
Abstract
Background
Rituximab is a chimeric anti-CD20 monoclonal antibody that induces sustained remission in children with steroid-dependent nephrotic syndrome. However, there is no consensus on the optimal regimen and monitoring of rituximab. In other autoimmune diseases, anti-rituximab antibodies (ARA) have been reported in 10–40% of patients, but their clinical relevance remains unclear. In nephrotic syndrome, data are scarce.
Methods
We report a single-center retrospective study with immuno- and pharmacological monitoring of rituximab treatment in children with frequent relapsing (FR) or steroid-dependent nephrotic syndrome (SDNS). We analyzed the monthly monitoring of 24 children, receiving a dose of rituximab (375 mg/m2) between December 2017 and April 2018 at the Pediatric Nephrology Department of Robert-Debré hospital, Paris.
Results
ARA were detected in 7/24 patients (29%), sometimes after the first infusion of rituximab. ARA were present at baseline in two patients previously treated with rituximab. Both displayed no B-cell depletion. ARA were also reported in 5/22 patients during follow-up, with antibodies always detected in the first month following B-cell recovery. An incomplete CD19+CD20− B-cell depletion at M1 (5–25/mm3) and low serum rituximab levels was predictive of developing ARA. The development of de novo ARA during follow-up was not associated with shorter B-cell depletion.
Conclusions
This study shows that ARA are frequent in children with FR/SDNS and that close immuno- and pharmacological monitoring may help personalizing rituximab treatment in patients needing repeated injections.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Idiopathic nephrotic syndrome (INS) is the most frequent acquired glomerular disease in children. Although most patients are steroid sensitive, 60% relapse frequently and/or become steroid dependent. These patients require additional immunosuppressive treatments to maintain remission, and experience a long lasting disease with a heavy burden of steroids and steroid-sparing treatments [1].
Rituximab is a chimeric anti-CD20 monoclonal antibody that induces peripheral B-cell depletion [2]. It was first developed in the treatment of B-cell malignancies and was further successfully used in the treatment of auto-immune diseases. Since the first report of the use of rituximab in a patient with both INS and idiopathic thrombocytopenic purpura [3], it is now well established that rituximab is able to induce long-lasting remission in patients with frequent relapsing (FR) or steroid-dependent (SD) nephrotic syndrome, even after B-cell recovery [4,5,6]. However, the optimal regimen and monitoring of rituximab remain to be determined. Data on rituximab pharmacokinetics in childhood INS are scarce and its monitoring most often relies on peripheral CD 19+ B-cell count. Indeed, when a strategy of long-term B-cell depletion is chosen, rituximab infusions are repeated when peripheral B-cells reconstitute. However, no specific biological marker of disease activity itself, besides recurrence of proteinuria, is available, preventing tailoring rituximab treatment to each patient.
In addition, as a chimeric monoclonal antibody, rituximab carries the theoretical risk of inducing the development of anti-drug antibodies, including Human Anti-Chimeric Antibodies (HACA). The development of anti-rituximab antibodies (ARA) in patients receiving repeated rituximab infusions has been described in other autoimmune conditions and may reduce circulating levels of rituximab and lead to shorter or absence of B-cell depletion [7,8,9,10]. Such anti-drug antibodies may promote resistance to rituximab and/or be involved in the development of severe adverse reactions [9,10,11,12].
The aim of this study was to assess the frequency of ARA and the pharmacokinetics of rituximab in children with FR/SDNS and to evaluate their impact on B-cell depletion and relapse-free survival.
Methods
Population
We report a single-center retrospective study of children with FR or SDNS, who received a rituximab infusion between December 2017 and April 2018 at the Pediatric Nephrology Department of Robert-Debré hospital (Paris, France) and their biological monthly monitoring. Patients could have received either a unique rituximab infusion or a reinfusion at the time of B-cell recovery. Some patients received five additional monthly infusions of intravenous immunoglobulins (IVIg) of 2 g/kg.
Patients received an infusion of rituximab (Rixathon®) at the dose of 375 mg/m2, after premedication with methylprednisolone 1 mg/kg and intravenous dexchlorpheniramine. All patients had negative proteinuria at rituximab infusion. When present at baseline, oral treatments (steroids and/or immunosuppressive drugs) were discontinued within 2 months.
