
Abstract
Key message
Genetic diversity of Monoon tirunelveliense is reported here from India to facilitate its conservation and to take up such studies for conserving threatened plants from different parts of the world.
Abstract
As endemic and threatened species are susceptible to loss of genetic variation due to stochastic factors, investigation was carried out to assess genetic diversity of Monoon tirunelveliense (Annonaceae), which is a steno endemic and a critically endangered species from the Kalakkad-Mundanthurai Tiger Reserve in India, using inter-simple sequence repeats (ISSRs). Thirty-five samples collected from seven populations were screened by 34 primers, wherein 11 primers produced 65 clear and reproducible bands that included 43.08 % of polymorphic bands (PPB), at the rate of 1–6 bands population−1 and 5.90 bands primer−1. Nei’s genetic diversity (H) was 0.1428, on an average. Analysis of Shannon information index (I) showed 0.2120, on an average, and total genetic diversity (H T) was moderate with 0.2651 and genetic diversity within populations (H S) was a low of 0.1428. Proportion of total diversity among populations (G ST) was 0.4613. Gene flow among populations (Nm) was 0.5839 individuals generation−1 indicating a low migration rate among populations. Mantel test showed no significant correlation among genetic differentiation and geographical distance (r = 0.489, p = 0.007). Phylogenetic relationship among Monoon species using rbcL and matK gene sequences derived by combined Bayesian analysis is given. Besides conserving all the existing populations, matured fruits should be collected and sown in debris-free soil to increase the percentage of seed germination as seeds are highly vulnerable to the attack of termites and microbial decay. Seeds from genetic diversity-rich Populations 2 and 3 are recommended for propagation to increase the extent and occurrence of the species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The relative importance of stochastic factors such as distribution among regions, habitat fragmentation, and/or population isolation, populations, individuals within populations, genetic drift, mating system, mutation, gene flow and natural selection varies among species and ecological groups. Therefore, contribution of these factors which vary from a few to more determines total genetic variation of a species. During the course of time, loss of genetic variation of the species leads to reduction in adaptability over changing environments and increases susceptibility to any kind of pressures including disease, pest and stress. In such cases, species with small population readily prone to local extinction compared to species with larger populations (Ellstrand and Elam 1993). Thus, assessment of genetic variability becomes imperative to formulate conservation practices (Gonzalez-Astorga and Castillo-Campos 2004) to define ideal appropriate units for in situ conservation and to develop effective sample collection strategies for ex situ conservation (Hogbin and Peakall 1999), especially to restore threatened populations (Hamrick and Godt 1996). Considering the longer SSR-based primers and its advantages, inter-simple sequence repeat (ISSR)s, which enable higher-stringency amplifications (Wolfe et al. 1998), are widely used (Wang 2004). The ISSRs which are hypervariable and dominant in molecular nature have the capacity to detect variability among allied or clonal individuals (Esselman et al. 1999). The primer sequences of the ISSRs are longer than RAPD primers and they can reproduce reliable bands even at higher annealing temperature (Culley and Wolfe 2001) and never require any kind of prior knowledge to target sequences (Fang and Roose 1997). Xue et al. (2012) analyzed sequences of three chloroplast markers of matK, rbcL and trnL-F in the genus of Polyalthia (Annonaceae) and resurrected the genus of Monoon with 56 species, wherein 21 species are distributed in Malaysia, 9 in Philippines, 5 each in India and Indonesia, 4 in Vietnam, 3 in Australia, 2 each in Thailand and Sri Lanka and 1 each in India and Sri Lanka, China, Japan, Papua New Guinea and Cambodia. Out of the five species reported from India, M. tirunelveliense (M.B. Viswan. and Manik.) B. Xue and R.M.K. Saunders, described by Viswanathan and Manikandan (2001), is a steno endemic and critically endangered species distributed in the Kalakkad-Mundanthurai Tiger Reserve (KMTR) in India. Therefore, ISSR, rbcL and matK markers were used to investigate M. tirunelveliense (1) to assess the relationship between the factors and genetic variations within and among populations; (2) to derive phylogenetic relationship among the Monoon species; (3) to select superior germ plasm materials from the genetic diversity-rich populations; and (4) to compare with similar endemic and threatened taxa to suggest suitable conditions for seed germination to increase both the individuals and populations.
