
Abstract
The sperm-derived oocyte activating factor, phospholipase C zeta (PLC ζ), is the only PLC isoform reported in cattle. The objectives were to (1) localize PLC ζ in fresh and capacitated bovine sperm and (2) investigate the activation of PLC ζ during bull sperm capacitation and contributions of PLC activity to this process. We confirmed interaction of testis-specific isoform of Na/K-ATPase (ATP1A4) with PLC ζ (immunolocalization and immunoprecipitation) and tyrosine phosphorylation (immunoprecipitation) of PLC ζ (a post-translational protein modification commonly involved in activation of PLC in somatic cells) during capacitation. Furthermore, incubation of sperm under capacitating conditions upregulated PLC-mediated hyperactivated motility, tyrosine phosphoprotein content, acrosome reaction, and F-actin formation (flow cytometry), implying that PLC activity is enhanced during capacitation and contributing to these capacitation processes. In conclusion, we inferred that PLC ζ is activated during capacitation by tyrosine phosphorylation through a mechanism involving ATP1A4, contributing to capacitation-associated biochemical events.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phospholipase C zeta (PLC ζ) is a sperm-specific phosphoinositide-phospholipase C (PI-PLC) protein, associated with male factor infertility (Saunders et al. 2002) and identified in several mammals (rat: Ito et al. 2008; mouse: Saunders et al. 2002; pig: Yoneda et al. 2006; cow: Ross et al. 2008; monkeys and humans: Cox et al. 2002). Both PLC ζ (Saunders et al. 2002) and post-acrosomal WW-domain binding protein (PAWP; Wu et al. 2007) are regarded as the sperm oocyte activation factors. Microinjection of the PAWP cRNA or recombinant PAWP into porcine, bovine, Xenopus, murine, and human oocytes caused calcium oscillations similar to those in ICSI, plus oocyte activation. Furthermore, calcium oscillations in human and murine oocytes were prevented by a competitive inhibitor for PAWP-derived PPGY peptide (Aarabi et al. 2014; Wu et al. 2007). However, when murine oocytes were microinjected with recombinant PAWP and PLC ζ, only the latter caused calcium oscillations similar to those during mammalian fertilization (Nomikos et al. 2014). Moreover, several laboratories have validated the role of PLC ζ as a sperm oocyte activation factor (SOAF) in a repeatable and reliable manner and confirmed its involvement in oocyte activation by initiating calcium oscillations (Heytens et al. 2009; Kashir et al. 2012; Knott et al. 2005; Miyazaki et al. 1993; Saunders et al. 2002; Yoon et al. 2008; Swann 1990; Swann and Yu 2008). Upon sperm entry into the oocyte, PLC ζ in the perinuclear theca (PT) region of sperm (Escoffier et al. 2015; Fujimoto et al. 2004) attaches to small vesicles inside the oocyte and catalyses hydrolysis of PIP2 to form DAG and IP3; the latter binds to IP3 receptors in the intracellular calcium reserves (endoplasmic reticulum), releasing calcium, leading to calcium oscillation and oocyte activation. There are species-specific relative changes in frequency and duration of calcium release, ranging from every 2 min to every hour (Fissore et al. 1992; Kline 1991; Nomikos et al. 2011). Furthermore, PLC ζ is immunolocalized to different regions of sperm in various mammals (Bedford-Guaus et al. 2011; Fujimoto et al. 2004; Kaewmala et al. 2012; Kashir et al. 2013; Mejía-Flores et al. 2017; Yoneda et al. 2006; Yoon et al. 2008). Furthermore, location of PLC ζ changes from acrosomal region to post-acrosomal region in human and mouse sperm during capacitation (Grasa et al. 2008; Young et al. 2009).
Capacitation is a maturation process undergone by ejaculated sperm in the female reproductive tract for a species-dependent interval in order to achieve fertilizing ability (Yanagimachi 1994). Various capacitation-associated physiological, biochemical, and molecular changes are involved in regulation of sperm function. This involves efflux of cholesterol (Salicioni et al. 2007), increases in intracellular pH and calcium (Martínez-López et al. 2009), membrane hyperpolarisation (Martínez-López et al. 2009), phosphorylation modification of proteins at serine (Grasa et al. 2009), threonine (Ramio‐Lluch et al. 2019) and tyrosine residues (Alvau et al. 2016; Jin and Yang 2017; Yanagimachi 1994; Zhao and Kan 2019), remodelling of actin, and hyperactivated motility (Salicioni et al. 2007). Various sperm proteins are engaged in regulation of specific sperm functions during capacitation, although they are not well characterized. Despite several studies on PLC ζ, the mechanism by which it is activated or kept inactive in sperm is unknown. A compromised release or activation of sperm oocyte factor is the putative cause of insufficiency in the calcium oscillation that precedes oocyte activation; a lack of oocyte activation causes fertilization failure (Malcuit et al. 2006) that contributes to relatively low efficiency of intracytoplasmic sperm injection (ICSI) in cattle compared to other species (Agulia et al. 2017; Hara et al. 2011; Morozumi and Yanagimachi 2005; Salamone et al. 2017). Perhaps PLC ζ undergoes protein interactions (Kurokawa et al. 2005) and activation during capacitation. Regardless, identifying capacitation conditions that promote PLC ζ activation could have applications for improving the efficiency of assisted reproductive technologies.
A sperm-specific protein Na/K-ATPase α4 (ATP1A4), involved in regulation of sperm motility (Jimenez et al. 2010, 2012) and capacitation (Newton et al. 2010; Thundathil et al. 2006) was co-localized with PLC ζ in the post-acrosomal region of capacitated bovine sperm (Thundathil et al. 2018). Furthermore, in somatic cells, the ATP1A1 subunit forms a signalling complex with PLC-γ1 and its effector IP3 receptors to form a scaffold (Yuan et al. 2005). In opossum kidney cells, stimulation of D1-like receptors coupled to Gsα proteins inhibited Na/K-ATPase activity, sequentially involving adenylyl cyclase-protein kinase A (AC-PKA) system and the PLC-PKA system (Gomes and Soares-da-Silva 2019). In addition, Ang-(1–7) [angiotensin-(1–7), a heptapeptide in heart and kidney with a role in maintaining renal homeostasis (Padda et al. 2015) induced inhibition of Na/K-ATPase activity that involved participation in a PI-PLC β pathway in MDCK cells (Lara et al. 2005). Moreover, PLC is activated by tyrosine phosphorylation in somatic cells (Kim et al. 1991; Sekiya et al. 2004; Tomes et al. 1996; Yu et al. 1998). Based on these reports, perhaps sperm-specific ATP1A4 activates PLC ζ by tyrosine phosphorylation. Furthermore, the activity of PIP2-PLC was higher in capacitated versus uncapacitated mouse sperm (Tomes et al. 1996). Therefore, we hypothesized that PLC ζ is activated by tyrosine phosphorylation during capacitation, and PLC activity contributes to bovine sperm capacitation.
Materials and methods
Preparation of reagents
Ouabain (100 μM) and heparin (1 mg/ml) stock solutions were prepared in sp-TALP (sperm Tyrode’s albumin lactate pyruvate) medium and stored at 4 °C. On the day of use, working solutions of ouabain (50 nM) and heparin (10 μg/ml) were prepared in final sp-TALP medium (1 mM pyruvate, 25 mM NaHCO3 and 2 mM Ca2 +), as described (Rajamanickam et al. 2017a). Stock solution of U73122 inhibitor (1.9 mM) was prepared by diluting it in dimethyl sulfoxide (DMSO) and storing it at −20 °C. Working solution of U73122 inhibitor (10 µM; Sigma-Aldrich, Oakville, ON, Canada) was prepared in final sp-TALP medium on the day of use.
Generation of bovine anti-PLC ζ antiserum
A bovine anti-PLC ζ antiserum against N-terminal amino acid sequences of PLC zeta 1 Bos taurus was developed in collaboration with Thermo Fisher Scientific, Antibody Services (Rockford, IL, USA). Bovine PLC zeta sequence (accession no. AAI14837; Fig. 1a) was used to identify a suitable N-terminal peptide sequence (RDDFKGGKITLEKALKLLEK) for peptide synthesis and immunization of rabbits. The antiserum from the terminal bleed was affinity-purified and both affinity-purified antiserum and rabbit pre-immune serum were used for the study.