Methods
According to our local practice of B-cell depletion monitoring, urine protein/creatinine ratio, serum albumin, plasma levels of immunoglobulins, white blood cells, and platelets and CD19/CD20 count were scheduled at baseline and monthly until B-cell recovery. In patients with a previous history of short B-cell depletion, biological monitoring could also be performed at day 7 to confirm complete B-cell depletion, defined by a CD19+ B-cell count < 5/mm3. Concentrations of rituximab and ARA were assessed with a commercial ELISA kit (Lisa Tracker Duo Rituximab, Theradiag). The cut-off values for positive results were 2 μg/ml and 5 ng/ml, respectively.
Data were collected from routine clinical and biological charts and an informed consent of care was collected for all patients.
Outcomes
Outcomes were detection of ARA, rituximab blood concentrations, duration of B-cell depletion, and time to relapse. Safety endpoints were frequency and severity of adverse events and abnormal values in biochemical and hematology assessments.
Statistical analysis
Results are presented as median and IQR for continuous variables and number and percentage for dichotomous variables. Comparison of patients’ characteristics was performed using the Mann–Whitney test and Fischer exact test for continuous and categorical variables, respectively. Kaplan–Meier method was used to study B-cell depletion duration and relapse-free survival, and log-rank tests were used to compare these between patients with or without ARA. The correlation between serum rituximab levels at M2 and B-cell depletion duration was assessed by estimating the Spearman correlation coefficient. The statistical significance was established at p < 0.05.
Results
Population at baseline
Twenty-eight patients received an infusion of rituximab during the study period; four were excluded because of incomplete monitoring. Characteristics at baseline of the remaining 24 patients are reported in Table 1. The median age at disease onset was 5 years (IQ 3–6) and male-to-female ratio was 2/1. The median age at baseline rituximab was 8.8 years (IQ 7.2–13.3). Thirteen patients (54%) had previously received one or more rituximab infusions. The median duration of B-cell depletion after the prior rituximab infusion was 3 months. Sixteen patients, receiving rituximab after a recent relapse, had oral steroids and/or immunosuppressive drugs at baseline, and eight patients receiving a reinjection after B-cell recovery had no oral treatment at baseline. Oral treatments were discontinued within 2 months in 14 patients and delayed at month 3 in two patients because of early relapse. Nine patients received additional intravenous immunoglobulins during follow-up.
Anti-rituximab antibodies
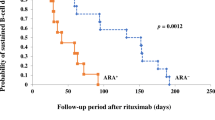
Anti-rituximab antibodies (ARA) were detected in a total of 7/24 patients (29%). The median age of patients with ARA was 12.7 (IQ 9–15.4) years at baseline, and 11.3 (IQ 8.2–14) years at first rituximab. ARA were detected at baseline in two patients with titers > 100 ng/ml. These two patients had previously received rituximab, respectively, 15 and 17 months before. De novo ARA were additionally detected during follow-up in 5/22 patients (23%) with negative baseline screening. Of note, three of them were receiving their first rituximab infusion (Fig. 1). There was no association of ARA development with age, baseline B-cell count, or Ig G level, nor with oral immunosuppressors or intravenous immunoglobulins during B-cell depletion (Table 1). ARA were always detected in the first month following B-cell recovery and remained detectable at 12 months in all patients but one (Fig. 2).