Methods
Plant material
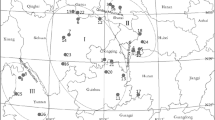
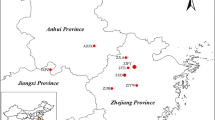
M. tirunelveliense is a lofty tree species that grows up to 25 m high. Samples of fresh, tender leaf tissues of 35 plants from seven natural populations in the KMTR (Fig. 1) were collected in vials and stored at 4 °C until DNA extraction (Fig. 1). Authentic voucher herbarium specimens (MBV, CR and PSK 1401–1407) were prepared for each population and deposited in the Herbarium of the Centre for Research and Development of Siddha-Ayurveda Medicines (CRDSAM), Department of Plant Science, Bharathidasan University, Tiruchirappalli, Tamil Nadu, India, for future reference.

Geographical position of seven natural populations of M. tirunelveliense in the KMTR
DNA extraction
Total genomic DNA was extracted following the modified CTAB method (Doyle 1991) from fresh leaf material (200 mg) which was ground with liquid nitrogen, transferred to a 1.5 ml Eppendorf tube holding 600 µl preheated 2 % CTAB extraction buffer containing 0.2 % mercaptoethanol and incubated at 60 °C for 60 min. Subsequently, a mixture of 600 µl cold chloroform: isoamylalchohol (24:1, v/v) was added, shaken gently for 3 min and spin down at 10,000 rpm for 10 min at room temperature. The supernatant thus collected was transferred into a new tube, mixed with 2/3 volume of ice-cold isopropanol, centrifuged at 12,000 rpm for 5 min, washed in 70 % ethanol and dried using hair dryer. The pellet of DNA thus recovered was re-suspended in 100 µl 1X TAE buffer, purified using spin column (Chromous Biotech, Bengaluru, India) and checked on 1.0 % agarose gel for quality and quantity.
ISSR amplification
For polymerase chain reaction (PCR) amplification, protocol of Wolfe et al. (1998) was performed with some modifications. After an initial screening of 34 (UBC-ISSR) primers from Chromous Biotech, 11 primers, each being 17–18 nucleotides in length, were selected for further analysis to record maximum genetic variation within and among populations of the species. Reactions were carried out in a total volume of 20 µl, comprising 10 µl Taq premix of Exprime Taq Premix (GeNet Bio, Chennai, India), 4 µl molecular biology grade water, 3 µl (µl/5 pmol) primer and 3 µl template (µl/50–100 ng) DNA. The PCR was performed with 96-well Thermal Cycler of Eppendorf Pro S, Hamburg, Germany, under the following conditions: 5 min at 94 °C, followed by 40 cycles of 1 min at 94 °C, 1 min annealing at 45–55 °C, and 2 min extension at 72 °C, and, a final extension cycle of 8 min at 72 °C. Amplified products were separated by electrophoresis on 1.5 % agarose gel buffered with 1X TAE and ethidium bromide. Fragment sizes were estimated based on 1 kb and 100 bp DNA ladders (Hi-Media, Mumbai, India) as size markers. To check the absence of contamination, DNA was replaced by water and performed experiment alongside of the every PCR amplification. DNA fragments were identified by image analysis software for gel documentation system (AlphaImager® EP Gel Documentation System).
Sequencing of chloroplast gene markers
The primers used for sequencing 2 chloroplast DNA regions by amplification were: forward–TAT CTT AGC GCC ATT CCG AGTA and reverse–CGC GGA TAA TTT CAT TAC CTT C (Hasebe et al. 1994) to rbcL (ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit); and, forward–ATT TGC GAT CTA TTC ATT CAA T and reverse–TGA GAT TCC GCA GGT CAT T (Manos and Steel 1997) to matK (maturase K gene). The PCR reactions were performed in a 60 μl total volume, containing 30 µl of Exprime Taq Premix (GeNet Bio, Chennai, India), 12 µl molecular biology grade water, 4.5 µl each forward and reverse primers (µl/5 pmol) and 9 µl template (µl/50–100 ng) DNA. Amplifications were performed by an initial denaturation at 94 °C for 3 min, 35 cycles at 94 °C for 30 s, annealing at 58 °C for 30 s, extension at 72 °C for 1 min, and an additional final extension at 72 °C for 5 min. The PCR products were separated on 1.2 % agarose gel and the purified PCR products were sequenced with the original amplification primers using the BigDye Terminator Cycle Sequencing Ready Reaction Kit and Genetic Analyzer Model No. ABI 3500, Life Technologies, USA.
