
Abstract
Patients with steroid-resistant nephrotic syndrome (SRNS) who develop resistance to immunosuppressive agents, defined as refractory SRNS, have poor renal outcomes. Although the chimeric anti-CD20 monoclonal antibody rituximab has shown efficacy for frequently relapsing nephrotic syndrome and steroid-dependent nephrotic syndrome, its efficacy for refractory SRNS remains uncertain due to limited data. According to previous case reports, 50.4% of patients with refractory SRNS showed clinical improvements after rituximab treatment. Remission rates in patients with initial steroid resistance and late steroid resistance were 43.9 and 57.7%, respectively, and 41.5 and 63.6% in patients with focal segmental glomerulosclerosis and minor glomerular abnormalities, respectively. However, various factors (race, disease severity, number of rituximab doses, concomitant treatments, and observation period) differed among these observational studies and their consensus may also have been affected by potential publication bias. Rituximab monotherapy may have some degree of efficacy and lead to satisfactory outcomes in a subset of patients with refractory SRNS. However, administration of concomitant treatments during rituximab-mediated B cell depletion, such as methylprednisolone pulse therapy, daily oral prednisolone therapy, and immunosuppressive agents, may lead to better outcomes in these patients. Large-scale, multi-center prospective studies are needed to evaluate the efficacy and safety of such regimens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Idiopathic nephrotic syndrome is the most common glomerular disease in children and is characterized by hypoalbuminemia and edema caused by heavy proteinuria. A recent nationwide analysis in Japan revealed an incidence rate of 6.5 cases per 100,000 children per year, with half of cases diagnosed in children younger than 5 years of age [1]. Idiopathic nephrotic syndrome occurs less frequently in Caucasian populations (two cases per 100,000 children per year). Almost 10% of cases develop steroid-resistant nephrotic syndrome (SRNS). Renal prognoses for children with SRNS are poor, and 10-year renal survival was reported at approximately 60% [2, 3]. Renal outcomes in patients who develop resistance to both steroids and immunosuppressive agents, defined as refractory SRNS, are especially poor. A cohort study of 1354 SRNS patients enrolled in the PodoNet Registry in Europe showed that 10-year end-stage renal disease (ESRD)-free survival rates were 94%, 72%, and 43% in children with complete remission, partial remission, and no remission, respectively [3]. Thus, achieving remission is crucial for optimal long-term renal prognosis.
The standard induction therapy for children with SRNS is a calcineurin inhibitor or combination treatment with calcineurin inhibitor and methyl prednisone pulse therapy (MPT) [4]. Several case series have reported the efficacy of MPT, renin-angiotensin system inhibitors, plasma exchange, and low-density lipoprotein in reducing proteinuria in children with SRNS, although few randomized controlled trials (RCTs) have been conducted [5,6,7,8,9,10,11,12]. Hamasaki et al. reported high remission rates in patients with minor glomerular abnormalities (MGAs) treated with a regimen of cyclosporine (CsA) and oral prednisolone (82.1%) and in patients with focal segmental glomerulosclerosis (FSGS) treated with a CsA, MPT, and oral prednisolone regimen (85.7%). A Cochrane review [4], the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines [13], and the 2013 Japanese guideline for pediatric idiopathic nephrotic syndrome [14] all designated calcineurin inhibitors as the first-line induction therapy for patients with SRNS. However, 20–30% of children with SRNS fail to achieve remission after first-line therapies, a condition defined as refractory SRNS. Standard treatment regimens for patients with refractory SRNS have not yet been established.
Rituximab is a chimeric anti-CD20 monoclonal antibody which depletes B-cells. Its ability to prevent or delay relapse in patients with frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome (FRNS/SDNS) has been demonstrated in several case series and RCTs [15, 16]. Rituximab was approved in Japan by the Ministry of Health, Labor, and Welfare for complicated FRNS/SDNS on August 29, 2014, based on the results of a multicenter, double-blind, placebo-controlled RCT [15, 16]. However, the efficacy of rituximab therapy for refractory SRNS remains uncertain and is based on limited reports, most of them observational studies. The aim of rituximab administration differs by clinical condition: the goal is to prevent relapse in patients with FRNS/SDNS and to induce remission in patients with refractory SRNS. In this review, we highlight recent studies of rituximab treatment for refractory SRNS in children, attempt to retrospectively assess its efficacy, and discuss future perspectives.
Case studies and case series of rituximab therapy for refractory SRNS
Supplementary Tables 1 and 2 show case reports [17,18,19,20,21,22,23,24,25,26,27,28,29] and case series [30,31,32,33,34,35,36,37,38] of rituximab therapy for children with refractory SRNS. Bagga et al. described excellent outcomes in five children (aged 2.8–16.0 years) receiving rituximab as a single weekly infusion of 375 mg/m2 for 4 weeks (four complete remissions and one partial remission) [30]. This was the first report of rituximab as induction therapy in pediatric or adult patients with refractory SRNS. Subsequently, reports of positive outcomes following rituximab therapy for refractory SRNS continued to be published [17,18,19,20].