Patient flowchart. Anti-rituximab antibodies in patients with first or prior rituximab and impact of intravenous immunoglobulins on their development (intravenous immunoglobulin given monthly from month 1 to 5 at the dose of 2 g/kg). ARA: anti-rituximab antibodies, RTX: rituximab, IVIg: intravenous immunoglobulins at the dose of 2 g/kg received monthly from month 1 to month 5

Clinical and biological course of the 7 patients with anti-rituximab antibodies. For patients 1 to 5 who developed ARA during follow up, ARA were always detected at the time of B-cell recovery or within the following month. For patients 6 and 7, ARA were detected at baseline before the rituximab infusion. ARA were still detected at month 12 in all patients with available data except for patient 2 who received a dose of obinutuzumab at month 4. Data at month 12 is unfortunately not available for patients 4 and 6. *CD19+ CD20− B-cells, with CD19 B-cell ranging from 5 to 25/mm3. **CD19+ CD20+ B-cells, with CD19 B-cell > 25/mm3
Pharmacokinetics of rituximab
Serum levels of rituximab are presented in Fig. 3. Rituximabemia was above the 2 μg/ml threshold in 82%, 61%, and 20% of patients at M1, M2, and M3, respectively. Rituximab was undetectable at M1 in three patients, two of whom had ARA at baseline. For the others, low concentrations of rituximab at M2 were associated with shorter B-cell depletion and more frequent development of ARA (Fig. 4a and b). In addition, rituximab was undetectable at M2 in 3/5 patients who developed de novo ARA compared to only 2/11 without ARA (p = 0.24). A trend toward lower rituximab concentration at M2 was also noticed in patients receiving intravenous immunoglobulins (Fig. 4c) (p = 0.19).

Serum rituximab levels after baseline rituximab infusion. Rituximab was detectable at M1 in all patients except three, of whom two had baseline anti-rituximab antibodies. Rituximabemia was above the 2 μg/ml threshold in 82%, 61%, and 20% of patients at M1, M2, and M3, respectively, with mean serum rituximab levels of 22, 5, and 1 μg/ml. At M4, rituximab was undetectable in all patients

Serum rituximab levels at Month 2. a Correlation with the duration of B-cell depletion, b serum rituximab levels in patients with anti-rituximab antibodies (ARA +) and without (ARA−), c serum rituximab levels in patients receiving rituximab alone (IVIg−) or rituximab in association to intravenous immunoglobulins (IVIg +). It should be noted that rituximab levels at M2 were missing in 6 out of 24 patients, of whom 4 out of the 9 patients were treated with IVIG
B-cell depletion
Rituximab was successful in inducing peripheral B-cell depletion in 22/24 patients (92%). The two “rituximab-resistant” patients had ARA at baseline and had previously received 1 and 2 rituximab infusions with a duration of B-cell depletion of 3 and 5 months after prior rituximab, respectively, (Fig. 2, patients 6 and 7). By contrast, B-cell depletion was achieved in all patients without ARA at baseline, with a median duration of 4 months (IQ 3.0–4.8). In addition, time to B-cell recovery was not significantly different whether ARA were detected or not during follow-up. Interestingly, early monitoring at day 7 or M1 showed detectable CD19+ but CD20− B-cells (ranging from 5 to 25/mm3) in 3 patients who developed ARA secondarily, whereas complete CD19-/ CD20− B-cell depletion (0/mm3) was recorded early (D7-M1) in all patients without ARA (Table 1 and Fig. 2) (p = 0.0002). These three patients further had a complete peripheral B-cell depletion with CD19+ B-cells at 0/mm3 at M2.
Time to first relapse after rituximab
After 12 months follow-up, 17/24 patients (71%) remained free of relapse and 7/24 patients relapsed, including 3 early relapses. One patient with baseline ARA and incomplete B-cell depletion relapsed at M1. Three others relapsed during the first month despite complete B-cell depletion (13% of the entire cohort) and had further negative ARA screening. Of note, one of them relapsed again one month after B-cell recovery. Two other patients relapsed after B-cell recovery, respectively, 2 and 4 months later. Concerning patients with ARA development, only 1/5 relapsed during follow-up, at 8 months after B-cell recovery and ARA positivation (Fig. 2). After excluding the two patients with ARA at baseline, no relationship was found between the time to relapse and the development of de novo ARA (Table 2), even after separating patients having received or not a second dose of anti-CD20 during the study. Of the 10 patients having received a second dose, three had relapsed before but no one relapsed after (Table 2).
Reinfusion of a second dose anti-CD20 and resistance to anti-CD20
Ten patients with no baseline ARA received a second dose of anti-CD20 (45%) during follow-up; three because of relapse after B-cell recovery and seven at the time of B-cell recovery in order to prolong B-cell depletion. Of note, 3 patients with positive ARA received a second line anti-CD20 monoclonal antibody: patient no. 7 had a successful B-cell depletion after a single injection of 750 mg/1.73 m2 of atumumab, while patient no. 6 had no B-cell depletion with both rituximab and ofatumumab, and successful B-cell depletion with a single injection of 300 mg/1.73 m2 of obinutuzumab. Patient no. 2, who developed ARA after 3 months, received a single injection of 300 mg/1.73 m2 of obinutuzumab resulting in a B-cell depletion of 5 months (Fig. 2).
