefore, we concluded that Nrf2/ASPM axis was activated under hypoxia, thus promoting EMT, CSCs and VM development, ultimately driving adverse outcomes in HCC patients.
Further transcriptome sequencing revealed that the retinol metabolism pathway was dysregulated in ASPM overexpression HCC cells. Data from the TCGA corroborated a negative relationship between ASPM expression and retinol metabolism. ATRA, a metabolite of retinol, is widely used in clinical therapy for acute promyelocytic leukemia (APL) and is known to induce cell differentiation in both stem cells and tumor cells [38, 39]. Recent research has suggested that ATRA effectively inhibits the growth, migration, and invasion of HCC cells by reversing EMT and inducing the differentiation of liver cancer stem cells [40,41,42]. Our study revealed that ATRA could suppress ASPM expression, attenuating the high expression of CD133 and EMT induced by ASPM. It also inhibited the enhanced clonality and VM tubule-forming capabilities driven by ASPM overexpression. This suggested that ATRA may inhibit HCC’s EMT phenotype and induce CSCs differentiation, preventing VM formation and malignant progression. Therefore, our study provided a new clue for HCC treatment. Nrf2/ASPM overexpression in HCC cells might be a plausible prerequisite for accepting ATRA treatment.
In summary, our research demonstrated that in a hypoxic environment, Nrf2 can directly regulate ASPM expression, which subsequently governs EMT, CSCs development, and VM formation. Retinol metabolism pathway was dysregulated in HCC cells with ASPM overexpression. Furthermore, ATRA treatment can reverse HCC malignant progression phenotype, possibly offering novel therapeutic point for HCC.
Abbreviations
- ASPM:
-
Abnormal spindle-like microcephaly-associated protein
- ATRA:
-
All-trans retinoic acid
- CSCs:
-
Cancer stem cells
- EMT:
-
Epithelial–mesenchymal transition
- FACS:
-
Flow cytometry
- HCC:
-
Hepatocellular carcinoma
- IHC:
-
Immunohistochemistry
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- qRT‒PCR:
-
Quantitative real-time PCR
- VM:
-
Vasculogenic mimicry
References
Rumgay H, Arnold M, Ferlay J, et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J Hepatol. 2022;77:1598–606.
Singal AG, Llovet JM, Yarchoan M, et al. AASLD practice guidance on prevention, diagnosis, and treatment of hepatocellular carcinoma. Hepatology. 2023;10:1097.
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Yuen VW, Wong CC. Hypoxia-inducible factors and innate immunity in liver cancer. J Clin Invest. 2020;130:5052–62.
Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–52.
Ren K, Yao N, Wang G, et al. Vasculogenic mimicry: a new prognostic sign of human osteosarcoma. Hum Pathol. 2014;45:2120–9.
Liu Y, Li F, Yang YT, et al. IGFBP2 promotes vasculogenic mimicry formation via regulating CD144 and MMP2 expression in glioma. Oncogene. 2019;38:1815–31.
Liu TJ, Sun BC, Zhao XL, et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene. 2013;32:544–53.
Li Y, Wu Z, Yuan J, et al. Long non-coding RNA MALAT1 promotes gastric cancer tumorigenicity and metastasis by regulating vasculogenic mimicry and angiogenesis. Cancer Lett. 2017;395:31–44.
Liu K, Sun B, Zhao X, et al. Hypoxia induced epithelial-mesenchymal transition and vasculogenic mimicry formation by promoting Bcl-2/Twist1 cooperation. Exp Mol Pathol. 2015;99:383–91.
Sun B, Zhang D, Zhao N, et al. Epithelial-to-endothelial transition and cancer stem cells: two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget. 2017;8:30502–10.
Wang H, Unternaehrer JJ. Epithelial-mesenchymal transition and cancer stem cells: at the crossroads of differentiation and dedifferentiation. Dev Dyn. 2019;248:10–20.
Bhuria V, Xing J, Scholta T, et al. Hypoxia induced Sonic Hedgehog signaling regulates cancer stemness, epithelial-to-mesenchymal transition and invasion in cholangiocarcinoma. Exp Cell Res. 2019;385: 111671.
He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. 2020;21:4777.
Tao S, de la Vega MR, Chapman E, et al. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol Carcinog. 2018;57:182–92.
Wang XJ, Sun Z, Villeneuve NF, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–43.
DeNicola GM, Chen PH, Mullarky E, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475–81.
Wakabayashi N, Skoko JJ, Chartoumpekis DV, et al. Notch-Nrf2 axis: regulation of Nrf2 gene expression and cytoprotection by notch signaling. Mol Cell Biol. 2014;34:653–63.
Li L, Pan H, Wang H, et al. Interplay between VEGF and Nrf2 regulates angiogenesis due to intracranial venous hypertension. Sci Rep. 2016;6:37338.
Pai VC, Hsu CC, Chan TS, et al. ASPM promotes prostate cancer stemness and progression by augmenting Wnt-Dvl-3-β-catenin signaling. Oncogene. 2019;38:1340–53.
Liao WY, Hsu CC, Chan TS, et al. Dishevelled 1-regulated superpotent cancer stem cells mediate Wnt heterogeneity and tumor progression in hepatocellular carcinoma. Stem Cell Reports. 2020;14:462–77.