Demonstration of the specificity of custom-made anti-PLC ζ antiserum. a Bovine PLC zeta 1 sequence (accession no. AAI14837), with the sequence used for antibody production highlighted in bold. b Sperm protein extract prepared by boiling sperm with sample buffer was electrophoresed, electrotransferred, and immunoblotted with a custom-made anti-PLC ζ antiserum. The anti-PLC ζ antiserum immunodetected protein bands at ~ 75, ~ 70, and ~ 30 kDa. However, anti-PLC ζ antiserum pre-adsorbed with its blocking peptide failed to recognize these bands, confirming its specificity. c The corresponding bands immunodetected by anti-PLC ζ antiserum were cut from a Coomassie blue-stained gel and subjected to mass spectrometry analysis. Amino acid sequences highlighted in bold represented bovine PLC ζ peptides (identified by mass spectrometry in the protein bands)
Confirmation of anti-PLC ζ antiserum specificity using blocking peptide and mass spectrometry analysis
The specificity of antiserum was confirmed using a blocking peptide (as described; Newton et al. 2009) and mass spectrometry analysis (as described; Ojaghi et al. 2017). In brief, a Percoll-washed fresh whole-sperm suspension (50 × 106/100 µL) was extracted by boiling with sample buffer and the resulting protein extracts were loaded on two 10% polyacrylamide gels and proteins resolved by SDS-PAGE. One gel was stored in TTBS at 4 °C, whereas the other gel was immunoblotted with custom-made anti-PLC ζ antiserum (1:10,000), and secondary goat anti-rabbit IgG, as described below. The gel stored at 4 °C was stained with Coomassie blue staining solution (50% methanol v/v, 40% dH2O v/v, 10% acetic acid v/v, 1 M Coomassie blue stain) on a rotating platform under the hood for a minimum of 2 h at room temperature. Gels were destained by soaking them in destaining solution (40% acetic acid v/v, 10% methanol v/v, 50% dH2O v/v) for 30 min on a rotating platform under the hood. The destaining solution was replaced with fresh solution once every 15 min. The protein bands corresponding to the immunoreactive bands detected by anti-PLC ζ antiserum were cut from the gel and sent for mass spectrometry analysis.
To confirm antiserum specificity, the immunoblotted membrane was stripped and re-probed with affinity-purified anti-PLC ζ antiserum pre-adsorbed to its blocking peptide. For this, affinity-purified antiserum (1.19 mg/ml) was incubated with its blocking peptide (BSA-peptide conjugate; 5 mg/ml) at a 1:2 dilution to provide a blocking peptide at a concentration approximately 10 times greater than the antiserum. The peptide and antiserum were incubated on a shaker at 4 °C overnight and used for immunoblotting, as described below.
Processing of fresh bull sperm for experiments
Semen samples were prepared as described (Ojaghi et al. 2017), with a slight modification. Fresh ejaculates from mature Holstein bulls were obtained from a local artificial insemination centre (Alta Genetics, Calgary, AB, Canada). Only samples with at least 70% progressively motile and 70% morphologically normal sperm were used. Semen was diluted 1:1 in sp-TALPH (sperm Tyrode’s albumin lactate pyruvate HEPES) and transported to the laboratory in a thermos maintained at 35 C. Percoll gradient (45–90%) washes of semen samples were done by centrifugation (700×g for 30 min). The resulting sperm pellet was washed with sp-TALPH to remove Percoll (400×g for 10 min, twice). The concentration of the resulting sperm pellet was determined using a hemocytometer and sperm concentration adjusted as required.
Sodium deoxycholate sperm protein extraction for immunodetection of ATP1A4
Sperm membrane proteins were extracted as described (Rajamanickam et al. 2017a). In brief, 500 µL of extraction buffer containing 250 mM sucrose, 50 mM imidazole, 1 mM EDTA, 0.1% sodium deoxycholate, pH 7.4, and 1× protease inhibitor (Roche Diagnostics, Mannheim, Germany) was incubated with 50 × 106 sperm on ice for 45 min, with occasional vortexing. Samples were centrifuged (10,000×g for 5 min at 4 °C) and the supernatant containing membrane proteins was used for immunoprecipitation of proteins interacting with ATP1A4.
Acetone precipitation of sperm proteins for immunoprecipitation of tyrosine phosphoproteins
Sperm proteins were extracted by re-suspending 50 × 106 sperm in 100 µL of 1× sp-TALPH, boiling with 25 µL of buffer containing 0.35 M DTT and 0.35 M SDS dissolved in 1 M Tris at 95 °C for 5 min and centrifuging at 10,000×g for 10 min. The resulting supernatant was subjected to acetone precipitation (Botelho et al. 2010) to remove SDS and DTT. Pre-chilled acetone (−20 °C) was added to the supernatant (four times the volume of supernatant), incubated at −20 °C for 1 h and centrifuged at 15,000×g for 15 min at 4 °C. The supernatant was removed and air dried to remove acetone (according to the ThermoFisher Scientific Co. protocol). The pellet was dissolved in 0.1% sodium deoxycholate buffer (prepared as described above) by sonication. The resulting protein mixture was used for immunoprecipitating tyrosine phosphoproteins.
SDS-PAGE, electrophoresis, and immunoblotting
Sperm protein extracts were incubated at 37 °C for 15 min (ATP1A4) or whole-sperm suspension (50 × 106 sperm/100 µL) boiled for 5 min (PLC ζ) with 25 μL sample buffer (5× sample buffer was prepared by dissolving 0.35 M DTT, 0.35 M SDS, and 1.8 mM Bromophenol Blue in 1 M Tris and mixing the resulting solution with glycerol, at a 1:1 ratio; Laemmli 1970) depending upon the experiment. These protein preparations were loaded on 10% polyacrylamide gel, electrophoresed, and electrotransferred at 100 V for 90 min. The nitrocellulose membranes were stained using 0.2% ponceau S in 3% acetic acid (to confirm protein transfer). Then, the membrane was blocked with 3% skim milk in 20 mM Tris-buffered saline containing Tween-20 (1 × TTBS) for 1 h and thereafter incubated with primary antibody [anti-ATP1A4 antiserum (custom-made at the University of Calgary, Calgary, AB, Canada; Newton et al. 2009) or anti-PLC ζ antiserum] at 4 °C overnight. After washing (in 1 × TTBS), the membrane was probed with secondary antibody conjugated to HRP (goat-rabbit IgG; Millipore, MA, USA) for 45 min. The membrane was washed and exposed to chemiluminescence reagents (prepared by mixing 5 mL 1.25 mM luminol, 50 μL 10% p-coumeric acid, 15 μL 3% hydrogen peroxide) and detected by capturing chemiluminescence on a Biorad Molecular Imager Chemi Doc™ XRS + imaging system.
Immunolocalization of ATP1A4 and PLC ζ in bull sperm
Percoll-washed sperm were adhered to poly-L-lysine-coated slides and fixed with 2.5% PFA for 15 min. Cells were permeabilized with Triton X-100 (0.1% prepared in PBS) for 20 min (in case of PLC ζ). Slides were washed in PBS and blocked with 10% normal chicken serum for 30 min and washed in PBS, then incubated with primary antibody [ATP1A4 (1:100) or PLC ζ (1:100)] diluted in 1% normal chicken serum for overnight at 4 C and slides were washed in PBS. Finally, preparations were incubated with chicken anti-rabbit Alexa 488 or chicken anti-rabbit cy3 antibody (1:1000 dilution in PBS; Santa Cruz, CA, USA) for 1 h at RT and washed in PBS. Sperm incubated with pre-immune serum and secondary antibody alone were used as controls. Slides were mounted with Vectashield mounting medium (Vector Laboratories Inc., Burlingame, CA, USA) containing DAPI and examined under epifluorescence microscopy (Leica DM 2500 or Zeiss Imager M2).
Immunoprecipitation
The protein A/G beads were washed three times (500× g, for 30 s at 4 °C). Then, the beads were conjugated with primary antibody by incubating at 4 °C for 1.5 h with slow agitation. After centrifugation at 500×g for 30 s at 4 C (× 3 times), the pellet was treated with 300 µL of sperm protein extracts. The contents were incubated for overnight at 4 °C with slow agitation. The preparations were centrifuged and thrice washed with 1% Tween in PBS at 500×g for 30 s at 4 °C. Finally, the pellet was prepared for immunoblotting by adding 100 µL of 1% Tween in PBS and 25 µL of sample buffer, then heated at 95 °C for 5 min and centrifuged at 10,000×g for 5 min, followed by SDS-PAGE, electrophoresis and immunoblotting, as described above.
Evaluation of the interaction of ATP1A4 and PLC ζ by immunoprecipitation
As described above, protein beads conjugated with anti-ATP1A4 antiserum (1:100 diluted in extraction buffer) or anti-PLC ζ antiserum (1:300 diluted in extraction buffer) were incubated with 300 µL of sperm protein extracts prepared from fresh and capacitated sperm, using sodium deoxycholate buffer, as described above. The final pellet obtained after washing was boiled with sample buffer and prepared for immunoblotting, as described above. The supernatant loaded was separated on 10% polyacrylamide gel, electrophoresed and electrotransferred and then blocked with skim milk. Thereafter, it was incubated with anti-PLC ζ antiserum (1:10,000 dilution in TTBS) or anti-ATP1A4 antiserum (1:4000), probed with secondary goat anti-rabbit IgG (1:4000 dilution in TTBS) and imaged for immunoreactive bands.