Adverse effects
Adverse effects were reported in seven patients (29%). Two patients (8%) presented mild infusion-related reactions consisting of nausea and vomiting. Of note, neither of the two patients with positive ARA at baseline experienced any infusion reaction. One patient presented hypogammaglobulinemia, with IgG level at 3.5 g/L 3 months after a second dose of rituximab, and two patients developed asymptomatic neutropenia (1 moderate and 1 severe) during B-cell depletion. Two patients required hospitalization for viral infections, including one patient who presented zona recurrence, during and after B-cell recovery.
Discussion
This study assessed the prevalence of ARA on the baseline and monthly biological monitoring of 24 children with FR/SDNS after one 375 mg/m2 rituximab infusion. ARA were positively detected in more than one-fourth of patients and preformed ARA were associated with resistance to peripheral B-cell depletion, while the development of de novo ARA during follow-up was not associated with shorter B-cell depletion. Moreover, rituximabemia monitoring suggested that low concentrations at month 2 were associated with shorter B-cell depletion and the development of de novo ARA.
The development of anti-rituximab antibodies (ARA) has been assessed in patients treated with rituximab for various underlying diseases. Consistently, ARA are rare in patients treated for B-cell malignancies (1%) [13], while they have been detected in up to 30–40% of patients with SLE [7, 14], or neuromyelitis optics spectrum disorders [8], 25% of patients with ANCA-vasculitis and Sjogren’s syndrome [15, 16], and 9–11% of patients with rheumatoid arthritis [9, 17]. In nephrotic syndrome, data are scarce, since only three pediatric studies have reported a screening for ARA. Iijima et al. reported a prevalence of 14% of ARA at 12 months in the rituximab group (n = 24) of their placebo controlled-randomized trial (RCT) [5], while Anh et al. detected ARA in 10% (2/19) of patients in a brief report [11]. The incidence in this second study has to be interpreted with caution because the reason for testing ARA was not given. In the present study, with a systematic monthly monitoring, we found a higher prevalence of 29%. The first hypothesis is that ARA could have been underestimated in previous studies based on a single screening at 12 months compared to systematic monthly screening. However, nearly all patients with early ARA detection in our cohort had persistent detectable ARA at M12 (Fig. 2). A second hypothesis for this higher prevalence might be the low dosage of rituximab, with a single 375 mg/m2 rituximab dose in this study, compared to 4 weekly infusions in the RCT of Iijima et al., while Anh et al. also report patients treated with a single infusion. Indeed, a lower dose of rituximab may favor incomplete or delayed depletion, and subsequently ARA development [7]. Interestingly, early B-cell monitoring showed that only patients developing ARA during follow–up had incomplete peripheral CD19+ CD20− B-cell depletion at first monitoring (Table 1). Additionally, incomplete peripheral CD19+ B-cell depletion highlights that a subset of remnant circulating or tissular B-cells might differentiate into anti-rituximab IgG-producing cells and consequently neutralize the circulating drug, lowering rituximabemia and its half-life. A third study retrospectively reported a high prevalence of ARA up to 38%, but a major bias was that screening was performed only in patients who had displayed an infusion-related reaction during a second or subsequent rituximab dose [18], possibly overestimating ARA incidence.
Although one may suppose that ARA are more likely to be detected after repeated exposure to rituximab, ARA were detected similarily in 3/11 (27%) and 4/13 (30%) of patients after a first RTX or a reinfusion (Fig. 1). This is consistent with data from studies in other autoimmune diseases showing that ARA mostly appear prior to the second infusion of rituximab [8, 17]. Unfortunately, we do not have the late monitoring for the nine patients who received a second dose of rituximab and were still under B-cell depletion at M12.
Furthermore, in our series, the monthly monitoring of B-cell count and ARA showed that ARA were first detected either at the time of B-cell recovery or within the following month (Fig. 2). Considering these results, one may propose a systematic screening for ARA one month after B-cell recovery, specifically in patients who display a delayed complete B-cell depletion.