Data analysis
The ISSR bands were scored as presence and absence by applying binary characters and the resultant data matrix was analyzed using POPGENE v. 1.31 (Yeh et al. 1999) to estimate two genetic diversity parameters–the percentage of polymorphic band (PPB) and the expected heterozygosity (H-). Total gene diversity (H T), and the coefficient of gene differentiation (G ST) were measured following Nei’s (1973). The genetic diversity (I) and genetic distance (D) among the populations were computed along with Nei’s (1972). Shannon information index was calculated by H O = −∑ p i log2 p i (Lewontin, 1972), wherein p i is the frequency of a given ISSR fragment. Total diversity (H sp) was measured by Shannon information index of phenotypic diversity and mean intra-population diversity (H pop). Then, the proportion of diversity between the populations was calculated as (H sp−H pop)/H sp. Gene flow (Nm) was estimated using Wright’s (1931) formula [Nm = (1−F ST)/4F ST, where F ST, the standardized variance among populations is considered equivalent to G ST, estimated by partitioning of the total gene diversity (Nei 1977)]. Tools for Population Genetic Analysis (Miller 1997) were applied in Mantel test (computing 1,000 permutations) to estimate correlations between geographical distances (km) and genetic distances among the populations. Unweighted pair-group method with arithmetic mean (UPGMA) dendrogram constructed based on Nei’s (1972). Total phenotypic variance among the individuals within populations and among populations was estimated by analyzing the input files created by GenAlex 6.5 version (Peakall and Smouse 2012) and used for the analysis of molecular variance (AMOVA). On the basis of Nei’s genetic distance values using software (XLSTAT 2013), Multidimensional Scaling Analysis (MDS) and Principal Coordinate Analysis (PCoA) were derived.
A Bayesian analysis with the software STRUCTURE (Pritchard et al. 2000) was applied to understand the relationships among populations, identify the number of genetic pools, assign individuals to population and identify migrants and admixed individuals. Several runs were carried out with default sampling parameters (burn-in = 5,000, the number of Markov-Chain Monte Carlo (MCMC) runs after burn-in = 5,000, using the admixture model and allele frequencies correlates). K was calculated by Evanno et al. (2005).
MEGA5 was used to align gene sequences and manual adjustments were performed to insert gaps for improving alignments (Tamura et al. 2011). Maximum Parsimony (MP) analyses of gene sequences of rbcL and matK were performed to 23 Monoon species by combined Bayesian analyses using MrBayes3.2 (Ronquist et al. 2012). The parameters of settings applied were: sampling frequency = 100; temp = 0.1; burn-in = 1,000 and the number of MCMC generations = 25,000.
Results
ISSR polymorphism and genetic diversity
Thirty-four ISSR primers were screened. At the end, 11 primers chosen for analysis produced a total of 65 clear and reproducible bands ranging in size from 300 to 2,000 bp (an average of 5.90 bands primer−1). Among the 65 bands, 28 bands (43.08 %) were polymorphic at species level. The PPB ranged from 10.71 (Population 6) to 64.29 (Population 2) with an average of 38.78 %. Population 2 and 3 had higher PPB value than the rest of the populations. Nei’s genetic diversity (H) was estimated to be 0.1428, on an average, at the population level, ranging from 0.0444 (Population 6) to 0.2384 (Population 2). Shannon information index (I) was 0.2120, on an average, at the population level, varying from 0.0648 (Population 6) to 0.3517 (Population 2). All the populations showed significant differences where the highest level of variability was recorded to Population 2 and the lowest level to Population 6. Total genetic diversity (H T) was a moderate of 0.2651 (Table 1).
Genetic differentiation
The Bayesian STRUCTURE analyses showed seven populations of M. tirunelveliense in two genetic pools, with migrants and admixed individuals. Genetic diversity within populations (H S) was a low of 0.1428. Genetic identities between populations varied from 0.7409 to 0.8976. Proportion of total diversity among populations (G ST) was higher being 0.4613. Gene flow among populations (Nm) was 0.5839 individuals generation−1.