Evaluation of tyrosine phosphorylation of PLC ζ during sperm capacitation
As described above, protein beads conjugated with anti-phosphotyrosine antibody (1:300 diluted in extraction buffer; Millipore, Billerica, MA, USA) were incubated with 300 µL of sperm protein extracts prepared from fresh and capacitated sperm by boiling with sample buffer, followed by acetone precipitation of proteins, as described above. The final pellet obtained after washing was boiled with sample buffer and prepared for immunoblotting. The supernatant loaded was separated on 10% polyacrylamide gel, electrophoresed, electrotransferred, and blocked with skim milk. Thereafter, it was incubated with anti-PLC ζ antiserum (1:10,000 dilution in TTBS) and probed with secondary goat anti-rabbit IgG (1:4000 dilution in TTBS). Finally, blots were imaged to detect immunoreactive bands.
Sperm capacitation
Capacitation was done as described (Rajamanickam et al. 2017a). Briefly, capacitated sperm was prepared by incubating the Percoll-washed fresh sperm sample (40 × 106/mL) in sp-TALP at 39 °C and 5% CO2 for 4 h under high humidity, with 50 nM ouabain as the capacitating agent. Capacitation status was confirmed by comparing tyrosine phosphorylation content of proteins among experimental groups by immunoblotting and evaluating sperm motility with computer-assisted sperm analysis (CASA), as described below.
Evaluation of capacitation status of sperm based on sperm motility patterns, acrosome reaction, and tyrosine phosphoprotein content
CASA (Sperm Vision Minitube, Canada) was used to evaluate motility. An aliquot (4 μL) from each experimental group was loaded into prewarmed (37 °C) Leija slide (Nieuw-Vennep, Netherlands) and seven fields per sample analysed using the bovine sperm motility program. Proportions of hyperactivated sperm were compared among experimental groups. All treatment groups were analysed for their ability to undergo an acrosome reaction. For this, 50 μL of sperm sample (40 × 106/mL) was incubated with either 100 μL /ml LPC (lysophosphatidylcholine; Sigma-Aldrich, Oakville, ON, Canada) or TALP alone (negative control) for 30 min at 39 °C, in 5% CO2 and high humidity. Then, 20 μL of diluted sperm from each treatment group was used to prepare smears and fixed using 100% ethanol (at −20 °C for 2 min). The smears were dried and stained with 20 μL of FITC-PSA (100 μL/mL; Sigma-Aldrich, Oakville, ON, Canada) for 10 min in a humidified chamber in a dark room (Galantino-Homer et al. 1997). Slides were washed and examined under a fluorescent microscope and 100 sperm per slide were evaluated for acrosomal integrity (acrosome-intact or acrosome-reacted). For evaluation of tyrosine phosphorylation of proteins, ouabain-capacitated sperm samples, along with the controls, were concentrated (10,000×g for 3 min at RT) and the pellet was washed (10,000×g for 5 min at RT) in 1 mL PBS containing 0.2 mM sodium vanadate (Na2VO3). The resulting pellet was boiled with sample buffer (containing 1 M Na2VO3) to extract total protein and used for immunoblotting with anti-phosphotyrosine antibody (1:10,000 dilution in TTBS). The membrane was stripped and re-probed with a monoclonal anti-β-tubulin antibody (1:10,000 dilution in TTBS; Sigma-Aldrich, Oakville, ON, Canada) and used for evaluating equal loading of proteins from various experimental groups.
Quantification of F-actin in bull sperm
A flow cytometry-based approach was used for quantification of F-actin formation in bull sperm during ouabain-induced capacitation. The experiment was done as described (Rajamanickam et al. 2017c), with modifications. The experimental design included sperm (20 × 106 sperm/mL) incubated ± PLC inhibitor (U73122; 10 μM) during ouabain (50 nM) mediated capacitation, plus control groups [(fresh uncapacitated sperm (designated as fresh) and sperm incubated in sp-TALP at 39 °C, 5% CO2 under high humidity for 4 h (incubation control)]. Sperm samples were treated with protamine sulfate (20 µg/mL) to detach agglutinated sperm and facilitate a single-cell suspension. All groups were subsequently washed in PBS to remove capacitation-associated reagents. Then, 1 µL of fixable live and dead cell stain was added to the sperm suspension and incubated for 30 min at RT. Sperm was fixed with 2.5% PFA for 15 min and then permeabilized with 0.5% Triton for 30 min. Alexa-488 FITC-Phalloidin stain (F-actin probe) was incubated (1:100) for 1 h at RT, and data were acquired using a BD LSR II cytometer (BD Biosciences, Mississauga, ON, Canada). The excitation source was a diode pumped solid state (DPSS) 488 nm laser. Voltage settings (log scale) used were as follows: FSC, 320; SSC, 180; FITC, 790; violet, 400. Negative control or auto fluorescent control (cells only) was used to adjust voltages and gates, whereas single-color controls (violet and Alexa 488) were used for compensation to minimize overlap of violet fluorescence detected in the green channel. Subsequently, Detector 1 (emission range of 450 ± 25 nm) was used for detecting violet fluorescence (viability status), whereas Detector 2 (emission range of 530 ± 15 nm) was used for detecting green (F-actin) fluorescence. A total of 20 × 103 events was recorded for each group in the form of a scatter plot and histogram. The resulting flow cytometric data were analysed by computing relative median fluorescence intensity (MFI) of each sample.
Statistical analyses
All statistical analyses were done in R studio software. Data were not normally distributed when assessed using a histogram. Therefore, we used a non-parametric multiple comparison test (Kruskal–Wallis). The F-actin content, phosphotyrosine content, percentage hyperactived motility, and acrosome reaction were analyzed by a Kruskal–Wallis test, followed by a Dunn test for multiple comparisons of groups. For all analyses, P < 0.05 was considered significant. ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to quantify the pixel intensities of the bands in immunoblots and GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA) was used for generating bar graphs. Sample size calculations for F-actin quantification and hyperactivated motility was done using OpenEpi Version 3.01 software.
Results
Generation of a custom-made anti-PLC ζ antiserum
Affinity-purified PLC ζ antiserum developed against a N-terminal sequence (RDDFKGGKITLEKALKLLEK; accession no. AAI14837) of 1-phospholipase C zeta of Bos taurus identified specific bands (∼ 75, ~ 70, and ~ 30 kDa) from bull sperm protein extracts (Fig. 1b). Re-probing the same membrane with anti-PLC ζ antiserum pre-adsorbed to its blocking peptide failed to detect the same protein bands (Fig. 1b), demonstrating antiserum specificity. Using mass spectrometry (LC/MS), we confirmed that ~ 75, ~ 70, and ~ 30 kDa protein bands included PLC ζ (Fig. 1c).
Expression of PLC ζ and ATP1A4 in bull sperm
Immunolocalization experiments determined that PLC ζ was expressed in the acrosomal region of fresh bull sperm. Control groups incubated with pre-immune serum and secondary antibody alone without primary antibody lacked a fluorescent signal. DAPI (blue) was used to counterstain the nucleus. PLC ζ was redistributed to post-acrosomal region of capacitated sperm (Fig. 2a, g). Furthermore, ATP1A4 was immunolocalized to the entire sperm head in fresh sperm, but only to the post-acrosomal region of capacitated sperm (Fig. 2i, j).

Expression of PLC ζ and ATP1A4 in bull sperm. Sperm were adhered to poly-L-lysine coated slides. Immunofluorescence staining was done using Alexa-fluor 488 or Cy3 after probing the sperm with anti-PLC ζ antiserum or anti-ATP1A4, respectively, with nucleus counterstained by DAPI (blue). PLC ζ was localized to acrosomal region of fresh sperm a Alexa-fluor 488 and counterstained with DAPI; b staining with Alexa-fluor alone and post-acrosomal region of capacitated sperm g Alexa-fluor 488 and counterstained with DAPI; h staining with Alexa-fluor alone. Negative controls included sperm incubated with pre-immune serum or secondary antibody alone d, f, respectively with corresponding DAPI-stained fields c, e, respectively. ATP1A4 was immunolocalized to entire fresh sperm head i and to the post-acrosomal region in capacitated sperm j
ATP1A4 and PLC ζ interact during capacitation
Immunoprecipitation of sodium deoxycholate extracted sperm protein with anti-ATP1A4 antiserum and immunoblotting with anti-PLC ζ antiserum demonstrated a specific immunoreactive band of PLC ζ at ~ 75 kDa. The positive control (sample buffer-treated total sperm protein extract) had multiple bands of PLC ζ at ~ 75, ~ 70, and ~ 30 kDa (indicated with arrows; Fig. 3a). Conversely, when anti-PLC ζ antiserum was used for immunoprecipitation and anti-ATP1A4 antiserum was used for immunoblotting, an immunoreactive band corresponding to ATP1A4 was identified at ~ 110 kDa (Fig. 3b).