However, the pathogenic effects of such anti-drug antibodies are unclear and the clinical relevance of ARA is still under debate. Data suggest that ARA could be associated with a shorter B-cell depletion duration and an increased risk of side effects such as infusion reactions or serum sickness disease [7,8,9, 15]. In childhood nephrotic syndrome, only case reports have linked ARA detection to rituximab intolerance [11, 19]. In this study, circulating ARA at baseline were associated in both patients with rituximab resistance, but not with infusion-related reactions (IRR), as in another prospective study in patients with SLE [14]. Limited data suggest that ARA may negatively influence treatment efficacy in lupus patients [7, 14], while no difference on clinical improvement is reported in patients with neuromyelitis optic spectrum disorder [8] or rheumatoid arthritis [9, 17] besides shorter B-cell depletion and increased frequency of rituximab reinfusions. In this small study, while preformed ARA were associated with an absence of B-cell depletion, the development of de novo ARA during follow-up was not associated with shorter B-cell depletion (Table 1 and 2). We assume that the heterogeneity of rituximab regimens, reinfusions, and total duration of B-cell depletion is a major bias, as rituximab regimen has an impact on B-cell depletion and relapse-free survival [20]. Moreover, three patients had an early relapse during the first 4 weeks despite complete peripheral CD19 B-cell depletion. Early relapses following rituximab have already been reported [21] and authors suggest the pathophysiology of these relapses may not rely directly on B-cells, but on indirect B-cell interactions or unknown mechanisms. These early relapses might be prevented by delayed discontinuation of oral suppressors, supporting our local practice. Interestingly none of the early relapsers developed further ARA.
One main difficulty in the management of nephrotic syndrome is the lack of clinical score and/or biological biomarkers of disease activity that could help to guide treatment. The current monitoring of rituximab treatment is based on peripheral B-cell count and its efficacy is only evaluated on the occurrence and/or delay of relapses. In our small study, serum levels of rituximab at M2 were correlated with the duration of B-cell depletion and the development of ARA (Fig. 4). Accordingly, in adults with membranous nephropathy and higher dose of rituximab, Boyer-Suavet et al. recently showed that serum rituximab level at month 3 was significantly correlated with a longer B-cell depletion [22]. In addition, our study also shows an association between rituximabemia at M2 and ARA development. This association was also highlighted in adult patients with SLE [14]. One may suppose that low rituximab levels could favor incomplete early CD19+ B-cell depletion and secondarily ARA development, as all three patients with incomplete early CD19+ B-cell depletion had low or undetectable rituximab levels at M2. Of note, the subset of patients receiving concomitant intravenous immunoglobulins displayed lower levels of rituximab at M2 (Fig. 4c). We hypothesize that polyclonal immunoglobulins may reduce rituximab half-life by a competitive effect on rituximab recycling by the FcRn recycling process on endothelial cells [23]. This subgroup also displayed a higher proportion of de novo ARA (3 of 9 vs. 2 of 13), suggesting again that a reduced efficacy of rituximab could favor ARA development. However, such results must be interpreted with caution because of the different treatment regimens and the impossibility of performing multivariate analysis in such small cohorts. By contrast, Einarsson et al. observed no significant difference in the frequency of ARA in patients receiving different doses of rituximab [9]. This could be explained by extremely variable inter-individual levels of rituximabemia in patients receiving the same rituximab dose [14]. In childhood nephrotic syndrome, again, few data on rituximab pharmacokinetics are available. Iijima reported, in patients treated with 4 weekly infusions, mean levels of rituximab of 28 μg/ml at day 85 and 2 μg/l at day 169, with a median duration of B-cell depletion of 148 days (4.8 months) [5]. Unfortunately, no link was specified between levels of rituximab and duration of B-cell depletion. Kamei et al. reported rituximab levels after a single dose of rituximab in 12 children with SDNS, showing a peak at 24 h and mean levels of 27, 18, and 3 μg/ml at M1, M2, and M3, respectively, and undetectable rituximab in all patients at M5 with a median duration of B-cell depletion of 119 days [24]. The mean values were lower in our study (22, 5, and 1 μg/ml at M1, M2, and M3, respectively, and undetectable rituximab in all patients at M4), which might be explained by the concomitant intravenous immunoglobulins in a subset of patients (Fig. 3).