Genetic relationships
Mantel test showed no significant correlation among genetic differentiation and geographical distance (r = 0.489, P = 0.007) for which separating distances were a meager of about 6.5 km only (Table 2; Fig. 2). The analysis of molecular variance (AMOVA) showed P < 0.01 significance (Table 3).

Geographical distance (km) and genetic distance (D) depicting affinity among seven populations in M. tirunelveliense
Multidimensional scaling (MDS) analysis clearly separated seven populations into two clusters comprising Populations 1, 4, 3 and 2 under cluster-I within 0.148 to 0.263 in dimension 1 and −0.019 to −0.093 in dimension 2, and Populations 5, 6 and 7 under cluster-II within 0.063 to 0.160 in dimension 1 and 0.025 to 127 in dimension 2. Thus, the use of a geographic pictorial representation of data using MDS also has clarified the genetic distance among population. The PCoA provided additional supportive evidence for the highly structured nature of the populations (Fig. 3). The percentage of variance accumulated on the two axes was 43.68 % (F1 = 30.12 %, F2 = 13.56 %).

Principal coordinate analysis (PCoA) among seven populations of Monoon tirunelveliense
Sequences of chloroplast markers
The phylogram constructed using MrBayes for the sequences of rbcL and matK gene revealed a close affinity among the Indian species (Fig. 4).

Combined Bayesian analyses of rbcL and matK gene sequences of 23 Monoon species and an out group of Cinnamomum camphora (50 % majority-rule consensus and 0.47 posterior probabilities)
Discussion
Genetic structure and differentiation
Low levels of genetic diversity within populations and relatively high levels of genetic differentiation among populations in M. tirunelveliense could be due to inbreeding, genetic drift and genetic isolation of the populations as postulated by Hogbin and Peakall (1999). Ellstrand and Elam (1993) stated that genetic structure and its differentiation increase due to the influence of genetic drift in small and isolated populations. The greater role played by genetic drift shows an absence of correlation between the (small and isolated) populations and geographic distances. This is a revelation in Monoon tirunelveliense. Further, the condition has been aggravated by the loss of genetic variability over the period due to reduced seedling establishment (Fischer and Matthies 1998). Hamrick et al. (1992) reported more variation in woody species than other life forms such as endemics have 25 % of the value for the widespread species and 39 and 29 % of the values for the narrowly and regionally distributed species, respectively. Further, they had a mean within population diversity (H S) such as 49 % of the widespread species and 56 % of the value for regional species. Among endemic species, species with gravity, gravity-attached and wind-dispersed seeds have less genetic diversity. The research findings in M. tirunelveliense more or less corroborate Hamrick et al. (1992) findings but some of the interesting deviations such as less total genetic diversity (H T) −0.2651 (reported value of 0.283), mean within population diversity (H S) −0.1428 (reported value of 0.253), more genetic diversity within populations −0.489 (reported value of 0.45) when correlated with geographic range (r), and higher total diversity among populations (G ST) −0.4613 (the reported values of 0.076 for gymnosperms and 0.084 for angiosperms) are recorded. The findings in M. tirunelveliense (Table 3) agree with the hypothesis of Hamrick et al. (1992) and Austerlitz et al. (2000) that woody species mostly tend to have high levels of variation within natural populations and less structure between populations. The Bayesian structure analyses showed seven populations of M. tirunelveliense in two genetic pools, with migrants and admixed individuals. Populations 6 and 7 share the same genetic pool as they are very closely located (<2 km) geographically on the river side. The remaining 5 populations are geographically close to each other (<6.5 km), wherein Populations 2 and 3 are so close (Table 2; Fig. 2). Hamrick and Godt (1996) reported that selfing with gravity-attached seed dispersal ones have an average of 0.533 (G ST) but in M. tirunelveliense it is less with 0.4613. More or less similar value of 0.340 was reported by Ratnayake et al. (2006) in Polyalthia korinti. This observation supports the hypothesis and concludes that species of Monoon and Polyalthia are animal-pollinated and have mixed breeding systems. The reason could be due to the restricted occurrence of the populations within a particular geographic locality. More so, the recorded value falls in between narrow (0.512) and widespread (0.446) selfing species in respect of breeding system and geographic range. Genetic variation is low in restrictedly distributed geographical species and widespread congeners have more genetic variation (Gitzendanner and Soltis 2000). In Monoon tirunelveliense, gene flow (Nm) of 0.5839 individuals generation−1 could be due to its steno endemic distribution within the northern part of the KMTR. Earlier findings by Schemske et al. (1994) and Alvarez-Buylla et