However, negative outcomes have also been reported following rituximab therapy, such as a case report showing no response to rituximab in refractory SRNS [21, 27, 31, 36]. Gulati et al. treated 33 patients with refractory SRNS (including three adult cases) with four doses of rituximab [31]. Nine patients (27.2%) achieved complete remission, 7 patients (21.2%) achieved partial remission, and 17 patients (51.5%) showed no response following rituximab therapy. The same group also published a similar result in 2015 [36]. Since the consensus of these case reports and case series was that the efficacy of rituximab for refractory SRNS, per se, might be unsatisfactory, concomitant treatments following rituximab administration have been considered.
We published a case series describing 10 patients with refractory SRNS treated with a combination of rituximab, MPT, and immunosuppressive agents [35]. Seven patients (70%) achieved complete remission, one patient (10%) achieved partial remission, and two patients (20%) showed no response. The two patients showing no response to treatment progressed to ESRD. Nakagawa et al. also reported excellent results using a similar protocol [28]. By contrast, Fujinaga and colleagues and Hirano et al. described three cases with refractory SRNS who achieved complete remission after receiving oral daily prednisolone therapy following rituximab therapy [24, 26, 29]. Basu et al. showed that administration of mycophenolate mofetil following rituximab treatment resulted in higher remission rates [37]. Rituximab therapy with additional subsequent treatments, such as MPT, oral daily prednisolone therapy, and immunosuppressive agents, might be more effective in patients with refractory SRNS than rituximab alone. However, one must be aware of potential publication bias among these observational reports. The efficacy and safety of such regimens should be investigated using large prospective studies.
RCTs examining rituximab therapy for refractory SRNS
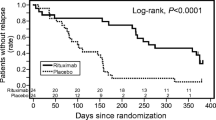
The only RCT examining rituximab therapy for children with refractory SRNS was published by Magnasco et al. in 2012 (Supplementary Table 3) [39]. Thirty-one children with refractory SRNS were randomized into either the rituximab group (n = 16) or the placebo group (n = 15). All patients were negative for NPHS2 and WT1 mutations. Age, serum albumin levels, daily urinary protein excretion, and renal biopsy results were comparable between both groups. Patients randomized to the rituximab group received two doses of rituximab. After 3 months of treatment, three patients in both groups achieved complete remission. Although proteinuria decreased in patients with delayed resistance, rituximab did not reduce proteinuria in early-resistant patients. This study concluded that addition of rituximab to standard regimens did not improve clinical outcomes for children with refractory SRNS.
However, the observation period of this study was only 3 months, which may be too short to evaluate the efficacy of treatments for refractory SRNS. Moreover, the impact of additional treatments such as MPT following rituximab therapy was not examined in this study. These factors might have served to dilute the effects of rituximab therapy in this study.
Response to rituximab therapy for refractory SRNS according to type of steroid resistance and renal pathology
Table 1 shows remission rates following rituximab therapy in patients with refractory SRNS. One must be aware of possible publication bias, as positive results are more likely to be more reported than negative results, especially in case reports. Among case series, remission rates varied from 18.8 to 80.0%. However, race, disease severity, number of rituximab doses, concomitant treatments after rituximab, and observational period differed among these studies.
Table 2 shows responses to rituximab therapy stratified by type of steroid resistance. In the studies by Gulati et al. [31], Sinha et al. [36], Basu et al. [37], and Magnasco et al. [39], patients with late steroid resistance showed higher remission rates, although other reports did not confirm this finding. The overall remission rates of patients with initial steroid resistance and late steroid resistance were 43.9 and 57.7%, respectively; the difference between these figures was not statistically significant. Although patients with congenital nephrotic syndrome were excluded in these studies, it is possible that some patients with initial steroid resistance might have carried genetic mutations leading to nephrotic syndrome, as the number of established susceptibility genes for nephrotic syndrome has been increasing.
Table 3 shows responses to rituximab therapy according to renal biopsy findings. In the studies by Gulati et al. [31], Sinha et al. [36], Basu et al. [37], patients with FSGS showed lower remission rates than those with MGAs. The overall remission rates were 41.5% for patients with FSGS and 63.6% for those with MGAs; this difference was statistically significant (p = 0.003). However, as pathological findings sometimes change in a single patient, there is a possibility that MGA and FSGS represent pathologically identical clinical entities. For example, if a patient fails to achieve remission and continues to experience proteinuria, renal biopsy finding sometimes change from MGAs to FSGS. Considering these phenomena, pathological findings in SRNS may simply result from persistent proteinuria.