In children with SD/FRNS, because of a high proportion of patients relapsing after B-cell recovery, strategies with rituximab reinfusions have been proposed to prolong B-cell depletion. Two strategies are presently possible: rituximab reinfusion at the time of peripheral B-cell recovery, which requires a monthly monitoring, or systematic reinfusion of rituximab. However, B-cell recovery might not be the most relevant biomarker to monitor rituximab and the optimal approach remains controversial. Our data suggest that monitoring rituximab concentrations at M2 could be helpful to guide rituximab reinfusions, as undetectable or low serum rituximab concentrations at M2 were associated with a shorter B-cell depletion (Fig. 4a). The good sensitivity of this ELISA technique and its reasonable price (around 20 Euros per test) makes its use conceivable in routine practice. However, additional data are needed to determine a threshold and evaluate how rituximabemia at M2 could help monitoring and treatment guidance.
Concerning the two patients with ARA at baseline and “rituximab-resistance,” both received a single injection of 750 mg/1.73 m2 of ofatumumab, a more-humanized anti-CD20 antibody recognizing a different epitope of the CD20 molecule. Indeed, case reports or small series of children resistant to rituximab showed successful B-cell depletion with ofatumumab [25, 26] or obinutuzumab [27], and in vitro studies do not argue for cross-reactions between ARA and new generation anti-CD20 antibodies [28]. However, one patient was also resistant to ofatumumab, and only showed successful peripheral B-cell depletion after obinutuzumab (Fig. 2), suggesting that anti-drug antibodies may cross-react and compromise alternative B-cell depletion attempts. Interestingly, longer follow-up of 2 patients with ARA who further received obinutuzumab showed that ARA were no longer detectable after obinutuzumab, and one patient could again receive rituximab successfully.
The main limitation of our study is the small sample size and the heterogeneity of the population and the rituximab regimens, which makes it difficult to interpret any correlation with de novo ARA and relapse-free survival. In addition, pharmacological monitoring did not enable detailed pharmacokinetics as blood samples were drawn only during routine monthly monitoring, though missing early data. However, several studies show that early measurements of rituximab do not correlate with the clinical outcome in diseases with available activity scores.
Conclusion
This study shows that the development of anti-drug antibodies to rituximab is common in children treated for FR/SDNS and may lead to rituximab resistance. Lower serum rituximab levels could also be associated with worse outcomes. Therefore, close immuno-monitoring of serum rituximab, B-cell count, and ARA development may help personalizing rituximab treatment in patients requiring repeated injections. However, larger studies are needed to confirm specific cut-offs to guide therapy and seek for the optimal rituximab regimen.
References
Dossier C, Delbet JD, Boyer O, Daoud P, Mesples B, Pellegrino B, See H, Benoist G, Chace A, Larakeb A, Hogan J, Deschênes G (2019) Five-year outcome of children with idiopathic nephrotic syndrome: the NEPHROVIR population-based cohort study. Pediatr Nephrol 34:671–678. https://doi.org/10.1007/s00467-018-4149-2
Johnson PW, Glennie MJ (2001) Rituximab: mechanisms and applications. Br J Cancer 85:1619–1623. https://doi.org/10.1054/bjoc.2001.2127
Benz K, Dötsch J, Rascher W, Stachel D (2004) Change of the course of steroid dependent nephrotic syndrome after rituximab therapy. Pediatr Nephrol 19:794–797. https://doi.org/10.1007/s00467-004-1434-z