al. (1996) state that tropical endemic trees are vulnerable to forest fragmentation because of their low densities, complex demographic dynamics, high genetic differentiation (q ≥ 1), and self-incompatibility systems. Also, tropical forest fragmentation is likely to decrease gene flow, increase inbreeding and therefore produce a high differentiation among remnant populations (Alvarez-Buylla et al. 1996). The findings in M. tirunelveliense also agree with these findings. Further, low levels of polymorphism in M. tirunelveliense may be attributed largely to inbreeding nature within populations. M. tirunelveliense is a protogyny and insect-pollinated species, thereby it promotes xenogamous pollination. However, insect-dispersed pollens in close proximities and gravity-disseminated seeds in the natural habitats never facilitate to extend natural populations to longer distances. Ashton (1969) investigated the claim that frequent occurrence of inbreeding which is caused by the low population density in tropical rain forests has accelerated species diversification. In Monoon tirunelveliense, low level of genetic variation could also be due to a low genetic drift amongst the seven populations distributed in between 932 and 1503 m MSL. Because of the impact of rains and floods, the geographical distances among populations vary from a minimum of 1 km to a maximum of 6.5 km (Table 2; Fig. 2).
Doyle and Le Thomas (1996, 1997a, b) have utilized morphological characters of flowers (van Heusden 1992) and pollen, fruits and seeds (van Setten and Koek-Noorman 1992) of Annonaceae for Cladistic analyses. Doyle et al. (2000) provided a comparison of morphological and molecular data. Couvreur et al. (2011) analyzed concatenating data of 93 of the 112 genera of Annonaceae using seven plastid markers and reported four main groups. Mols et al. (2004a, b) combined matK, rbcl and trnl-F sequence data of Polyalthia species and showed five distinct clades after analyses by maximum parsimony (MP), maximum likelihood (ML) and Bayesian method. Similar analyses by Saunders et al. (2011) recognized 123 Annonaceae species under four clades and restored the Madagascan endemic genus Fenerivia. Xue et al. (2011) analyzed 123 Annonaceae species, reported four clades and reestablished the genus Marsypopetalum with five species. Chaowasku et al. (2012) characterized and segregated Hubera, an allied to Miliusa, from Polyalthia on the basis of molecular phylogenetics using seven plastid markers (rbcL exon, trnL intron, trnL-F spacer, matK exon, ndhF exon, psbA-trnH spacer, ycf1 exon), pollen morphology (light microscopy, scanning electron microscopy and transmission electron microscopy) and macromorphology (base of leaf, its venation and domatia, position of inflorescence, pollen infratectum, number of ovules per ovary, seed raphe and endosperm ruminations). Xue et al. (2012) reported 126 Annonaceae species under three clades wherein clade two species are characterized by eucamptodromous leaf venation. In the present study, rbcL and matK gene sequences of 22 Monoon species were collected from Genbank, combined with that of the sequences of M. tirunelveliense and analyzed for phylogenetic relationship (Table 4; Fig. 4). The findings strongly agree with that of Xue et al. (2012), place M. tirunelveliense in the clade II and reveal more affinity to Indian and Sri Lankan species on one hand and Malaysian species of Monoon cupulare and another Philippines species of Monoon oblongifolium on other hand. Further, the genetic diversity of M. tirunelveliense was compared with two species of Annonaceae and four other threatened species belonging to different families (Table 5).
Potentials of conservation
The primary objective in nature conservation is to preserve evolutionary potential of the species by maintaining as much genetic diversity as possible. Low intra-population genetic variation with relatively high spatial differentiation is a biological and evolutionary characteristic of Monoon species (Ratnayake et al. 2006). Hamrick and Godt (1996) suggested that five strategically placed population should be sufficient enough to maintain 99 % of the total genetic diversity. But, Deshpande et al. (2001), who recorded an average genetic diversity among populations in Symplocos laurina as 53.7 %, recommended conservation of more populations to maintain genetic diversity within a species. As M. tirunelveliense is represented only by seven extant populations in the northern part of the KMTR, it is imperative to conserve all for preserving existing variations of the species. Being a montane species, it is deterrent to any change in the existing environmental conditions. Matured fruits are highly vulnerable to termites and microbial decay. Therefore, seeds should be freed from debris or collected on time and sown in debris-free soil. This timely intervention by humans is of paramount importance. For propagation, seeds of genetic diversity-rich Populations 2 and 3 are recommended. These conservation measures will certainly increase both the individuals and populations of the species and facilitate to extend its occurrence and range of distribution.