Rituximab therapy for recurrence of nephrotic syndrome after kidney transplantation
Patients with SRNS who progress to ESRD are at high risk of recurrence of nephrotic syndrome after kidney transplantation. Approximately 30% of these patients develop recurrence after the first allograft. Young age (< 15 years) at onset of nephrotic syndrome, rapid progression to ESRD (< 3 years), late steroid-resistance, MGA (rather than FSGS), live donor allograft, and non-genetic forms of nephrotic syndrome were reported as risk factors for recurrence [40,41,42]. Patients with relapsed nephrotic syndrome after kidney transplantation are at increased risk of allograft failure. Moreover, after the loss of the allograft, the risk of recurrence of nephrotic syndrome following subsequent kidney transplantation was reported at 80–100% [43]. Traditionally, plasma exchange and high-dose cyclosporine were administered for the treatment of this disease.
Nozu et al. reported a case of a 12-year-old boy who suffered from recurrence of nephrotic syndrome and posttransplant lymphoproliferative disorder (PTLD). The patient was treated with rituximab (four doses, once weekly) resulting in the improvement of proteinuria and PTLD [44]. This case study was the first description of rituximab treatment in a patient with recurrence of nephrotic syndrome after kidney transplantation. Subsequently, several case series describing rituximab treatment for this condition were reported [32, 45, 46]. Combined therapy with plasma exchange and rituximab resulted in complete or partial remission in 60% of patients enrolled in the IPNA multicenter study [45] and 64.1% according to a meta-analysis of 18 reports comprising 39 patients in total [46]. However, it is difficult to evaluate the efficacy of rituximab treatment per se in patients with recurrence of nephrotic syndrome following kidney transplantation because other treatments, such as plasma exchange, were also administered to most patients.
Adverse events due to rituximab therapy
The most well-known adverse events (AEs) resulting from rituximab therapy are infusion reactions (IRs) and are typically observed within 24 h of treatment. The mechanism underlying IRs to rituximab is not fully understood, although the release of cytokines or chemokines due to B cell apoptosis is the most likely cause. Although IRs were reported in about 80% of non-Hodgkin lymphoma patients treated with rituximab, the frequency of IRs was lower (165 of 309 infusions, 53.4%) and their severity was milder in patients with nephrotic syndrome in our center [47]. Sixty-eight percent of AEs following rituximab therapy were classified as grade 1 and the remainder were classified as grade 2. Only 18% of AEs required medical intervention and no severe AEs were observed. Respiratory symptoms were the most common AE (66% of all events), 95% of which were observed within 3 h of initiation of rituximab infusion. B cell counts in patients with IRs were significantly higher than in patients without IRs. Patients who experienced IRs during their first rituximab treatment were more likely to experience recurrent IRs with subsequent treatments (odds ratio 3.64; p < 0.001).
Late-onset AEs to rituximab therapy can be problematic in some patients. Agranulocytosis has been reported as a late-onset complication of rituximab therapy. In our center, agranulocytosis occurred in 9.6% of all patients and was associated with 5.2% of all rituximab infusions [48]. The median time of onset of agranulocytosis was 66 days (range: 54–161 days) following rituximab therapy. Most patients experienced acute infections and received antibiotics. The median age of patients who developed agranulocytosis following rituximab therapy was 6.4 years at the time of treatment, and significantly younger than the median age of patients who did not develop agranulocytosis (median 12.5 years; p = 0.0009). It is important for patients to recognize this complication and to visit an emergency clinic as soon as possible upon developing fever.
Fatal pulmonary fibrosis in patients with refractory SRNS and pleural effusion after rituximab therapy has been reported [22]. Patients with cardiovascular or respiratory complications are categorized as at high risk of severe AEs after rituximab infusion. We recommend that patients be examined by routine chest radiograph and electrocardiography prior to rituximab therapy. The indication for rituximab treatment should be thoroughly discussed if patients with refractory SRNS are suffering from severe pleural effusion or their general condition has deteriorated due to severe edema [49].
Pneumocystis pneumonia [50], immune-mediated ulcerative colitis [51], and fulminant myocarditis [52] have been reported during rituximab-mediated B cell depletion in patients with nephrotic syndrome. Patients with persistent hypogammaglobulinemia have also been reported [53]. Progressive multifocal leukoencephalopathy due to JC virus [54] and reactivation of hepatitis B virus [55] were also reported following rituximab treatment, although not in patients with nephrotic syndrome. It is important that the possibility of rare but severe AEs following rituximab therapy be explained to patients and their caregivers. Decreased efficiency of vaccination during rituximab-mediated B cell depletion can also be a problem for children.
Future perspectives
In two recent large studies, 20–30% of pediatric SRNS cases were classified as genetic nephrotic syndrome [56, 57]. More than 50 disease-associated genes have been described, and more might be identified in the near future. In patients with genetic SRNS, the response rate to immunosuppressive agents is lower than in patients with non-genetic SRNS [56]. Buscher et al. reported a remission rate of 19% in patients with congenital SRNS compared with 78% in patients with non-genetic SRNS [58]. Thus, because genetic markers are useful for predicting responses to immunosuppressive agents, genetic analysis of patients with refractory SRNS should be conducted prior to administration of rituximab. If a patient is positive for a disease-associated gene, immunosuppressive treatments should be discontinued. Moreover, in patients with certain disease-associated genes (CDK20, DLC1, EMP2, ITSN1, ITSN2, KANK2, MAGI2, PLCE1, and TNS2), steroid and immunosuppressive agents have been reported to be effective [59,60,61,62,63]. The efficacy of rituximab therapy in patients with these mutations requires further evaluation.