Ravani P, Rossi R, Bonanni A, Quinn RR, Sica F, Bodria M, Pasini A, Montini G, Edefonti A, Belingheri M, De Giovanni D, Barbano G, Degl'Innocenti L, Scolari F, Murer L, Reiser J, Fornoni A, Ghiggeri GM (2015) Rituximab in children with steroid-dependant nephrotic syndrome: a multicenter, open-label, noninferiority, randomized controlled trial. J Am Soc Nephrol 26:2259–2266. https://doi.org/10.1681/ASN.2014080799
Iijima K, Sako M, Nozu K, Mori R, Tuchida N, Kamei K, Miura K, Aya K, Nakanishi K, Ohtomo Y, Takahashi S, Tanaka R, Kaito HH, Ishikura K, Ito S, Ohashi Y, Rituximab for Childhood-onset Refractory Nephrotic Syndrome (RCRNS) Study Group (2014) Rituximab for childhood-onset, complicated, frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet 384:1273–1281. https://doi.org/10.1016/S0140-6736(14)60541-9
Basu B, Sander A, Roy B, Preussler S, Barua S, Mahapatra TKS, Schaefer F (2018) Efficacy of rituximab vs tacrolimus in pediatric corticosteroid-dependent nephrotic syndrome: a randomized clinical trial. JAMA Pediatr 172:757–764. https://doi.org/10.1001/jamapediatrics.2018.1323
Albert D, Dunham J, Khan S, Stansberry J, Kolasinski S, Tsai D, Pullman-Mooar S, Barnack F, Striebich C, Looney RJ, Prak ET, Kimberly R, Zhang Y, Eisenberg R (2008) Variability in the biological response to anti-CD20 B cell depletion in systemic lupus erythaematosus. Ann Rheum Dis 67:1724–1731. https://doi.org/10.1136/ard.2007.083162
Li T, Zhang LJ, Zhang QX, Yang CS, Zhang C, Li YJ, Shi FD, Yang L (2018) Anti-rituximab antibody in patients with NMOSDs treated with low dose rituximab. J Neuroimmunol 316:107–111. https://doi.org/10.1016/j.jneuroim.2017.12.021
Einarsson JT, Evert M, Geborek P, Saxne T, Lundgren M, Kapetanovic MC (2017) Rituximab in clinical practice: dosage, drug adherence, Ig levels, infections, and drug antibodies. Clin Rheumatol 36:2743–2750. https://doi.org/10.1007/s10067-017-3848-6
Wincup C, Menon M, Smith E, Schwartz A, Isenberg D, Jury EC, Mauri C, ABIRISK Consortium (2019) Presence of anti-rituximab antibodies predicts infusion-related reactions in patients with systemic lupus erythematosus. Ann Rheum Dis 78:1140–1142. https://doi.org/10.1136/annrheumdis-2019-215200
Ahn YH, Kang HG, Lee JM, Choi HJ, Ha IS, Cheong HI (2014) Development of antirituximab antibodies in children with nephrotic syndrome. Pediatr Nephrol 29:1461–1464. https://doi.org/10.1007/s00467-014-2794-7
Vendramin C, Thomas M, Westwood JP, McGuckin S, Scully M (2019) Rituximab-induced acute and delayed serum sickness in thrombotic thrombocytopenic purpura: the role of anti-rituximab antibodies. Br J Haematol 184:858–861. https://doi.org/10.1111/bjh.15177
Maloney DG, Grillo-López AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, Janakiraman N, Foon KA, Liles TM, Dallaire BK, Wey K, Royston I, Davis T, Levy R (1997) IDEC-C2B8 (Rituximab) anti CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood Sep 90:2188–2195
Looney RJ, Anolik JH, Campbell D, Felgar RE, Young F, Arend LJ, Sloand JA, Rosenblatt J, Sanz I (2004) B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum 50:2580–2589. https://doi.org/10.1002/art.20430
Pijpe J, van Imhoff GW, Spijkervet FK, Roodenburg JL, Wolbink GJ, Mansour K, Vissink A, Kallenberg CG, Bootsma H (2005) Rituximab treatment in patients with primary Sjögren's syndrome: an open-label phase II study. Arthritis Rheum 52:2740–2750. https://doi.org/10.1002/art.21260
Smith KG, Jones RB, Burns SM, Jayne DR (2006) Long-term comparison of rituximab treatment for refractory systemic lupus erythematosus and vasculitis: Remission, relapse, and re-treatment. Arthritis Rheum 54:2970–2982. https://doi.org/10.1002/art.22046