References
Alvarez-Buylla ER, Garcia-Barrios R, Lara-Moreno C, Martinez-Ramos M (1996) Demographic and genetic models in conservation biology: applications and perspectives for Tropical Rain Forest tree species. Annu Rev Ecol Evol Syst 27:387–421
Ashton PS (1969) Speciation among tropical forest trees: some deductions in the light of recent evidence. Biol J Linn Soc 1:155–196
Austerlitz F, Mariette S, Machon N, Gouyon P-H, Godelle B (2000) Effects of colonization processes on genetic diversity: differences between annual plants and tree species. Genetics 154:1309–1321
Cao PJ, Yao QF, Ding BY, Zeng HY, Zhong YX, Fu CX, Jin XF (2006) Genetic diversity of Sinojackia dolichocarpa (Styracaceae), a species endangered and endemic to China, detected by inter-simple sequence repeat (ISSR). Biochem Sys Ecol 34:231–239
Chaowasku T, Johnson DM, Van der Ham RWJM, Chatrou LW (2012) Characterization of Hubera (Annonaceae), a new genus segregated from Polyalthia and allied to Miliusa. Phytotaxa 69:33–56
Couvreur TLP, Pirie MD, Chatrou LW, Saunders RMK, Su YCF, Richardson JE, Erkens RHJ (2011) Early evolutionary history of the flowering plant family Annonaceae: steady diversification and boreotropical geodispersal. J Biogeogr 38:664–680
Culley TM, Wolfe AD (2001) Population genetic structure of the cleistogamous plant species Viola pubescens Aiton (Violaceae), as indicated by allozyme and ISSR molecular markers. Heredity 86:545–556
Deshpande AU, Apte GS, Bahulikar RA, Lagu MD, Kulkarni BG, Suresh HS, Singh NP, Rao MKV, Gupta VS, Pant A, Ranjekar PK (2001) Genetic diversity across natural populations of three montane plant species from the Western Ghats, India revealed by inter simple sequence repeats. Mol Ecol 10:2397–2408
Doyle J (1991) DNA protocols for plants–CTAB total DNA isolation. In: Hewitt GM, Johnston A (eds) Molecular techniques in taxonomy. Springer-Verlag, Berlin, pp 283–293
Doyle JA, Le Thomas A (1996) Phylogenetic analysis and character evolution in Annonaceae. Bull Mus Natl Hist Nat, B, Adansonia 3–4:279–334
Doyle JA, Le Thomas A (1997a) Significance of palynology for phylogeny of Annonaceae: experiments with removal of pollen characters. Plant Syst Evol 206:133–159
Doyle JA, Le Thomas A (1997b) Phylogeny and geographic history of Annonaceae. Geogr Phys Quatern 51:252–361
Doyle JA, Bygrave P, Le Thomas A (2000) Implications of molecular data for pollen evolution in Annonaceae. In: Harley MM, Morton CM, Blackmore S (eds) Pollen and spores: morphology and biology. Royal Botanical Gardens, Kew, pp 259–284
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242
Esselman EJ, Jianqiang L, Crawford DJ, Windus JL, Wolfe AD (1999) Clonal diversity in the rare Calamagrostis porteri ssp. insperata (Poaceae): comparative results for allozymes and random amplified polymorphic DNA (RAPD) and inter simple sequence repeat (ISSR) markers. Mol Ecol 8:443–451
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fang DQ, Roose ML (1997) Identification of closely related citrus cultivars with inter-simple sequence repeat markers. Theor Appl Genet 95:408–417
Fischer M, Matthies D (1998) Effects of population size on performance in the rare plant Gentianella germanica. J Ecol 86:195–204
Gitzendanner MA, Soltis PS (2000) Patterns of genetic variation in rare and widespread plant congeners. Am J Bot 87:783–792
González-Astorga J, Castillo-Campos G (2004) Genetic variability of the narrow endemic tree Antirhea aromatica Castillo-Campos and Lorence (Rubiaceae, Guettardeae) in a Tropical Forest of Mexico. Ann Bot 93:521–528
Hamrick JL, Godt MJW (1996) Conservation genetics of endemic plant species. In: Avise JC, Hamrick JL (eds) Conservation genetics: case histories from nature. Chapman and Hall, New York, pp 281–304
