
Abstract
Intrauterine growth restriction (IUGR) is a disease affecting 10% of all pregnancies. IUGR is associated with maternal, fetal, or placental abnormalities. Studies investigating the effects of secondhand smoke (SHS) exposure and IUGR are limited. The receptor for advanced glycation end-products (RAGE) is a pro-inflammatory transmembrane receptor increased by SHS in the placenta. We tested the hypothesis that inhibition of RAGE during SHS exposure protects from smoke-induced IUGR. C57BL/6 mice were exposed to SHS or SHS + semi-synthetic glycosaminoglycan ethers (SAGEs) known to inhibit RAGE signaling. Trophoblast cells were treated with cigarette smoke extract (CSE) with or without SAGEs in order to address the effects of RAGE inhibition during trophoblast invasion in vitro. SHS-treated mice demonstrated a significant reduction in fetal weight (7.35-fold, P ≤ 0.0001) and placental weight (1.13-fold, P ≤ 0.0001) compared with controls. Mice co-treated with SHS and SAGEs were protected from SHS-induced fetal weights decreases. SHS treatment of C57BL/6 mice activated placental extracellular signal-regulated kinase (ERK) (3.0-fold, P ≤ 0.05), JNK (2.4-fold, P ≤ 0.05) and p38 (2.1-fold, P ≤ 0.05) and the expression of inflammatory mediators including TNF-α (1.34-fold, P ≤ 0.05) and IL-1β (1.03-fold, P ≤ 0.05). SHS-mediated activation of these molecules was reduced to basal levels when SAGE was co-administered. Invasion of trophoblast cells decreased 92% (P < 0.002) when treated with CSE and CSE-mediated invasion was completely reversed by SAGEs. We conclude that RAGE inhibition protects against fetal weight loss during SHS-induced IUGR. These studies provide insight into tobacco-mediated IUGR development and clarify avenues that may be helpful in the alleviation of placental complications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human pregnancy is characterized by complex temporal and spatial signaling processes that culminate in the creation of a healthy newborn. Disruption of these carefully orchestrated events occurs in obstetric complications such as intrauterine growth restriction (IUGR), that regularly impacts fetal and neonatal morbidity and mortality. Many complications have been associated with restricted fetal growth such as perinatal hypoxia and asphyxia, cerebral palsy and persistent pulmonary hypertension of the newborn (Brar and Rutherford 1988; Gray et al. 1999; Pollack and Divon 1992). Furthermore, several studies have reported long-term sequelae of IUGR complications including adult hypertension, heart disease, stroke and diabetes (Barker 1993; Smith et al. 2016; Barker et al. 1993; Holemans et al. 1993; Phipps et al. 1993; Reusens-Billen et al. 1989). Placental abnormalities associated with the IUGR placenta include reduced syncytiotrophoblast surface area (DiFederico et al. 1999), increased thickness of the exchange barrier formed by the trophoblast and fetal capillary endothelium (Ishihara et al. 2002), decreased trophoblast invasion (Mayhew et al. 2003; Pijnenborg et al. 1991), increased mTOR protein (Roos et al. 2007) and an increase in placental trophoblast apoptosis (Allaire et al. 2000; Hung et al. 2002). Although research has discovered many processes that contribute to the development of these pathologies, much more research is required to fully elucidate the underlying mechanisms associated with placental insufficiency in the context of IUGR.