Keystone E, Fleischmann R, Emery P, Furst DE, van Vollenhoven R, Bathon J, Dougados M, Baldassare A, Ferraccioli G, Chubick A, Udell J, Cravets MW, Agarwal S, Cooper S, Magrini F (2007) Safety and efficacy of additional courses of rituximab in patients with active rheumatoid arthritis. Arthritis Rheum 56:3896–3908. https://doi.org/10.1002/art.23059
Fujinaga S, Nishino T, Endo S, Umeda C, Watanabe Y, Nakagawa M (2020) Unfavorable impact of anti-rituximab antibodies on clinical outcomes in children with complicated steroid-dependent nephrotic syndrome. Pediatr Nephrol 35:2003–2008. https://doi.org/10.1007/s00467-020-04629-w
Fujinaga S, Nishino T (2018) Is cytokine-release syndrome the cause of rituximab treatment-related infusion reactions in children with nephrotic syndrome? Impact of anti-rituximab antibodies. Pediatr Nephrol 33:1097–1098. https://doi.org/10.1007/s00467-018-3960-0
Chan EY, Webb H, Yu E, Ghiggeri GM, Kemper MJ, Ma AL, Yamamura T, Sinha A, Bagga A, Hogan J, Dossier C, Vivarelli M, Liu ID, Kamei K, Ishikura K, Saini P, Tullus K (2020) Both the rituximab dose and maintenance immunosuppression in steroid-dependent/frequently-relapsing nephrotic syndrome have important effects on outcomes. Kidney Int 97:393–401. https://doi.org/10.1016/j.kint.2019.09.033
Kamei K, Ishikura K, Sako M, Aya K, Tanaka R, Nozu K, Kaito H, Nakanishi K, Ohtomo Y, Miura K, Takahashi S, Morimoto T, Kubota W, Ito S, Nakamura H, Iijima K, Rituximab for Childhood-Onset Refractory Nephrotic Syndrome (RCRNS) Study Group (2017) Long-term outcome of childhood-onset complicated nephrotic syndrome after a multicenter, double-blind, randomized, placebo-controlled trial of rituximab. Pediatr Nephrol 32:2071–2078. https://doi.org/10.1007/s00467-017-3718-0
Boyer-Suavet S, Andreani M, Cremoni M, Brglez V, Benzaken S, Bernard G, Nachman P, Esnault V, Seitz-Polski B (2019) Rituximab bioavailability in primary membranous nephropathy. Nephrol Dial Transplant 34:1423–1425. https://doi.org/10.1093/ndt/gfz041
Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H (2002) Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99:754–758. https://doi.org/10.1182/blood.v99.3.754
Kamei K, Ito S, Nozu K, Fujinaga S, Nakayama M, Sako M, Saito M, Yoneko M, Iijima K (2009) Single dose of rituximab for refractory steroid-dependent nephrotic syndrome in children. Pediatr Nephrol 24:1321–1328. https://doi.org/10.1007/s00467-009-1191-0
Fujinaga S, Sakuraya K (2018) Single infusion of low-dose ofatumumab in a child with complicated nephrotic syndrome with anti-rituximab antibodies. Pediatr Nephrol Mar 33(3):527–528. https://doi.org/10.1007/s00467-017-3866-2
Wang CS, Liverman RS, Garro R, George RP, Glumova A, Karp A, Jernigan S, Warshaw B (2017) Ofatumumab for the treatment of childhood nephrotic syndrome. Pediatr Nephrol 32:835–841. https://doi.org/10.1007/s00467-017-3621-8
Dossier C, Prim B, Moreau C, Kwon T, Maisin A, Nathanson S, De Gennes C, Barsotti K, Bourrassi A, Hogan J, Deschênes G (2020) A global antiB cell strategy combining obinutuzumab and daratumumab in severe pediatric nephrotic syndrome. Pediatr Nephrol. https://doi.org/10.1007/s00467-020-04811-0
Boyer S, Benzaken S, Bernard G, Esnault V, Seitz-Polski B (2017) Réactivité croisée des anticorps anti-rituximab neutralisants avec les anti-CD20 de troisième génération : une alternative thérapeutique dans la glomérulopathie extramembraneuse? Oral communication. Nephrol Ther 5:267. https://doi.org/10.1016/j.nephro.2017.08.040
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bertrand, Q., Mignot, S., Kwon, T. et al. Anti-rituximab antibodies in pediatric steroid-dependent nephrotic syndrome. Pediatr Nephrol 37, 357–365 (2022). https://doi.org/10.1007/s00467-021-05069-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-021-05069-w