Hamrick JL, Godt MJW, Sherman-Broyles SL (1992) Factors influencing levels of genetic diversity in woody plant species. New Forest 6:95–124
Hasebe M, Omori T, Nakazawa M, Sano T, Kato M, Iwatsuki K (1994) rbcL gene sequences provide evidence for the evolutionary lineages of leptosporangiate ferns. Proc Natl Acad Sci USA 91:5730–5734
Hogbin PM, Peakall R (1999) Evaluation of the conservation of genetic research to the management of endangered plant Zieria prostrata. Conserv Biol 13:514–522
Hollingsworth P, Forrest L, Spouge J et al (2009) Polyalthia oblongifolia voucher WP2A0446: ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit (rbcL) gene, partial cds; chloroplast. GenBank:GQ248677.1 & maturase K (matK) gene, partial cds; chloroplast. GenBank:GQ248183.1
Lewontin RC (1972) The apportionment of human diversity. Evolution Biol 6:381–398
Manos PS, Steel KP (1997) Phylogenetic analysis of “higher” Hamamelididae based on plastid sequence data. Am J Bot 84:1407–1419
Miller MP (1997) Tools for Population Genetic Analyses (TFPGA), ver. 1.3. A Windows program for the analysis of allozyme and molecular population genetic data. Computer software distributed by author. http://www.marksgeneticsoftware.net/_vti _bin/shtml.exe/tfpga.html
Mols JB, Co DLV, Gravendeel B, Chatrou LW, Pirie MD, van der Ham RWJM, van Marle EJ, Keßler PJA (2004a) Morphological character evolution in the miliusoid clade (Annonaceae). In: Mols JB (ed) From Miliusa to Miliuseae to Miliusoid: identifying clades in Asian Annonaceae, Ph.D. Thesis, Leiden University, Leiden, pp 37–75
Mols JB, Gravendeel B, Chatrou LW, Pirie MD, Bygrave PC, Chase MW, Keßler PJA (2004b) Identifying clades in Asian Annonaceae: monophyletic genera in the polyphyletic Miliuseae. Am J Bot 91:590–600
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Nei M (1977) F-Statistics and analysis of gene diversity in subdivided populations. Ann Hum Genet 41:225–233
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28:2537–2539
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qiu YX, Hong DY, Fu CX, Cameron KM (2004) Genetic variation in the endangered and endemic species Changium smyrnioides (Apiaceae). Biochem Sys Ecol 32:583–596
Qiu YX, Li JH, Liu HL, Chen YY, Fu CX (2006) Population structure and genetic diversity of Dysosma versipellis (Berberidaceae), a rare endemic from China. Biochem Sys Ecol 34:745–752
Ratnayake RMCS, Su YCF, Gunatilleke IAUN, Wijesundara DSA, Saunders RMK (2006) Reproductive biology of two sympatric species of Polyalthia (Annonaceae) in Sri Lanka. II. Breeding systems and population genetic structure. Int J Plant Sci 167:495–502
Ronquist F, Teslenko M, Van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, John P, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Saunders RMK, Yuonne CFS, Xue B (2011) Phylogenetic affinities of Polyalthia species (Annonaceae) with columellar-sulcate pollen: enlarging the Madagascan endemic genus Fenerivia. Taxon 60:1407–1416
Schemske DW, Husband BC, Ruckelshaus MH, Goodwillie C, Parker IM, Bishop JG (1994) Evaluation approaches to the conservation of rare and endangered plants. Ecology 75:584–606
Su YC, Smith GJ, Saunders RM (2008) Phylogeny of the basal angiosperm genus Pseuduvaria (Annonaceae) inferred from five chloroplast DNA regions, with interpretation of morphological character evolution. Mol Phylogenet Evol 48:188–206
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Phylogenet Evol 28:2731–2739
van Heusden ECH (1992) Flowers of Annonaceae: Morphology, Classification, and Evolution. Blumea, Suppl 7. Leiden