Interaction of ATP1A4 and PLC ζ during sperm capacitation. a Sodium deoxycholate sperm protein extract was immunoprecipitated with a custom anti-ATP1A4 antiserum and immunoblotted with a custom anti-PLC ζ antiserum. Fresh and ouabain-capacitated sperm demonstrated PLC ζ at ~ 75 kDa; positive control (total sperm protein extract). b Sodium deoxycholate sperm protein extract was immunoprecipitated with custom anti-PLC ζ antiserum and immunoblotted with custom-made anti-ATP1A4 antiserum. Fresh sperm and ouabain-capacitated sperm demonstrated ATP1A4 at ~ 110 kDa; positive control (sodium deoxycholate sperm protein extract). Negative control (anti-PLC ζ antiserum or anti-ATP1A4 antiserum conjugated with protein A beads without sperm protein extract) demonstrated only HC and LC; HC: heavy chain, ~ 50 kDa; LC: light chain, ~ 25 kDa
PLC ζ undergoes tyrosine phosphorylation during sperm capacitation
Our immunoprecipitation studies demonstrated that PLC ζ (~ 75 kDa) underwent tyrosine phosphorylation and the content of tyrosine phosphorylated PLC ζ was higher in ouabain-capacitated sperm compared to uncapacitated fresh sperm (Fig. 4a, b).

Tyrosine phosphorylation of PLC ζ during capacitation. Proteins were acetone-precipitated from sperm extracts prepared by boiling sperm with sample buffer and used for immunoprecipitation. a Tyrosine phosphoproteins immunoprecipitated with antiphosphotyrosine antibody and immunoblotted with anti-PLC ζ antiserum demonstrated PLC ζ ~ 75 kDa from fresh and capacitated sperm; positive control (acetone-precipitated sperm protein extract); negative control (anti-phosphotyrosine antibody conjugated with protein A beads without sperm protein extract); HC: heavy chain, ~ 50 kDa; LC: light chain, ~ 25 kDa. b Relative pixel intensities of PLC ζ bands from fresh and capacitated sperm were quantified after normalizing the data with β- tubulin. abp < 0.05; n = 3
PLC activity required for hyperactivated motility and achieving the ability to undergo an acrosome reaction
The percentage of sperm with hyperactivated motility (Fig. 5a) in the ouabain-capacitated group was higher (16.4%; p < 0.05) than in the control groups: uncapacitated sperm (fresh); sperm incubated in sp-TALP for 4 h (3.7 or 10.5%, respectively), or the group preincubated with U73122, followed by ouabain capacitation (4.5%). The ability of sperm to undergo an acrosome reaction in the group preincubated with PLC inhibitor U73122, followed by incubation under capacitating conditions, was lower (21.3%; p < 0.05) than the ouabain-capacitated group (53.7%; Fig. 5b).

Involvement of PLC ζ in regulation of sperm capacitation. Percoll-washed sperm preparations were incubated in medium alone for 0 (fresh) or 4 h with or without ouabain (incubation control); or preincubated with U73122, followed by incubation with ouabain for 4 h at 39 C in 5% CO2 under high humidity. These experimental groups were incubated in triplicate and processed concurrently to assess hyperactivation and tyrosine phosphoprotein content. a Hyperactivated motility, assessed using computer-assisted sperm analysis (CASA). Percentage hyperactivated motility was calculated based on amplitude of lateral head displacement > 7 μm, linearity < 60%, and curvilinear velocity > 120 μm. a−cValues without a common superscript differed (p < 0.05, n = 3). b Acrosome reaction test was induced using LPC and percentage acrosome-reacted sperm from each experimental group was calculated. a,bValues without a common superscript differed (p < 0.05, n = 3). c Total sperm proteins extracted by boiling with sample buffer were immunoblotted with antiphosphotyrosine antibody (upper panel) and re-probed with anti-β-tubulin antibody (lower panel) for equal protein loading. c Pixel intensities for 270, 250, 100, 75, and 45 kDa phosphotyrosine bands (indicated by arrows) were quantified and normalized to corresponding β-tubulin and compared among the groups. Zero values along the x-axis represent groups for which pixel intensity could not be detected in the immunoblots. a−cWithin a band, pixel intensity values without a common superscript differed (p < 0.05, n = 3)
PLC activity required for tyrosine phosphorylation of sperm proteins during capacitation
Phosphotyrosine content of sperm proteins in the group preincubated with the PLC inhibitor U73122, followed by incubation under capacitating conditions, was lower (p < 0.05) than that of the ouabain-capacitated group (Fig. 5c, d).
F-actin content increased in sperm under capacitating conditions
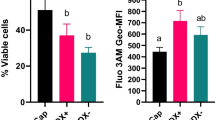
Flow cytometry-based quantification of F-actin content (Fig. 6a) demonstrated that content of F- actin increased during capacitation (Fig. 6b, c).

F-actin quantification in sperm using flow cytometry. a P1: total cells analyzed; P3: live cells; P4: dead cells; P5: F-actin fluorescence from viable sperm cells; Q3 and Q4: viable sperm cells with low and high F-actin fluorescence, respectively; Q1 and Q2: dead sperm cells with low and high F-actin fluorescence, respectively. b Represents fluorescence intensity histogram from uncapacitated (fresh) and ouabain-capacitated sperm. A shift towards the right represents the increase in the content of F-actin evidenced by increased fluorescence intensity. c The sperm sample from the experimental groups; uncapacitated group (after percoll wash) and rest of the experimental group (at end of capacitation) were stained with FITC-Phalloidin stain (F-actin probe) and relative median fluorescence intensity was quantified using flow cytometry. The experiment was replicated using four samples of bull semen. a,bValues without a common superscript differed (p < 0.05)
PLC activity required for F-actin formation under capacitating conditions in sperm
Ouabain-capacitated sperm had increased F-actin content and ouabain-capacitated sperm preincubated with U73122 (PLC inhibitor) had decreased F-actin content, as evidenced by the histogram on the FITC log scale on the x-axis (Fig. 7a, b).

Involvement of PLC ζ in F-actin formation during capacitation. a Sperm samples from the experimental groups were stained with FITC-Phalloidin stain (F-actin probe) and relative median fluorescence intensity was quantified using flow cytometry. a−cValues without a common superscript differed (p < 0.05). The experiment was replicated with semen samples from four bulls. b Fluorescence intensity histogram from each group. The shift of the histogram towards right represents an increase in the F-actin content, as indicated by an increase in FITC-Phalloidin fluorescence intensity
Discussion
Bovine PLC ζ was expressed in acrosomal region of fresh bull sperm and localized to the post-acrosomal region of capacitated sperm. The IP studies confirmed interaction of PLC ζ with ATP1A4 and tyrosine phosphorylation of PLC ζ during ouabain-mediated capacitation. Unfortunately, we could not quantify a capacitation-associated change in PLC activity, due to the lack of a suitable assay. However, based on studies from somatic cells (Li et al. 2009; Yu et al. 1998) and our previous study (Rajamanickam 2017b), we expected that capacitation-associated tyrosine phosphorylation of PLC ζ upregulates its activity through a mechanism involving ATP1A4, EGFR, which is critical for the successful completion of tyrosine phosphorylation of a cohort of sperm proteins, hyperactivated motility, F-actin formation and ability to undergo an acrosome reaction. This study provided evidence for the potential involvement of PLC ζ in the regulation of these processes.
A commercial PLC ζ antibody was available from MyBiosource Co., San Diego, CA, USA. The antibody was developed in rabbits against the N-terminal sequence of human PLC ζ. Using mass spectrometry, we confirmed that the protein bands detected by the antibody were PLC ζ. However, potency of this antibody was inconsistent following storage. Therefore, we generated an affinity purified PLC ζ antiserum in rabbit against the N-terminal sequence (RDDFKGGKITLEKALKLLEK; accession no. AAI14837) of bovine PLC ζ and used it in our subsequent experiments. The antiserum detected immunoreactive protein bands at ~ 75, ~ 70, and ~ 30 kDa from total sperm protein extracts prepared by boiling sperm with sample buffer. The X–Y catalytic domain, the linker region of PLC, is susceptible to proteolysis (Ellis et al. 1993; Fernald et al. 1994). The lower molecular bands (~ 70 and ~ 30 kDa) may have been due to proteolytic degradation (Kurokawa et al. 2007) of PLC ζ. Moreover, immunoblotting using this antiserum pre-adsorbed to the peptide sequence used for developing the antiserum (blocking peptide) failed to detect the above-described protein bands, confirming antiserum specificity. Furthermore, mass spectrometry analysis confirmed these immunoreactive bands (~ 75, ~ 70, and ~ 30 kDa) identified by our custom anti-PLC ζ antiserum also included PLC ζ.
We were unable to demonstrate PLC ζ from sperm protein extracts prepared with sodium deoxycholate detergent, a detergent used by our laboratory for extracting ATP1A4 from sperm. Similarly, cytosolic sperm extracts prepared using sperm buffer (containing 75 mM KCl and 1 mM DTT) and high-pH soluble sperm extracts prepared using alkaline carbonate (100 mM Na2CO3, pH 11.5) from pig sperm with PLC activity failed to detect 72 kDa immunoreactive bands of PLC ζ (Kurokawa et al. 2005, 2007). However, a specific PLC ζ band was demonstrated at ~ 75 kDa by IP of sperm proteins from sodium deoxycholate-extracted sperm proteins. Extracting PLC ζ is difficult, due to its unique localization to the PT of sperm (Fujimoto et al. 2004). However, IP could concentrate low-abundant protein from protein extracts (Michielsen et al. 2005), demonstrating PLC ζ in the immunoprecipitate extracted through this approach. Briefly, sodium deoxycholate extracted sperm proteins were incubated with anti-PLC ζ antibody conjugated with beads, resulting in immunoprecipitation of PLC ζ.