To date, most reports of rituximab therapy for patients with refractory SRNS have been retrospective observational studies, excepting one RCT [39]. Thus, one must be aware of publication bias when considering these data. Rituximab treatment protocols, concomitant and subsequent treatments, and observation period differed among these studies. It remains possible that patients with genetic nephrotic syndrome were not completely excluded from these studies, as the number of known responsible genes for genetic nephrotic syndrome is increasing. All of these factors make evaluating the precise efficacy of rituximab treatment in patients with refractory SRNS from previous reports extremely challenging.
However, synthesizing the results of previous reports, the efficacy of rituximab treatment per se for patients with refractory SRNS appears to be limited. Patients with refractory SRNS, including patients who do not respond to CsA and MPT, should be treated not only with rituximab but also with concomitant treatments during B cell depletion, such as MPT, oral daily prednisolone therapy, and immunosuppressive agents. Further studies are needed to evaluate the efficacy and safety of rituximab for children with refractory SRNS. A large-scale, multicenter prospective study including a long-term observational period is needed to clarify these issues.
References
Kikunaga K, Ishikura K, Terano C, Sato M, Komaki F, Hamasaki Y, Sasaki S, Iijima K, Yoshikawa N, Nakanishi K, Nakazato H, Matsuyama T, Ando T, Ito S, Honda M, Japanese Pediatric Survey Holding Information of Nephrotic syndrome (JP-SHINE) study of the Japanese Study Group of Renal Disease in Children (2017) High incidence of idiopathic nephrotic syndrome in East Asian children: a nationwide survey in Japan (JP-SHINE study). Clin Exp Nephrol 21:651–657
Mekahli D, Liutkus A, Ranchin B, Yu A, Bessenay L, Girardin E, Van Damme-Lombaerts R, Palcoux JB, Cachat F, Lavocat MP, Bourdat-Michel G, Nobili F, Cochat P (2009) Long-term outcome of idiopathic steroid-resistant nephrotic syndrome: a multicenter study. Pediatr Nephrol 24:1525–1532
Trautmann A, Schnaidt S, Lipska-Ziętkiewicz BS, Bodria M, Ozaltin F, Emma F, Anarat A, Melk A, Azocar M, Oh J, Saeed B, Gheisari A, Caliskan S, Gellermann J, Higuita LMS, Jankauskiene A, Drozdz D, Mir S, Balat A, Szczepanska M, Paripovic D, Zurowska A, Bogdanovic R, Yilmaz A, Ranchin B, Baskin E, Erdogan O, Remuzzi G, Firszt-Adamczyk A, Kuzma-Mroczkowska E, Litwin M, Murer L, Tkaczyk M, Jardim H, Wasilewska A, Printza N, Fidan K, Simkova E, Borzecka H, Staude H, Hees K, Schaefer F, PodoNet Consortium (2017) Long-term outcome of steroid-resistant nephrotic syndrome in children. J Am Soc Nephrol 28:3055–3065
Hamasaki Y, Yoshikawa N, Hattori S, Sasaki S, Iijima K, Nakanishi K, Matsuyama T, Ishikura K, Yata N, Kaneko T, Honda M, Japanese Study Group of Renal Disease (2009) Cyclosporine and steroid therapy in children with steroid-resistant nephrotic syndrome. Pediatr Nephrol 24:2177–2185
Yorgin PD, Krasher J, Al-Uzri AY (2001) Pulse methylprednisolone treatment of idiopathic steroid-resistant nephrotic syndrome. Pediatr Nephrol 16:245–250
Shenoy M, Plant ND, Lewis MA, Bradbury MG, Lennon R, Webb NJ (2010) Intravenous methylprednisolone in idiopathic childhood nephrotic syndrome. Pediatr Nephrol 25:899–903
Mori K, Honda M, Ikeda M (2004) Efficacy of methylprednisolone pulse therapy in steroid-resistant nephrotic syndrome. Pediatr Nephrol 19:1232–1236
Bagga A, Mudigoudar BD, Hari P, Vasudev V (2004) Enalapril dosage in steroid-resistant nephrotic syndrome. Pediatr Nephrol 19:45–50
Yi Z, Li Z, Wu XC, He QN, Dang XQ, He XJ (2006) Effect of fosinopril in children with steroid-resistant idiopathic nephrotic syndrome. Pediatr Nephrol 21:967–972
Feld SM, Figueroa P, Savin V, Nast CC, Sharma R, Sharma M, Hirschberg R, Adler SG (1998) Plasmapheresis in the treatment of steroid-resistant focal segmental glomerulosclerosis in native kidneys. Am J Kidney Dis 32:230–237