Cigarette smoking during pregnancy has been a subject of intense study for decades (Arffin et al. 2012). In fact, many researchers have posited that smoking cigarettes throughout pregnancy may be the single most important avoidable cause of adverse pregnancy outcomes (Bickerstaff et al. 2012; Akkar et al. 2015). Indeed, even exposure to low levels of nicotine, the primary addictive chemical compound found in cigarettes, has been demonstrated to affect normal brain development, increase infant mortality rates and lead to the development of IUGR (Guan et al. 2009; Wahabi et al. 2013). Overall, exposure to cigarettes has been shown to have devastating effects on the developing fetus; however, reports detailing the consequences of passive secondhand smoke (SHS) exposure have yet to produce conclusive findings. Evidence suggests that mothers who are exposed to SHS may experience an increase in the risk of newborn orofacial clefting, (Kummet et al. 2016), elevated risks of wheeze development in newborns (Vardavas et al. 2016) and even learning difficulties (Jorge et al. 2016). While data continue to demonstrate the deleterious effects of tobacco smoke, there are conflicting reports as to its correlation with IUGR. A study by Wahabi et al. (2013) found a link between SHS and the induction of IUGR (Wahabi et al. 2013), yet, in contrast, Subramoney et al. (2010) reported no correlation between IUGR incidence and pregnant women exposed to smoke (Subramoney et al. 2010). Despite the conflicting reports, studies have suggested that SHS significantly increases the risk for spontaneous abortion, preterm delivery, sudden infant death syndrome and still births (Dempsey and Benowitz 2001; Engel et al. 2013; Khader et al. 2011; Lee et al. 2012a, b). More research is needed to fully understand the relationship between placental dysfunction and SHS exposure. Furthermore, an investigation into the possible mechanisms that may be contributing to the development of these adverse outcomes is warranted, as it may lead to pioneering insights and possible therapeutic improvements.
The receptor for advanced glycation end-products (RAGE) is a pattern recognition cell surface receptor increased during SHS exposure (Winden et al. 2014; Robinson et al. 2012). RAGE is capable of binding a host of ligands such as advanced glycation end-products (AGEs), HMGB1, S100/calgranulins and an additional variety of substances (Morbini et al. 2006). RAGE signaling is implicated in the pathogenesis of many inflammatory diseases such as Alzheimer’s (Lubitz et al. 2016), diabetes (Nelson et al. 2015), atherosclerosis (Belmokhtar et al. 2016), COPD (Reynolds et al. 2011) and diverse rheumatological disorders (Wu et al. 2016). Some products generated via RAGE signaling include NF-kB, Cox-2, IL-1ß and TNF-a, thus RAGE signaling likely influences apoptosis and the mediating of potent pro-inflammatory responses found in many chronic inflammatory pathologies (Bianchi et al. 2010). RAGE has been discovered in many cell types including smooth muscle, endothelium, macrophages and epithelium but has its highest expression in the lung and the placenta (Buckley and Ehrhardt 2010; Holmlund et al. 2007). Because the RAGE signaling axis has been linked with pro-inflammatory cascade events, novel drug therapies that target RAGE or its family of ligands are currently being investigated. One such recent modality with promise is semi-synthetic glycosaminoglycan ethers (SAGEs). SAGEs are partially lipophilic sulfated polysaccharide derivatives that modulate inflammation via the abrogation of RAGE signaling (Zhang et al. 2011). In this research endeavor, we examine the potential protective effects of SAGEs, specifically through blocking RAGE signaling in the lessening of downstream pro-inflammatory targets that contribute to the development of IUGR.
Materials and methods
Animals and tissue preparation
C57 Black 6 (C57BL/6) mice were obtained from Charles River Laboratories (Wilmington, MA, USA). Mice were housed in a conventional animal facility, supplied with food and water ad libitum and maintained on a 12-h light:dark cycle. Animals were separated into three groups: wild-type room air (RA; WT + RA), wild-type and SHS (WT + SHS) and wild-type, SHS and SAGEs injections (WT + SHS + SAGEs). To obtain timed pregnancies, females were caged with males overnight. Mice were allowed to undergo normal gestational development until embryonic day 14.5 (E14.5) at which time mice were either restrained and exposed to room air, or restrained and exposed to SHS. On E14.5, pregnant mice were then given either PBS or SAGEs injections until E18.5, at which time they were euthanized. Only live birth animals were considered for the study and any mice with absorbed fetuses were noted and excluded from the datasets. The first five conceptuses proximal to the ovaries from each uterine horn were collected and placentae and fetuses were weighed at this stage of pregnancy. Placental tissues were then snap-frozen in liquid nitrogen for protein analysis. For immunohistochemistry analysis, whole conceptuses were frozen in dry ice-cooled heptane. Mice were housed and utilized in accordance with protocols approved by the IACUC at Brigham Young University.