There are differences among species in distribution of PLC ζ in sperm: in acrosomal, equatorial, and post-acrosomal regions of head and in the tail region of human sperm (Kashir et al. 2011a, 2011b, 2012, 2013; Yoon et al. 2008); in acrosomal and post-acrosomal regions of murine sperm (Fujimoto et al. 2004); in acrosomal and post-acrosomal regions and tail region of porcine sperm (Fujimoto et al. 2004; Kaewmala et al. 2012; Yoneda et al. 2006); in acrosomal, equatorial segment, mid-piece, as well as principle piece of flagellum of equine sperm (Bedford-Guaus et al. 2011); and in equatorial region of bovine sperm (Mejía-Flores et al. 2017). In the present study, PLC ζ was detected in the acrosomal region of fresh bovine sperm, using immunolocalization and an affinity-purified antiserum developed in rabbits. The pre-immune serum used as a control failed to detect any similar pattern, indicating the specificity of this antibody. This differed from previous reports (Mejía-Flores et al. 2017), but the pattern was more consistent with reports from other species. Variation in the localization of PLC ζ was reported in murine (Fujimoto et al. 2004; Young et al. 2009) and human sperm (Grasa et al. 2008; Kashir et al. 2011a, 2011b, 2013; Yoon et al. 2008) and capacitation-associated re-localization of this protein was reported in both of these species (Grasa et al. 2008; Young et al. 2009). Similarly, our results demonstrated re-localization of PLC ζ to the post-acrosomal region in capacitated bovine sperm. Consistent with our previous reports (Newton et al. 2010; Rajamanickam et al. 2017a), ATP1A4 was immunolocalized to the entire head in fresh sperm. Moreover, PLC ζ and ATP1A4 was co-localized to the post-acrosomal region in capacitated sperm (Thundathil et al. 2018). Similarly, re-localization of phospho-tyrosine containing protein (Cormier and Bailey 2003), heat shock protein (Kamaruddin et al. 2004), ATP1A4 (Newton et al., 2010), and tACE (Ojaghi et al. 2017) occurs during capacitation in bovine sperm. However, underlying mechanisms of protein redistribution remain unknown. During synthesis, proteins will be targeted to specific locations (Counillon and Pouyssegur 2000; Hubbard et al. 1989) through various mechanisms, including passive diffusion with trapping and active translocation or active transport directed by attachment of membrane proteins to actin cytoskeleton (Cowan et al. 1991). The protein, which has binding sites on actin filaments, can move by indirectly binding to them with intermittent attachments to glycoproteins (Cowan et al. 1991; Kucik et al. 1989; Ouyang et al. 2005). The lipid raft as a molecular protein transport system has reported for heat shock protein (HSP70); it is transported to the lipid droplet, then folded on to the lipid monolayer and transported across the membrane (Elmallah et al. 2020). These suggested mechanisms could facilitate redistribution of sperm proteins during capacitation.
The molecular mechanisms of oocyte activation by PLC ζ are clearly defined. PLC ζ from sperm, when released into the oocyte, activates the PIP2 pathway, resulting in increased intracellular calcium, leading to calcium oscillation and oocyte activation (Fissore et al. 1992; Nomikos et al. 2011; Swann et al. 2006; Xu et al. 1994). However, the immediate oocyte activation induced by PLC ζ in sperm following oocyte penetration suggests its potential activation during sperm capacitation. Consistent with this hypothesis, the activity of PIP2-PLC was higher in capacitated versus uncapacitated mouse sperm (Tomes et al. 1996). However, molecular mechanisms of capacitation-associated PLC ζ activation are unknown.
In somatic cells, Na+/K+-ATPase and PLC interaction (Gomes and Soares-da-Silva 2019; Lara et al. 2005; Yuan et al. 2005) leads to its activation by tyrosine phosphorylation (Wang et al. 2004; Yuan et al. 2005). Again, PLC is activated by tyrosine phosphorylation (Rodríguez-Fragoso et al. 2009; Wahl et al. 1989; Yuan et al. 2005) by EGFR, which in turn is activated by Src (Tice et al. 1999; Liu et al. 2004; Nair and Sealfon 2003). Furthermore, Src is activated by ouabain-mediated Na+/K+-ATPase signaling complex (Wang et al. 2004). In addition, ouabain-induced activation of EGFR (Rajamanickam et al. 2017b) and involvement of EGFR in the activation of PLC (Finkelstein et al. 2010) have been reported in sperm during capacitation. Furthermore, tyrosine phosphorylation of PLC is involved in several cellular processes, including chemotaxis, cell proliferation and migration (Asokan et al. 2014; de Gorter et al. 2007; Jones et al. 2005; Kim et al. 1991). Our immunoprecipitation studies confirmed the interaction of ATP1A4 and PLC ζ in bovine sperm. Based on studies from somatic cells and previous research from our lab (Thundathil et al. 2018), we hypothesized that PLC ζ is activated by tyrosine phosphorylation and its activation promotes capacitation-associated biochemical changes in sperm. To test this hypothesis, we used immunoprecipitation (IP) studies to evaluate tyrosine phosphorylation of PLC ζ during capacitation. In that regard, since SDS, a component of the sample buffer, interfered with the IP experiments, we used acetone to precipitate sperm proteins from sample buffer-extracted sperm proteins, as described (Botelho et al. 2010). As expected, our IP results demonstrated tyrosine phosphorylation of PLC ζ in capacitated sperm. Furthermore, mass spectrometry analysis of the bands from SDS-PAGE gel (~ 75, ~ 70, and ~ 30 kDa) had specified variable modifications (mascot best match using possible arrangements of modifications that may or may not be present; www.matrixscience.com) at phospho groups of serine, threonine, and tyrosine in the peptide. In somatic cells, PLC γ1 isoform undergoes phosphorylation on tyrosine residues Try-771, 783, 1253, 1254 (Kim et al. 1991; Sekiya et al. 2004). However, further studies are required to identify specific tyrosine phosphorylation sites on PLC ζ. Although PLC ζ and ATP1A4 interacts and tyrosine phosphorylation of PLC ζ occurs during capacitation, further studies are required to confirm involvement of an ATP1A4-mediated mechanism in tyrosine phosphorylation and activation of PLC ζ. However, based on information from other cell systems, ATP1A4 signalling initiated by ouabain interaction leads to EGFR activation, tyrosine phosphorylation of PLC ζ and its activation during capacitation.
We investigated involvement of PLC activity in tyrosine phosphorylation of sperm proteins, hyperactivation, ability to undergo acrosome reaction, and F-actin formation during capacitation. Pre-incubation of sperm with the PLC inhibitor U73122 (Alonso et al. 2017), followed by induction of capacitation using 50 nM ouabain, inhibited phosphotyrosine content of a cohort of sperm proteins (45 to 270 kDa range); proportion of sperm undergoing hyperactivated motility and acrosome reaction; and actin polymerization. The direct involvement of PLC in tyrosine phosphorylation of other proteins remains unknown. Regardless, PLC contributes to the upstream regulation in activation of PKC, which triggers activation of multiple signalling pathways involved in tyrosine phosphorylation of proteins (Thundathil et al. 2012). Therefore, we inferred that an increase in PLC activity contributed to an increase in tyrosine phosphoprotein content of sperm during capacitation, through the above-described mechanisms. This interpretation was further supported by the finding that the presence of a PLC inhibitor during capacitation decreased tyrosine phosphoprotein content of sperm proteins.
Actin (G-actin monomer) is present in the sperm head, connecting piece in the neck or tail regions, with species-specific variations in their location (Flaherty et al. 1998; Fouquet et al. 1992). The major location of F-actin in mammalian species is in the sub-acrosomal region (Clarke et al. 1982; Fouquet et al. 1990; Peterson et al. 1990). F-actin, present in the flagellum of guinea pig sperm, is involved in sperm motility (Azamar et al. 2007). Furthermore, gelsolin, an actin-severing protein is translocated to the sperm head during capacitation. As gelsolin prevented actin polymerization, this translocation facilitated an increase in F-actin in sperm tail during capacitation essential for hyperactivated motility (Breitbart and Finkelstein 2015; Itach et al. 2012). In somatic cells, tyrosine phosphorylation of PLC γ1 mediated by growth factor receptor has an important role in cytoskeletal (actin) organization (Yu et al. 1998). Perhaps activation of PLC by tyrosine phosphorylation is involved in increased F-actin formation in the sperm tail and head; the former contributes to hyperactivated motility and the latter prevents spontaneous acrosome reaction. Consistent with this hypothesis, there was reduced hyperactivated motility and decreased acrosome reaction when PLC inhibitor was used during capacitation, suggesting the involvement of PLC activity in the regulation of hyperactivated motility and acrosome reaction.