Franke D, Zimmering M, Wolfish N, Ehrich JH, Filler G (2000) Treatment of FSGS with plasma exchange and immunadsorption. Pediatr Nephrol 14:965–969
Hattori M, Chikamoto H, Akioka Y, Nakakura H, Ogino D, Matsunaga A, Fukazawa A, Miyakawa S, Khono M, Kawaguchi H, Ito K (2003) A combined low-density lipoprotein apheresis and prednisone therapy for steroid-resistant primary focal segmental glomerulosclerosis in children. Am J Kidney Dis 42:1121–1130
Kidney Disease: Improving global outcomes (KDIGO) (2012) Steroid-resistant nephrotic syndrome in children. Kidney Int Suppl 2:172–176
Ishikura K, Matsumoto S, Sako M, Tsuruga K, Nakanishi K, Kamei K, Saito H, Fujinaga S, Hamasaki Y, Chikamoto H, Ohtsuka Y, Komatsu Y, Ohta T, Nagai T, Kaito H, Kondo S, Ikezumi Y, Tanaka S, Kaku Y, Iijima K, Japanese Society for Pediatric Nephrology; Japanese Society for Pediatric Nephrology (2015) Clinical practice guideline for pediatric idiopathic nephrotic syndrome 2013: medical therapy. Clin Exp Nephrol 19:6–33
Iijima K, Sako M, Nozu K, Mori R, Tuchida N, Kamei K, Miura K, Aya K, Nakanishi K, Ohtomo Y, Takahashi S, Tanaka R, Kaito H, Nakamura H, Ishikura K, Ito S, Ohashi Y, Rituximab for Childhood-onset Refractory Nephrotic Syndrome (RCRNS) Study Group (2014) Rituximab for childhood-onset, complicated, frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet 384:1273–1281
Iijima K, Sako M, Kamei K, Nozu K (2018) Rituximab in steroid-sensitive nephrotic syndrome: lessons from clinical trials. Pediatr Nephrol 33:1449–1455
Nakayama M, Kamei K, Nozu K, Matsuoka K, Nakagawa A, Sako M, Iijima K (2008) Rituximab for refractory focal segmental glomerulosclerosis. Pediatr Nephrol 23:481–485
Peters HP, van de Kar NC, Wetzels JF (2008) Rituximab in minimal change nephropathy and focal segmental glomerulosclerosis: report of four cases and review of the literature. Neth J Med 66:408–415
Suri M, Tran K, Sharma AP, Filler G, Grimmer J (2008) Remission of steroid-resistant nephrotic syndrome due to focal and segmental glomerulosclerosis using rituximab. Int Urol Nephrol 40:807–810
Sharma AP, Filler G (2009) Role of mycophenolate mofetil in remission maintenance after a successful response to rituximab. Pediatr Nephrol 24:423–424
Fernandez-Fresnedo G, Segarra A, González E, Alexandru S, Delgado R, Ramos N, Egido J, Praga M, Trabajo de Enfermedades Glomerulares de la Sociedad Española de Nefrología (GLOSEN) (2009) Rituximab treatment of adult patients with steroid-resistant focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 4:1317–1323
Chaumais MC, Garnier A, Chalard F, Peuchmaur M, Dauger S, Jacqz-Agrain E, Deschênes G (2009) Fatal pulmonary fibrosis after rituximab administration. Pediatr Nephrol 24:1753–1755
Kaito H, Kamei K, Kikuchi E, Ogura M, Matsuoka K, Nagata M, Iijima K, Ito S (2010) Successful treatment of collapsing focal segmental glomerulosclerosis with a combination of rituximab, steroids and ciclosporin. Pediatr Nephrol 25:957–959
Hirano D, Fujinaga S, Nishizaki N (2012) The uncertainty of rituximab and steroid dosing in refractory steroid-resistant nephrotic syndrome. Clin Nephrol 77:510–512
Janardan J, Ooi K, Menahem S (2014) Sustained complete remission of steroid- and cyclophosphamide-resistant minimal-change disease with a single course of rituximab therapy. Clin Kidney J 7:293–295
Fujinaga S, Hara T (2014) Re-treatment with high-dose prednisolone after rituximab infusion for childhood-onset steroid-resistant nephrotic syndrome. Pediatr Nephrol 29:1291–1292
Counsilman CE, Jol-van der Zijde CM, Stevens J, Cransberg K, Bredius RG, Sukhai RN (2015) Pharmacokinetics of rituximab in a pediatric patient with therapy-resistant nephrotic syndrome. Pediatr Nephrol 30:1367–1370
Nakagawa T, Shiratori A, Kawaba Y, Kanda K, Tanaka R (2016) Efficacy of rituximab therapy against intractable steroid-resistant nephrotic syndrome. Pediatr Int 58:1003–1008