Secondhand smoke (SHS) exposure
Mice were exposed to SHS generated from 3R4F research cigarettes from Kentucky Tobacco Research and Development Center, University of Kentucky, via a nose-only exposure system (InExpose System; Scireq, Montreal, Canada). Mice were individually placed in soft restraints and connected to an exposure tower, wherein a computer-controlled puff of smoke generated every minute resulted in 10 s of SHS followed by 50 s of fresh air. Six mice in each group where exposed to SHS from six cigarettes over 10 min. This procedure was repeated from E14.5 to E18.5 at which time the mice were euthanized. To generate RA controls, mice were similarly restrained for a period of 10 min but were only exposed to RA. The SHS challenge was determined to be acceptable and was tolerated without evidence of toxicity. Furthermore, this nose-only model of smoke exposure yielded chronic blood carboxyhemoglobin levels of ~5%, a value similarly observed in human smokers (Wright et al. 2008).
SAGEs treatment
SAGEs (GM-111) were used in these studies, kindly gifted by Dr. Glenn D. Prestwich from the University of Utah Medical Center. All animal groups were administered an intraperitoneal injection (IP) with 20 μg/day (Zhang et al. 2011) of either SAGEs or saline (PBS) from E13.5 to E18.5 and exposed to either SHS or RA as previously explained.
Immunoblotting
Western blot analysis was used to assess the expression of a collection of cell signaling proteins in both control, control + SHS exposed and control + SHS + SAGEs treated animals. Briefly, tissues were homogenized in protein lysis buffer (RIPA; Fisher Scientific, Pittsburg, PA, USA). Protein lysates (20 μg) were separated on Mini-PROTEAN® TGX™ Precast gel (Bio-Rad Laboratories) by electrophoresis and transferred to nitrocellulose membranes. Membranes were incubated overnight with antibodies for RAGE, TNF-α, IL-1β, phospho and total extracellular signal-regulated kinase (ERK), JNK, or p38 (all from Cell Signaling, Danvers, MA, USA). A secondary horseradish peroxidase antibody (1:1000; Santa Cruz Biotechnology) was incubated in 5% milk for 1 h at room temperature. The membranes were incubated with chemiluminescent substrate (Pierce, Rockford, IL, USA) for 5 min and the emission of light was digitally recorded using a C-DiGit® Blot Scanner (LI-COR, Lincoln, NE, USA). Quantification of proteins was performed by densitometry and normalization with actin provided comparisons between treated and control samples.
Invasive trophoblast cell culture, cigarette smoke extract (CSE) and SAGEs
A first-trimester cytotrophoblast cell line Swan71 (SW71; n = 9) was used for invasive cytotrophoblast studies. SW71 cells were maintained and cultured in RPMI medium (Mediatech, Manassas, VA, USA). Cell medium was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. Cigarette smoke extract (CSE) was generated as previously described by Reynolds et al. (2006, 2008). Briefly, two 2RF4 research cigarettes (University of Kentucky, Lexington, KY, USA) were continuously smoked with a vacuum pump into 20 ml of DMEM medium (Mediatech). The smoke-bubbled medium was filtered through a 0.22-μm filter to remove large particles. The resulting medium was defined as 100% CSE. Dilutions were made using DMEM medium to a stock concentration of 10% CSE.
Cell treatments
SW71 cells were detached and 20,000 cells/ml were incubated in 2% FBS medium alone, medium supplemented with 0.5% CSE or 0.5% CSE and 50 μg/ml SAGEs. All treatments were carried out for 24 h. Real-time cell invasion was determined following these treatments.