We used a flow cytometry-based approach to quantify F-actin content in capacitated sperm. There was significant decrease in F-actin content after pre-incubation of sperm with the PLC inhibitor U73122 (Alonso et al. 2017), followed by induction of capacitation using 50 nM ouabain. Phosphorylation-related activation of PLC by the signalling complex of Na+/K+-ATPase-EGFR through ouabain interaction (Haas et al. 2000; Ullrich and Schlessinger 1990) results in PIP2 pathway activation, which in turn activates PKC. Polymerization of G-actin to F-actin is facilitated by PKC through other mediator proteins (PLD, CaMKII; Rajamanickam et al. 2017b). The capacitation-associated increase in F-actin content and the inhibition of this process in presence of a PLC inhibitor implicated a capacitation-associated increase in PLC activity and its involvement in F-actin formation.
Altogether, based on the above studies, we inferred that PLC activity is crucial for capacitation. Since the PLC inhibitor used in this study was not specific for PLC ζ and other PLC isoforms are likely to be present in bull sperm as reported in the mouse (Choi et al. 2001; Fukami et al. 2003) and boar (Parrington et al. 2002), specific contributions of PLC ζ to bull sperm capacitation remain unknown. Therefore, further studies using knockout models are required to confirm the role of PLC ζ activity in this process and elucidate the functional significance of localization of PLC ζ in the acrosomal region of bull sperm, as reported in other species (Young et al. 2009).
In conclusion, this study established the relevance of PLC family during capacitation. We inferred that ATP1A4 in sperm interacts with ouabain and results in the formation of a signal plex with EGFR, followed by activation of Src, which in turn results in tyrosine phosphorylation and activation of PLC ζ. This activation increased PLC activity, contributing to upregulation of capacitation-associated biochemical modifications such as tyrosine phosphorylation of proteins, hyperactivated motility, acrosome reaction, and F-actin formation. In addition, this increase in capacitation-associated PLC activity may be relevant for oocyte activation immediately following sperm penetration of the oocyte.
References
Aarabi M, Balakier H, Bashar S, Moskovtsev SI, Sutovsky P, Librach CL, Oko R (2014) Sperm-derived WW domain-binding protein, PAWP, elicits calcium oscillations and oocyte activation in humans and mice. Faseb J 28:4434–4440
Alonso C, Osycka-Salut CE, Castellano L, Cesari A, Di Siervi N, Mutto A, Johannisson A, Morrell JM, Davio C, Perez-Martinez S (2017) Extracellular cAMP activates molecular signalling pathways associated with sperm capacitation in bovines. Mol Hum Reprod 23:521–534
Alvau A, Battistone MA, Gervasi MG, Navarrete FA, Xu X, Sánchez-Cárdenas C, De la Vega-Beltran JL, Da Ros VG, Greer PA, Darszon A, Krapf D, Salicioni AM, Cuasnicu PS, Visconti PE (2016) The tyrosine kinase FER is responsible for the capacitation-associated increase in tyrosine phosphorylation in murine sperm. Dev 143:2325–2333
Aguila L, Felmer R, Arias ME, Navarrete F, Martin-Hidalgo D, Lee HC, Visconti P, Fissore R (2017) Defective sperm head decondensation undermines the success of ICSI in the bovine. Reprod 154:307–318
Asokan SB, Johnson HE, Rahman A, King SJ, Rotty JD, Lebedeva IP, Haugh JM, Bear JE (2014) Mesenchymal chemotaxis requires selective inactivation of myosin II at the leading edge via a noncanonical PLCγ/PKCα pathway. Dev Cell 31:747–760
Azamar Y, Uribe S, Mújica A (2007) F-actin involvement in guinea pig sperm motility. Mol Reprod Dev 74:312–320
Bedford-Guaus SJ, McPartlin LA, Xie J, Westmiller SL, Buffone MG, Roberson MS (2011) Molecular cloning and characterization of phospholipase C zeta in equine sperm and testis reveals species-specific differences in expression of catalytically active protein. Biol Reprod 85:78–88
Botelho D, Wall MJ, Vieira DB, Fitzsimmons S, Liu F, Doucette A (2010) Top-down and bottom-up proteomics of SDS-containing solutions following mass-based separation. J Proteome Res 9:2863–2870
Breitbart H, Finkelstein M (2015) Regulation of sperm capacitation and the acrosome reaction by PIP 2 and actin modulation. Asian J Androl 17:597–600
Choi D, Lee E, Hwang S, Jun K, Kim D, Yoon BK, Shin HS, Lee JH (2001) The biological significance of phospholipase C beta 1 gene mutation in mouse sperm in the acrosome reaction, fertilization, and embryo development. J Assist Reprod Genet 18:305–310
Clarke GN, Clarke FM, Wilson S (1982) Actin in human spermatozoa. Biol Reprod 26:319–327
Cormier N, Bailey JL (2003) A differential mechanism is involved during heparin- and cryopreservation-induced capacitation of bovine spermatozoa. Biol Reprod 69:177–185
Counillon L, Pouysségur J (2000) The expanding family of eucaryotic Na (+)/H (+) exchangers. J Biol Chem 275:1–4
Cowan AE, Myles DG, Koppel DE (1991) Migration of the guinea pig sperm membrane protein PH-20 from one localized surface domain to another does not occur by a simple diffusion-trapping mechanism. Dev Biol 144:189–198
Cox LJ, Larman MG, Saunders CM, Hashimoto K, Swann K, Lai FA (2002) Sperm phospholipase C zeta from humans and cynomolgus monkeys’ triggers Ca2+ oscillations, activation and development of mouse oocytes. Reproduction 124:611–623
de Gorter DJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, Pals ST, Spaargaren M (2007) Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immun 26:93–104
Elmallah MIY, Cordonnier M, Vautrot V, Chanteloup G, Garrido C, Gobbo J (2020) Membrane-anchored heat-shock protein 70 (Hsp70) in cancer. Cancer Lett 469:134–141
Ellis MV, Carne A, Katan M (1993) Structural requirements of phosphatidylinositol-specific phospholipase C delta 1 for enzyme activity. Eur J Biochem 213:339–347
Escoffier J, Yassine S, Lee HC, Martinez G, Delaroche J, Coutton C, Karaouzène T, Zouari R, Metzler-Guillemain C, Pernet-Gallay K, Hennebicq S, Ray PF, Fissore R, Arnoult C (2015) Subcellular localization of phospholipase Cζ in human sperm and its absence in DPY19L2-deficient sperm are consistent with its role in oocyte activation. Mol Hum Reprod 21:157–168
Fernald AW, Jones GA, Carpenter G (1994) Limited proteolysis of phospholipase C-gamma 1 indicates stable association of X and Y domains with enhanced catalytic activity. Biochem J 302:503–509
Finkelstein M, Etkovitz N, Breitbart H (2010) Role and regulation of sperm gelsolin prior to fertilization. J Biol Chem 285:39702–39709
Fissore RA, Dobrinsky JR, Balise JJ, Duby RT, Robl JM (1992) Patterns of intracellular Ca2+ concentrations in fertilized bovine eggs. Biol Reprod 47:960–969
Flaherty SP, Winfrey VP, Olson GE (1998) Localization of actin in human, bull, rabbit, and hamster sperm by immunoelectron microscopy. Anat Rec 221:599–610
Fouquet JP, Kann ML, Dadoune JP (1990) Immunoelectron microscopic distribution of actin in hamster spermatids and epididymal, capacitated and acrosome-reacted spermatozoa. Tissue Cell 22:291–300
Fouquet JP, Kann ML (1992) Species-specific localization of actin in mammalian spermatozoa: fact or artifact? Microsc Res Tech 20:251–258
Fujimoto S, Yoshida N, Fukui T, Amanai M, Isobe T, Itagaki C, Izumi T, Perry AC (2004) Mammalian phospholipase C zeta induces oocyte activation from the sperm perinuclear matrix. Dev Biol 274:370–383
Fukami K, Yoshida M, Inoue T, Kurokawa M, Fissore RA, Yoshida N, Mikoshiba K, Takenawa T (2003) Phospholipase Cdelta4 is required for Ca2+ mobilization essential for acrosome reaction in sperm. J Cell Biol 161:79–88
Galantino-Homer HL, Visconti PE, Kopf GS (1997) Regulation of protein tyrosine phosphorylation during bovine sperm capacitation by a cyclic adenosine 3’5’-monophosphate-dependent pathway. Biol Reprod 56:707–719
Gomes P, Soares-da-Silva P (2019) Role of cAMP-PKA-PLC signaling cascade on dopamine-induced PKC-mediated inhibition of renal Na(+)-K(+)-ATPase activity. Am J Physiol Renal Physiol 282:1084–1096