Fujinaga S, Sakuraya K (2017) Repeated administrations of rituximab along with steroids and immunosuppressive agents in refractory steroid-resistant nephrotic syndrome. Indian Pediatr 54:49–50
Bagga A, Sinha A, Moudgil A (2007) Rituximab in patients with the steroid-resistant nephrotic syndrome. N Engl J Med 356:2751–2752
Gulati A, Sinha A, Jordan SC, Hari P, Dinda AK, Sharma S, Srivastava RN, Moudgil A, Bagga A (2010) Efficacy and safety of treatment with rituximab for difficult steroid-resistant and -dependent nephrotic syndrome: multicentric report. Clin J Am Soc Nephrol 5:2207–2212
Prytuła A, Iijima K, Kamei K, Geary D, Gottlich E, Majeed A, Taylor M, Marks SD, Tuchman S, Camilla R, Ognjanovic M, Filler G, Smith G, Tullus K (2010) Rituximab in refractory nephrotic syndrome. Pediatr Nephrol 25:461–468
Kari JA, El-Morshedy SM, El-Desoky S, Alshaya HO, Rahim KA, Edrees BM (2011) Rituximab for refractory cases of childhood nephrotic syndrome. Pediatr Nephrol 26:733–737
Ito S, Kamei K, Ogura M, Udagawa T, Fujinaga S, Saito M, Sako M, Iijima K (2013) Survey of rituximab treatment for childhood-onset refractory nephrotic syndrome. Pediatr Nephrol 28:257–264
Kamei K, Okada M, Sato M, Fujimaru T, Ogura M, Nakayama M, Kaito H, Iijima K, Ito S (2014) Rituximab treatment combined with methylprednisolone pulse therapy and immunosuppressants for childhood steroid-resistant nephrotic syndrome. Pediatr Nephrol 29:1181–1187
Sinha A, Bhatia D, Gulati A, Rawat M, Dinda AK, Hari P, Bagga A (2015) Efficacy and safety of rituximab in children with difficult-to-treat nephrotic syndrome. Nephrol Dial Transplant 30:96–106
Basu B, Mahapatra TK, Mondal N (2015) Mycophenolate mofetil following rituximab in children with steroid-resistant nephrotic syndrome. Pediatrics 136:e132–e139
Hoseini R, Sabzian K, Otukesh H, Zafaranloo N, Panahi P, Rahimzadeh N, Nakhaie S, Akhavan Sepehi M (2018) Efficacy and safety of rituximab in children with steroid- and cyclosporine-resistant and steroid- and cyclosporine-dependent nephrotic syndrome. Iran J Kidney Dis 12:27–32
Magnasco A, Ravani P, Edefonti A, Murer L, Ghio L, Belingheri M, Benetti E, Murtas C, Messina G, Massella L, Porcellini MG, Montagna M, Regazzi M, Scolari F, Ghiggeri GM (2012) Rituximab in children with resistant idiopathic nephrotic syndrome. J Am Soc Nephrol 23:1117–1124
Pelletier JH, Kumar KR, Engen R, Bensimhon A, Varner JD, Rheault MN, Srivastava T, Straatmann C, Silva C, Davis TK, Wenderfer SE, Gibson K, Selewski D, Barcia J, Weng P, Licht C, Jawa N, Kallash M, Foreman JW, Wigfall DR, Chua AN, Chambers E, Hornik CP, Brewer ED, Nagaraj SK, Greenbaum LA, Gbadegesin RA (2018) Recurrence of nephrotic syndrome following kidney transplantation is associated with initial native kidney biopsy findings. Pediatr Nephrol 33:1773–1780
Francis A, Trnka P, McTaggart SJ (2016) Long-term outcome of kidney transplantation in recipients with focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 11:2041–2046
Ding WY, Koziell A, McCarthy HJ, Bierzynska A, Bhagavatula MK, Dudley JA, Inward CD, Coward RJ, Tizard J, Reid C, Antignac C, Boyer O, Saleem MA (2014) Initial steroid sensitivity in children with steroid-resistant nephrotic syndrome predicts post-transplant recurrence. J Am Soc Nephrol 25:1342–1348
Bierzynska A, Saleem MA (2018) Deriving and understanding the risk of post-transplant recurrence of nephrotic syndrome in the light of current molecular and genetic advances. Pediatr Nephrol 33:2027–2035
Nozu K, Iijima K, Fujisawa M, Nakagawa A, Yoshikawa N, Matsuo M (2005) Rituximab treatment for posttransplant lymphoproliferative disorder (PTLD) induces complete remission of recurrent nephrotic syndrome. Pediatr Nephrol 20:1660–1663
Araya CE, Dharnidharka VR (2011) The factors that may predict response to rituximab therapy in recurrent focal segmental glomerulosclerosis: a systematic review. J Transp Secur 2011:374213