Tsai KK, Bae BI, Hsu CC, et al. Oncogenic ASPM Is a regulatory hub of developmental and stemness signaling in cancers. Cancer Res. 2023;83:2993–3000.
Yuan Y, Hilliard G, Ferguson T, et al. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J Biol Chem. 2003;278:15911–6.
Wang WY, Hsu CC, Wang TY, et al. A gene expression signature of epithelial tubulogenesis and a role for ASPM in pancreatic tumor progression. Gastroenterology. 2013;145:1110–20.
Yao XH, Ping YF, Bian XW. Contribution of cancer stem cells to tumor vasculogenic mimicry. Protein Cell. 2011;2:266–72.
Vange P, Bruland T, Beisvag V, et al. Genome-wide analysis of the oxyntic proliferative isthmus zone reveals ASPM as a possible gastric stem/progenitor cell marker over-expressed in cancer. J Pathol. 2015;237:447–59.
Brown G. Targeting the retinoic acid pathway to eradicate cancer stem cells. Int J Mol Sci. 2023;24:2373.
McKeown SR. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br J Radiol. 2014;87:20130676.
Zheng J, Kim SJ, Saeidi S, et al. Overactivated NRF2 induces pseudohypoxia in hepatocellular carcinoma by stabilizing HIF-1α. Free Radic Biol Med. 2023;194:347–56.
Zhang M, Zhang C, Zhang L, et al. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer. 2015;15:531.
Arfmann-Knübel S, Struck B, Genrich G, et al. The crosstalk between Nrf2 and TGF-β1 in the epithelial-mesenchymal transition of pancreatic duct epithelial cells. PLoS ONE. 2015;10: e0132978.
Shen H, Yang Y, Xia S, et al. Blockage of Nrf2 suppresses the migration and invasion of esophageal squamous cell carcinoma cells in hypoxic microenvironment. Dis Esophagus. 2014;27:685–92.
Park J, Kim SK, Hallis SP, et al. Role of CD133/NRF2 axis in the development of colon cancer stem cell-like properties. Front Oncol. 2022;11: 808300.
Fish JL, Kosodo Y, Enard W, et al. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA. 2006;103:10438–43.
Lin SY, Pan HW, Liu SH, et al. ASPM is a novel marker for vascular invasion, early recurrence, and poor prognosis of hepatocellular carcinoma. Clin Cancer Res. 2008;14:4814–20.
Wu B, Hu C, Kong L. ASPM combined with KIF11 promotes the malignant progression of hepatocellular carcinoma via the Wnt/β-catenin signaling pathway. Exp Ther Med. 2021;22:1154.
Suetsugu A, Nagaki M, Aoki H, et al. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006;351:820–4.
Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–21.
Napoli JL, Posch KP, Fiorella PD, et al. Physiological occurrence, biosynthesis and metabolism of retinoic acid: evidence for roles of cellular retinol-binding protein (CRBP) and cellular retinoic acid-binding protein (CRABP) in the pathway of retinoic acid homeostasis. Biomed Pharmacother. 1991;45:131–43.
Cui J, Gong M, He Y, et al. All-trans retinoic acid inhibits proliferation, migration, invasion and induces differentiation of hepa1-6 cells through reversing EMT in vitro. Int J Oncol. 2016;48:349–57.
Zhang Y, Guan DX, Shi J, et al. All-trans retinoic acid potentiates the chemotherapeutic effect of cisplatin by inducing differentiation of tumor initiating cells in liver cancer. J Hepatol. 2013;59:1255–63.
Yan T, Yu L, Zhang N, et al. The advanced development of molecular targeted therapy for hepatocellular carcinoma. Cancer Biol Med. 2022;19:802–17.
Acknowledgements
This work was partly supported by a Grant from Key project of the National Natural Science Foundation of China (no. 82172874).
Funding
This work was funded by the National Natural Science Foundation of China (no. 82172874).
Author information
Authors and Affiliations
Contributions
Y.Z. designed and performed the experiments, compiled and analyzed the data, and assisted in writing the manuscript. N.C. designed and performed the experiments, compiled and analyzed the data, and assisted in writing the manuscript. S.W. performed image analysis, western blot repeats for revision. J.M. assisted in vivo studies. N.Z. assisted with mouse model. J.H performed RT-PCR assays. X.D. assisted in western blot and in vivo studies. Y.L. and J.M. performed Immunohistochemistry staining. X.Z. conceptualized the idea, designed the experiments, analyzed the data, provided the resources, and wrote the manuscript. T.L. conceptualized the idea, provided the resources, assisted in manuscript writing and supervised the overall study.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Ethical approval
This study protocol was reviewed and approved by the Ethics Committee of Tianjin Medical University, (approval number 8157111462).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
535_2024_2140_MOESM1_ESM.tif
Supplementary file1 HIF-1α promotes the expression of the Nrf2/ASPM axis proteins in HCC cells. (a) Overexpression of HIF-1α enhances the expression of Nrf2 and ASPM in HCCLM3 cells. Conversely, under hypoxic conditions, knockdown of HIF-1α reduces the protein levels of Nrf2 and ASPM in HCCLM3 cells (TIF 166 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Y., Che, N., Wang, S. et al. Nrf2/ASPM axis regulated vasculogenic mimicry formation in hepatocellular carcinoma under hypoxia. J Gastroenterol 59, 941–957 (2024). https://doi.org/10.1007/s00535-024-02140-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-024-02140-9