van Setten AK, Koek-Noorman J (1992) Fruits and seeds of Annonaceae. Morphology and its significance for classification and identification. Bibliotheca Botanica, Heft 142. E. Schweizerbart’sche Verlagsbuchhandlung, Stuttgart
Viswanathan MB, Manikandan U (2001) Polyalthia tirunelveliensis (Annonaceae), a new species from Peninsular India. Kew Bull 56:217–221
Viswanathan MB, Rajasekar C, Sathish Kumar P, Rajesh R (2014) Monoon tirunelveliense voucher BDUT 1402: ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit (rbcL) gene, partial cds; chloroplast. GenBank:KF887447.1 & maturase K (matK) gene, partial cds; chloroplast. GenBank:KF887448.1. http://www.ncbi.nlm.nih.gov/nuccore/KF887448.1
Wang J (2004) Estimating pairwise relatedness from dominant genetic markers. Mol Ecol 13:3169–3178
Wolfe AD, Xiang QY, Kephart SR (1998) Assessing hybridization in natural populations of Penstemon (Scrophulariaceae) using hypervariable inter simple sequence repeat (ISSR) bands. Mol Ecol 7:1107–1125
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Xiao LQ, Ge XJ, Gong X, Hao G, Zheng SX (2004) ISSR Variation in the endemic and endangered plant Cycas guizhouensis (Cycadaceae). Ann Bot 94:133–138
XLSTAT (2013). Addinsoft Inc., Paris, France. http://www.xlstat.com
Xue B, Yvonne CFU, Mols JB, Keßler PJA, Saunders RMK (2011) Further fragmentation of the polypheletic genus Polyalthia (Annonaceae): molecular phylogenetic support for a broader delimitation of Marsypopetalum. Syst Biodivers 9:17–26
Xue B, Su YCF, Thomas DC, Saunders RMK (2012) Pruning the polyphyletic genus Polyalthia (Annonaceae) and resurrecting the genus Monoon. Taxon 61:1021–1039
Yeh FC, Yang RC, Boyle T (1999) POPGENE ver. 1.3.1. Microsoft Windows® - based freeware for population genetic analysis. Available: (www.ualberta.ca/~fyeh/) University of Alberta and the Centre for International Forestry Research, Edmonton
Author contribution statement
M.B. Viswanathan Principal Investigator of the Project entitled, “Studies on Population Ecology and Micropropagation Techniques of Selected Endemic and Threatened Plants of Peninsular India” sponsored by Department of Biotechnology, Government of India, New Delhi, for fund assistance (BT/PR-9368/BCE/08/563/2007 dt.25.06.2008). Major Contributor of the paper. C. Rajasekar: Senior Research Fellow of the Project. Next to first author, he contributed by field work and provided basic data. P. Sathish Kumar: Technical Assistant of the Project. He assisted the Senior Research Fellow in the field work and looked after the financial aspects of the Project.
Acknowledgments
The authors are grateful to the Department of Biotechnology, Government of India, New Delhi, for fund assistance (BT/PR-9368/BCE/08/563/2007 dt.25.06.2008), Dr. K.S. Charak, Adviser and Dr. Onkar N. Tiwari, Scientist ‘C’ for their administrative help, the Chief Wildlife Warden, Principal Chief Conservator of Forests, Chennai, and Chief Conservator of Forests and Field Director, Project Tiger, Tirunelveli, for permission to field work.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Carlson.
Electronic supplementary material
Below is the link to the electronic supplementary material.
468_2014_1122_MOESM1_ESM.tif
UPGMA dendrogram of the seven population of M. tirunelveliense constructed by ISSR and compared to Bayesian admixture proportion (K=7) (TIFF 144 kb)
Rights and permissions
About this article

Cite this article
Viswanathan, M.B., Rajasekar, C. & Sathish Kumar, P. Genetic diversity of steno endemic and critically endangered Monoon tirunelveliense (Annonaceae) from India as revealed by ISSRs. Trees 29, 437–447 (2015). https://doi.org/10.1007/s00468-014-1122-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00468-014-1122-y