Kumar J, Shatat IF, Skversky AL, Woroniecki RP, Del Rio M, Perelstein EM, Johnson VL, Mahesh S (2013) Rituximab in post-transplant pediatric recurrent focal segmental glomerulosclerosis. Pediatr Nephrol 28:333–338
Kamei K, Ogura M, Sato M, Ito S, Ishikura K (2018) Infusion reactions associated with rituximab treatment for childhood-onset complicated nephrotic syndrome. Pediatr Nephrol 33:1013–1018
Kamei K, Takahashi M, Fuyama M, Saida K, Machida H, Sato M, Ogura M, Ito S (2015) Rituximab-associated agranulocytosis in children with refractory idiopathic nephrotic syndrome: case series and review of literature. Nephrol Dial Transplant 30:91–96
Kamei K, Ito S, Iijima K (2010) Severe respiratory adverse events associated with rituximab infusion. Pediatr Nephrol 25:1193
Sato M, Ito S, Ogura M, Kamei K, Miyairi I, Miyata I, Higuchi M, Matsuoka K (2013) Atypical Pneumocystis jiroveci pneumonia with multiple nodular granulomas after rituximab for refractory nephrotic syndrome. Pediatr Nephrol 28:145–149
Ardelean DS, Gonska T, Wires S, Cutz E, Griffiths A, Harvey E, Tse SM, Benseler SM (2010) Severe ulcerative colitis after rituximab therapy. Pediatrics 126:e243–e246
Sellier-Leclerc AL, Belli E, Guérin V, Dorfmüller P, Deschênes G (2013) Fulminant viral myocarditis after rituximab therapy in pediatric nephrotic syndrome. Pediatr Nephrol 28:1875–1879
Delbe-Bertin L, Aoun B, Tudorache E, Lapillone H, Ulinski T (2013) Does rituximab induce hypogammaglobulinemia in patients with pediatric idiopathic nephrotic syndrome? Pediatr Nephrol 28:447–451
Boren EJ, Cheema GS, Naguwa SM, Ansari AA, Gershwin ME (2008) The emergence of progressive multifocal leukoencephalopathy (PML) in rheumatic diseases. J Autoimmun 30:90–98
Tsutsumi Y, Kanamori H, Mori A, Tanaka J, Asaka M, Imamura M, Masauzi N (2005) Reactivation of hepatitis B virus with rituximab. Expert Opin Drug Saf 4:599–608
Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, Anarat A, Caliskan S, Emma F, Gellermann J, Oh J, Baskin E, Ksiazek J, Remuzzi G, Erdogan O, Akman S, Dusek J, Davitaia T, Özkaya O, Papachristou F, Firszt-Adamczyk A, Urasinski T, Testa S, Krmar RT, Hyla-Klekot L, Pasini A, Özcakar ZB, Sallay P, Cakar N, Galanti M, Terzic J, Aoun B, Caldas Afonso A, Szymanik-Grzelak H, Lipska BS, Schnaidt S, Schaefer F, PodoNet Consortium (2015) Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol 10:592–600
Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA, SRNS Study Group, Hildebrandt F (2015) A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 26:1279–1289
Büscher AK, Beck BB, Melk A, Hoefele J, Kranz B, Bamborschke D, Baig S, Lange-Sperandio B, Jungraithmayr T, Weber LT, Kemper MJ, Tönshoff B, Hoyer PF, Konrad M, Weber S, German Pediatric Nephrology Association (GPN) (2016) Rapid response to cyclosporin a and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 11:245–253
Ashraf S, Kudo H, Rao J, Kikuchi A, Widmeier E, Lawson JA, Tan W, Hermle T, Warejko JK, Shril S, Airik M, Jobst-Schwan T, Lovric S, Braun DA, Gee HY, Schapiro D, Majmundar AJ, Sadowski CE, Pabst WL, Daga A, van der Ven AT, Schmidt JM, Low BC, Gupta AB, Tripathi BK, Wong J, Campbell K, Metcalfe K, Schanze D, Niihori T, Kaito H, Nozu K, Tsukaguchi H, Tanaka R, Hamahira K, Kobayashi Y, Takizawa T, Funayama R, Nakayama K, Aoki Y, Kumagai N, Iijima K, Fehrenbach H, Kari JA, El Desoky S, Jalalah S, Bogdanovic R, Stajić N, Zappel H, Rakhmetova A, Wassmer SR, Jungraithmayr T, Strehlau J, Kumar AS, Bagga A, Soliman NA, Mane SM, Kaufman L, Lowy DR, Jairajpuri MA, Lifton RP, Pei Y, Zenker M, Kure S, Hildebrandt F (2018) Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat Commun 9:1960