Grasa P, Coward K, Young C, Parrington J (2008) The pattern of localization of the putative oocyte activation factor, phospholipase C zeta, in uncapacitated, capacitated, and ionophore-treated human spermatozoa. Hum Reprod 23:2513–2522
Grasa P, Colas C, Gallego M, Monteagudo L, Muiño-Blanco T, Cebrián-Pérez JA (2009) Changes in content and localization of proteins phosphorylated at tyrosine, serine and threonine residues during ram sperm capacitation and acrosome reaction. Reprod 137:655–667
Haas M, Askari A, Xie Z (2000) Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem 275:27832–27837
Hara H, Abdalla H, Morita H, Kuwayama M, Hirabayashi M, Hochi S (2011) Procedure for bovine ICSI, not sperm freeze-drying, impairs the function of the Microtubule-Organizing Center. J Reprod Dev 57:10–14
Heytens E, Parrington J, Coward K, Young C, Lambrecht S, Yoon SY, Fissore RA, Hamer R, Deane CM, Ruas M, Grasa P, Soleimani R, Cuvelier CA, Gerris J, Dhont M, Deforce D, Leybaert L, De Sutter P (2009) Reduced amounts and abnormal forms of phospholipase C zeta (PLCzeta) in spermatozoa from infertile men. Hum Reprod 24:2417–2428
Hubbard AL, Stieger B, Bartles JR (1989) Biogenesis of endogenous plasma membrane proteins in epithelial cells. Annu Rev Physiol 51:755–770
Itach SB, Finklestein M, Etkovitz N, Breitbart H (2012) Hyper-activated motility in sperm capacitation is mediated by phospholipase D-dependent actin polymerization. Dev Biol 362:154–161
Ito M, Shikano T, Oda S, Horiguchi T, Tanimoto S, Awaji T, Mitani H, Miyazaki S (2008) Difference in Ca2+ oscillation-inducing activity and nuclear translocation ability of PLCZ1, an egg-activating sperm factor candidate, between mouse, rat, human, and medaka fish. Biol Reprod 78:1081–1090
Jimenez T, Sánchez G, Blanco G (2012) Activity of the Na, K-ATPase α4 isoform is regulated during sperm capacitation to support sperm motility. J Androl 33:1047–1057
Jimenez T, Sánchez G, Wertheimer E, Blanco G (2010) Activity of the Na, K-ATPase alpha4 isoform is important for membrane potential, intracellular Ca2+, and pH to maintain motility in rat spermatozoa. Reprod 139:835–845
Jin SK, Yang WX (2017) Factors and pathways involved in capacitation: how are they regulated? Oncotarget 8:3600–3627
Jones NP, Peak J, Brader S, Eccles SA, Katan M (2005) PLCgamma1 is essential for early events in integrin signalling required for cell motility. J Cell Sci 118:2695–2706
Kaewmala K, Uddin MJ, Cinar MU, Große-Brinkhaus C, Jonas E, Tesfaye D, Phatsara C, Tholen E, Looft C, Schellander K (2012) Investigation into association and expression of PLCz and COX-2 as candidate genes for boar sperm quality and fertility. Reprod Domest Anim 47:213–223
Kamaruddin M, Kroetsch T, Basrur PK, Hansen PJ, King WA (2004) Immunolocalization of heat shock protein 70 in bovine spermatozoa. Andrologia 36:327–334
Kashir J, Heynen A, Jones C, Durrans C, Craig J, Gadea J, Turner K, Parrington J, Coward K (2011a) Effects of cryopreservation and density-gradient washing on phospholipase C zeta concentrations in human spermatozoa. Reprod Biomed 23:263–267
Kashir J, Jones C, Lee HC, Rietdorf K, Nikiforaki D, Durrans C, Ruas M, Tee ST, Heindryckx B, Galione A, De Sutter P, Fissore RA, Parrington J, Coward K (2011b) Loss of activity mutations in phospholipase C zeta (PLCζ) abolishes calcium oscillatory ability of human recombinant protein in mouse oocytes. Hum Reprod 26:3372–3387
Kashir J, Jones C, Mounce G, Ramadan WM, Lemmon B, Heindryckx B, de Sutter P, Parrington J, Turner K, Child T, McVeigh E, Coward K (2013) Variance in total levels of phospholipase C zeta (PLC-ζ) in human sperm may limit the applicability of quantitative immunofluorescent analysis as a diagnostic indicator of oocyte activation capability. Fertil Steril 99:107–117
Kashir J, Konstantinidis M, Jones C, Lemmon B, Lee HC, Hamer R, Heindryckx B, Deane CM, De Sutter P, Fissore RA, Parrington J, Wells D, Coward K (2012) A maternally inherited autosomal point mutation in human phospholipase C zeta (PLCζ) leads to male infertility. Hum Reprod 27:222–231
Kim HK, Kim JW, Zilberstein A, Margolis B, Kim JG, Schlessinger J, Rhee SG (1991) PDGF stimulation of inositol phospholipid hydrolysis requires PLC-gamma 1 phosphorylation on tyrosine residues 783 and 1254. Cell 65:435–441
Kline D, Kline JT (1991) Repetitive calcium transients and the role of calcium in exocytosis and cell cycle activation in the mouse egg. Dev Biol 149:80–89
Knott JG, Kurokawa M, Fissore RA, Schultz RM, Williams CJ (2005) Transgenic RNA interference reveals role for mouse sperm phospholipase C zeta in triggering Ca2+ oscillations during fertilization. Biol Reprod 72:992–996
Kucik DF, Elson EL, Sheetz MP (1989) Forward transport of glycoproteins on leading lamellipodia in locomoting cells. Nature 340:315–317
Kurokawa M, Sato K, Wu H, He C, Malcuit C, Black SJ, Fukami K, Fissore RA (2005) Functional, biochemical, and chromatographic characterization of the complete [Ca2+]i oscillation-inducing activity of porcine sperm. Dev Biol 285:376–392
Kurokawa M, Yoon SY, Alfandari D, Fukami K, Sato K, Fissore RA (2007) Proteolytic processing of phospholipase Czeta and [Ca2+]i oscillations during mammalian fertilization. Dev Biol 312:407–418
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lara LS, De Carvalho T, Leão-Ferreira LR, Lopes AG, Caruso-Neves C (2005) Modulation of the (Na(+)+K+)ATPase activity by angiotensin-(1–7) in MDCK cells. Regul Pept 129:221–226
Li S, Wang Q, Wang Y, Chen X, Wang Z (2009) PLC-gamma1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol Endocrinol 23:901–913
Liu J, Liao Z, Camden J, Griffin KD, Garrad RC, Santiago-Pérez LI, González FA, Seye CI, Weisman GA, Erb L (2004) Src homology 3 binding sites in the P2Y2 nucleotide receptor interact with Src and regulate activities of Src, proline-rich tyrosine kinase 2, and growth factor receptors. J Biol Chem 279:8212–8218
Malcuit C, Maserati M, Takahashi Y, Page R, Fissore RA (2006) Intracytoplasmic sperm injection in the bovine induces abnormal [Ca2+]i responses and oocyte activation. Reprod Fertil Dev 18:39–51
Martínez-López P, Santi CM, Treviño CL, Ocampo-Gutiérrez AY, Acevedo JJ, Alisio A, Salkoff LB, Darszon A (2009) Mouse sperm K+ currents stimulated by pH and cAMP possibly coded by Slo3 channels. Biochem Biophys Res Commun 381:204–209
Mejía-Flores I, Chiquete-Félix N, Palma-Lara I, Uribe-Carvajal S, de Lourdes J-M (2017) During capacitation in bull spermatozoa, actin and PLC-ζ undergo dynamic interactions. Zygote 25:558–566
Michielsen EC, Diris JH, Hackeng CM, Wodzig WK, Van Dieijen-Visser MP (2005) Highly sensitive immunoprecipitation method for extracting and concentrating low-abundance proteins from human serum. Clin Chem 51:222–224
Miyazaki S, Shirakawa H, Nakada K, Honda Y (1993) Essential role of the inositol 1,4,5-trisphosphate receptor/Ca2+ release channel in Ca2+ waves and Ca2+ oscillations at fertilization of mammalian eggs. Dev Biol 158:62–78
Morozumi K, Yanagimachi R (2005) Incorporation of the acrosome into the oocyte during intracytoplasmic sperm injection could be potentially hazardous to embryo development. PNAS 102(40):14209–14214
Nair VD, Sealfon SC (2003) Agonist-specific transactivation of phosphoinositide 3-kinase signaling pathway mediated by the dopamine D2 receptor. J Biol Chem 278:47053–47061
Newton LD, Kastelic JP, Wong B, van der Hoorn F, Thundathil J (2009) Elevated testicular temperature modulates expression patterns of sperm proteins in Holstein bulls. Mol Reprod Dev 76:109–118
Newton LD, Krishnakumar S, Menon AG, Kastelic JP, van der Hoorn FA, Thundathil JC (2010) Na+/K+ATPase regulates sperm capacitation through a mechanism involving kinases and redistribution of its testis-specific isoform. Mol Reprod Dev 77:136–148
Nomikos M, Swann K, Lai FA (2011) Starting a new life: sperm PLC-zeta mobilizes the Ca2+ signal that induces egg activation and embryo development: an essential phospholipase C with implications for male infertility. Bioessays 34:126–134