Gee HY, Ashraf S, Wan X, Vega-Warner V, Esteve-Rudd J, Lovric S, Fang H, Hurd TW, Sadowski CE, Allen SJ, Otto EA, Korkmaz E, Washburn J, Levy S, Williams DS, Bakkaloglu SA, Zolotnitskaya A, Ozaltin F, Zhou W, Hildebrandt F (2014) Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet 94:884–890
Gee HY, Zhang F, Ashraf S, Kohl S, Sadowski CE, Vega-Warner V, Zhou W, Lovric S, Fang H, Nettleton M, Zhu JY, Hoefele J, Weber LT, Podracka L, Boor A, Fehrenbach H, Innis JW, Washburn J, Levy S, Lifton RP, Otto EA, Han Z, Hildebrandt F (2015) KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest 125:2375–2384
Bierzynska A, Soderquest K, Dean P, Colby E, Rollason R, Jones C, Inward CD, McCarthy HJ, Simpson MA, Lord GM, Williams M, Welsh GI, Koziell AB, Saleem MA, Nephro S, UK study of nephrotic syndrome (2017) MAGI2 mutations cause congenital nephrotic syndrome. J Am Soc Nephrol 28:1614–1621
Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nürnberg G, Garg P, Verma R, Chaib H, Hoskins BE, Ashraf S, Becker C, Hennies HC, Goyal M, Wharram BL, Schachter AD, Mudumana S, Drummond I, Kerjaschki D, Waldherr R, Dietrich A, Ozaltin F, Bakkaloglu A, Cleper R, Basel-Vanagaite L, Pohl M, Griebel M, Tsygin AN, Soylu A, Müller D, Sorli CS, Bunney TD, Katan M, Liu J, Attanasio M, O'toole JF, Hasselbacher K, Mucha B, Otto EA, Airik R, Kispert A, Kelley GG, Smrcka AV, Gudermann T, Holzman LB, Nürnberg P, Hildebrandt F (2006) Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 38:1397–1405
Acknowledgements
We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
KK has received lecture fees from Chugai Pharmaceutical Co., Ltd. and AbbVie GK. KIs has received lecture fees from Asahi Kasei Pharma, Chugai Pharmaceutical Co., Ltd., Zenyaku Kogyo Co., Ltd., and Novartis Pharma K.K. and grants from Asahi Kasei Pharma, Chugai Pharmaceutical Co., Ltd., and NovartisPharma K.K. MS has received a consulting fee from Zenyaku Kogyo Co. Ltd. SI has received grants from AbbVie GK., Asahi kasei Pharma Corporation, Astellas Pharma Inc., Chugai Pharmaceutical Co., Ltd., CSL Behring, Eisai Co. Ltd., Maruho Co. Ltd., Japan Blood Product Organization, Kyowa Hakko Kirin Co., Ltd., Phizer Co. Ltd. and Teijin Pharma Ltd. SI has received lecture fees and/or consultant fees from AbbVie LLC, Astellas Pharma Inc., Alexion Pharma LLC, Chugai Pharmaceutical Co., Ltd., Eisai Co. Ltd., Daiichi Sankyo, Co., Ltd., Novartis Pharma K.K., Japan Blood Product Organization, JCR Pharmaceuticals Co., Ltd., Takeda Pharmaceutical Co., Ltd., Teijin Pharma Ltd., Sanofi Genzyme Co. Ltd., Tanabe Mitsubishi Pharma, Novo Nordisk Pharma Ltd., Takeda Pharmaceutical Co., Ltd. and Zenyaku Kogyo Co. Ltd.. KN has received lecture fees from Chugai Pharmaceutical Co., Ltd., Novartis Pharma K.K. and Otsuka Pharmaceutical Co., Ltd. KIi has received grants from Novartis Pharma K.K., Japan Blood Product Organization, AbbVie LLC, JCR Pharmaceuticals Co., Ltd., Daiichi Sankyo, Co., Ltd., Teijin Pharma Ltd., CSL Behring, Novo Nordisk Pharma Ltd., Air Water Medical Inc., Astellas Pharma Inc., Takeda Pharmaceutical Co., Ltd., Taisho Toyama Pharmaceutical Co., Ltd., Eisai Co. Ltd.,Biofermin Pharmaceutical Co., Ltd., and Zenyaku Kogyo Co. Ltd., and lecture fees and/or consulting fees from Zenyaku Kogyo Co., Ltd., Novartis Pharma K.K., Chugai Pharmaceutical Co., Ltd., Astellas Pharma Inc., Springer Japan K.K., Meiji Seika Pharma Co., Ltd., Asahi kasei Pharma Corporation, Medical Review Co.,Ltd., Nippon Boehringer Ingelheim Co., Ltd., Baxter Limited, Ono Pharmaceutical Co., Ltd., Sanwa Kagaku Kenkyusho Co.,Ltd., Sanofi K.K., Alexion Pharma LLC., and Kyowa Hakko Kirin Co., Ltd.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PPTX 69 kb)
Rights and permissions
About this article

Cite this article
Kamei, K., Ishikura, K., Sako, M. et al. Rituximab therapy for refractory steroid-resistant nephrotic syndrome in children. Pediatr Nephrol 35, 17–24 (2020). https://doi.org/10.1007/s00467-018-4166-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-018-4166-1