Nomikos M, Sanders JR, Theodoridou M, Kashir J, Matthews E, Nounesis G, Lai FA, Swann K (2014) Sperm-specific post-acrosomal WW-domain binding protein (PAWP) does not cause Ca2+ release in mouse oocytes. Mol Hum Reprod 20:938–947
Ojaghi M, Kastelic J, Thundathil J (2017) Testis-specific isoform of angiotensin-converting enzyme (tACE) is involved in the regulation of bovine sperm capacitation. Mol Reprod Dev 84:376–388
Ouyang Y, Wong M, Capani F, Rensing N, Lee CS, Liu Q, Neusch C, Martone ME, Wu JY, Yamada K, Ellisman MH, Choi DW (2005) Transient decrease in F-actin may be necessary for translocation of proteins into dendritic spines. Eur J Neurosci 22:2995–3005
Padda RS, Shi Y, Lo CS, Zhang SL, Chan JS (2015) Angiotensin-(1–7): A novel peptide to treat hypertension and nephropathy in diabetes? Int J Diabs Metabol https://doi.org/10.4172/2155-6156.1000615
Parrington J, Jones ML, Tunwell R, Devader C, Katan M, Swann K (2002) Phospholipase C isoforms in mammalian spermatozoa: potential components of the sperm factor that causes Ca2+ release in eggs. Reprod 123:31–39
Peterson RN, Bozzola JJ, Hunt WP, Darabi A (1990) Characterization of membrane-associated actin in boar spermatozoa. J Exp Zool 253:202–214
Rajamanickam GD, Kastelic JP, Thundathil JC (2017a) Content of testis-Specific Isoform of Na/K-ATPase (ATP1A4) is increased during bovine sperm capacitation through translation in mitochondrial ribosomes. Cell Tissue Res 368:187–200
Rajamanickam GD, Kastelic JP, Thundathil JC (2017b) Na/K-ATPase regulates bovine sperm capacitation through raft- and non-raft-mediated signaling mechanisms. Mol Reprod Dev 84:1168–1182
Rajamanickam GD, Kroetsch T, Kastelic JP, Thundathil JC (2017c) Testis-specific isoform of Na/K-ATPase (ATP1A4) regulates sperm function and fertility in dairy bulls through potential mechanisms involving reactive oxygen species, calcium and actin polymerization. Andrology 5:814–823
Ramió-Lluch L, Prieto OB, Ramírez A, Fernández-Novell JM, Peña A, Rodríguez-Gil JE (2019) In vitro” capacitation and further progesterone-induced acrosome exocytosis are linked to specific changes in the expression and location of threonine phosphorylation of boar spermatozoa. Reprod Domest Anim 54:1085–1094
Rodríguez-Fragoso L, Melendez K, Hudson LG, Lauer FT, Burchiel SW (2009) EGF-receptor phosphorylation and downstream signaling are activated by benzo[a]pyrene 3,6-quinone and benzo[a]pyrene 1,6-quinone in human mammary epithelial cells. Toxicol Appl Pharmacol 235:321–328
Ross PJ, Beyhan Z, Iager AE, Yoon SY, Malcuit C, Schellander K, Fissore RA, Cibelli JB (2008) Parthenogenetic activation of bovine oocytes using bovine and murine phospholipase C zeta. BMC Dev Biol 8:16
Salamone DF, Canel NG, Rodríguez MB (2017) Intracytoplasmic sperm injection in domestic and wild mammals. Reprod 154:111–124
Salicioni AM, Platt MD, Wertheimer EV, Arcelay E, Allaire A, Sosnik J, Visconti PE (2007) Signalling pathways involved in sperm capacitation. Soc Reprod Fert Supp 65:245–259
Saunders CM, Larman MG, Parrington J, Cox LJ, Royse J, Blayney LM, Swann K, Lai FA (2002) PLC zeta: a sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Dev 129:3533–3544
Sekiya F, Poulin B, Kim YJ, Rhee SG (2004) Mechanism of tyrosine phosphorylation and activation of phospholipase C-gamma 1. Tyrosine 783 phosphorylation is not sufficient for lipase activation. J Biol Chem 279:32181–32190
Swann K (1990) A cytosolic sperm factor stimulates repetitive calcium increases and mimics fertilization in hamster eggs. Dev 110:1295–1302
Swann K, Saunders CM, Rogers NT, Lai FA (2006) PLCzeta(zeta): a sperm protein that triggers Ca2+ oscillations and egg activation in mammals. Semin Cell Dev Biol 17:264–273
Swann K, Yu Y (2008) The dynamics of calcium oscillations that activate mammalian eggs. Int J Dev Biol 52:585–594
Thundathil JC, Anzar M, Buhr MM (2006) Na+/K+ATPase as a signaling molecule during bovine sperm capacitation. Biol Reprod 75:308–317
Thundathil JC, Rajamanickam GD, Kastelic JP, Newton LD (2012) The effects of increased testicular temperature on testis-specific isoform of Na+/K+ -ATPase in sperm and its role in spermatogenesis and sperm function. Reprod Domest Anim 47:170–177
Thundathil JC, Rajamanickam GD, Kastelic JP (2018) Na/K-ATPase and regulation of sperm function. Anim Reprod 15:711–720
Tice DA, Biscardi JS, Nickles AL, Parsons SJ (1999) Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Cell Bio 96:1415–1420
Tomes CN, McMaster CR, Saling PM (1996) Activation of mouse sperm phosphatidylinositol-4,5 bisphosphate-phospholipase C by zona pellucida is modulated by tyrosine phosphorylation. Mol Reprod Dev 204:196–204
Ullrich A, Schlessinger J (1990) Signal transduction by receptors with tyrosine kinase activity. Cell 61:203–212
Wahl MI, Nishibe S, Suh PG, Rhee SG, Carpenter G (1989) Epidermal growth factor stimulates tyrosine phosphorylation of phospholipase C-II independently of receptor internalization and extracellular calcium. Cell Bio 86:1568–1572
Wang H, Haas M, Liang M, Cai T, Tian J, Li S, Xie Z (2004) Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem 279:17250–17259
Wu AT, Sutovsky P, Manandhar G, Xu W, Katayama M, Day BN, Park KW, Yi YJ, Xi YW, Prather RS, Oko R (2007) PAWP, a sperm-specific WW domain-binding protein, promotes meiotic resumption and pronuclear development during fertilization. J Biol Chem 282:12164–12175
Xu Z, Kopf GS, Schultz RM (1994) Involvement of inositol 1,4,5-trisphosphate-mediated Ca2+ release in early and late events of mouse egg activation. Dev 120:1851–1859
Yanagimachi R (1994) Mammalian Fertilisation. In: Knobil E, Neill J (eds) Physiol Reprod. Raven, New York, pp 189–299
Yoneda A, Kashima M, Yoshida S, Terada K, Nakagawa S, Sakamoto A, Hayakawa K, Suzuki K, Ueda J, Watanabe T (2006) Molecular cloning, testicular postnatal expression, and oocyte-activating potential of porcine phospholipase Czeta. Reprod 132:393–401
Yoon SY, Jellerette T, Salicioni AM, Lee HC, Yoo MS, Coward K, Parrington J, Grow D, Cibelli JB, Visconti PE, Mager J, Fissore RA (2008) Human sperm devoid of PLC, zeta 1 fail to induce Ca(2+) release and are unable to initiate the first step of embryo development. J Clin Investig 118:3671–3681
Young C, Grasa P, Coward K, Davis LC, Parrington J (2009) Phospholipase C zeta undergoes dynamic changes in its pattern of localization in sperm during capacitation and the acrosome reaction. Fertil Steril 91:2230–2242
Yu H, Fukami K, Itoh T, Takenawa T (1998) Phosphorylation of phospholipase Cgamma1 on tyrosine residue 783 by platelet-derived growth factor regulates reorganization of the cytoskeleton. Exp Cell Res 243:113–122
Yuan Z, Cai T, Tian J, Ivanov AV, Giovannucci DR, Xie Z (2005) Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mole Biol of Cell 16:4034–4045
Zhao Y, Kan FWK (2019) Human OVGP1 enhances tyrosine phosphorylation of proteins in the fibrous sheath involving AKAP3 and increases sperm-zona binding. J Assist Reprod Genet 36:1363–1377
Acknowledgements
This study received funding from the Natural Sciences and Engineering Research Council (NSERC) of Canada (Grant # RGPN-2020-04585 to JT). We thank Alta Genetics Inc., Calgary, AB, for providing semen samples for the study.
Funding
This study received funding from the Natural Sciences and Engineering Research Council (NSERC) of Canada (Grant No. RGPIN-2020-04585).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
The study reported herein was approved by the University of Calgary Institutional Animal Care and Use Committee (protocol number: AC170119).
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Unnikrishnan, V., Kastelic, J.P. & Thundathil, J.C. Ouabain-induced activation of phospholipase C zeta and its contributions to bovine sperm capacitation. Cell Tissue Res 385, 785–801 (2021). https://doi.org/10.1007/s00441-021-03455-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-021-03455-2