Real-time cell invasion determination
An xCELLigence was utilized to determine real-time invasion of trophoblast cells. Protocol was followed as suggested by the manufacturer. Briefly, invasion was assessed in 16-well CIM-Plates (n = 9). The top wells were coated with a 1:40 matrigel concentration and trophoblast cells were plated in the top chamber at a concentration of 20,000 cells/well in 2% FBS RPMI in a total volume of 100 μL, in the presence or absence of CSE or CSE and SAGEs. The bottom chamber wells were filled with 160 μL of 10% FBS RPMI. The cells were then placed in the RTCA DP instrument and invasion readings were carried out every 15 min for 24 h.
Statistical analysis
Data were assessed by one- or two-way analysis of variance (ANOVA). When ANOVA indicated significant differences, Student’s t test was used with the Bonferroni correction for multiple comparisons. Results were checked for normality and data were shown as means ± SE. Significant differences between groups were noted at P < 0.05.
Results
Inhibition of RAGE by SAGEs and placental and fetal weights
We first conducted in vivo studies in order to investigate the effects of SHS on fetal and placental weights. Compared to RA-exposed controls, animals treated with SHS developed significantly lighter placentas (1.13-fold, P ≤ 0.0001) and fetal weights were significantly less (7.35-fold, P ≤ 0.0001) following 4 days of SHS treatment (Fig. 1a, b).

Placental and fetal weight differences during secondhand smoke (SHS) exposure. A significant decrease in placental (a 1.4-fold, P ≤ 0.0003) and fetal weights (-: 2.3-fold, P ≤ 0.0002) was observed in SHS-treated wild-type (WT) animals compared to room air (RA) controls. No significant recovery was observed in placental weights when comparing placenta from animals co-treated with SHS and SAGEs to those exposed to SHS alone (a). A significant recovery in fetal weights (4.22-fold, P ≤ 0.0001) was observed in SHS-exposed mice with SAGEs when compared with animals treated with SHS alone (b)
We next sought to inhibit RAGE in wild-type mice during SHS exposure. This round of in vivo studies involved the treatment of mice with SAGEs, recently developed sulfated polysaccharide derivatives shown to significantly inhibit RAGE signaling. There was no significant recovery in placental weights from animals co-treated with SHS and SAGEs when compared to those exposed to SHS alone (Fig. 1a). However, treatment of SHS-exposed mice with SAGEs caused a significant increase in fetal weights (4.22-fold, P ≤ 0.0001) when compared with animals treated with SHS alone (Fig. 1b). Despite significant protection, fetal weights were not completely restored by SAGEs treatment when compared to RA-exposed controls (1.74-fold, P ≤ 0.0001; Fig. 1b). Importantly, no weight changes were observed when room-air control animals were treated with SAGEs when compared to non-treated controls (not shown).
RAGE expression and influence of IUGR following SHS exposure and SAGEs treatment
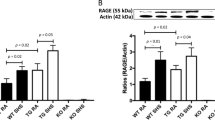
After examining placental and fetal weight changes stemming from SHS exposure, RAGE expression was evaluated to determine whether maternal SHS exposure elicited changes in placental protein expression. RAGE protein levels were elevated (1.13-fold, P ≤ 0.05) with the addition of SHS when compared to RA controls (Fig. 2). This elevation was reduced when animals were treated with SHS and SAGEs (Fig. 2).

Placental RAGE expression during SHS-induced IUGR. A characteristic RAGE western blot is shown in (a). RAGE was increased (1.13-fold, P ≤ 0.05) with SHS in the placenta of treated animals when compared to room air (RA) controls. This increase was reversed when animals were treated with SHS and SAGEs
RAGE signaling molecules in the placenta of SHS exposed animals with or without SAGEs treatment
To further characterize the effects of SAGEs in the placenta, we studied cell signaling molecules associated with RAGE activation. Increased TNF-α and IL-1β expression is known to be controlled, at least in part, by RAGE signaling (Xian et al. 2015; Li et al. 2015). A characteristic western blot for TNF-α and IL-1β is shown in Fig. 3a. SHS treatment increased both TNF-α (1.34-fold, P ≤ 0.05) and IL-1β (1.03-fold, P ≤ 0.05) in the placenta of SHS-treated animals when compared to RA controls (Fig. 3b, c). Interestingly, treatment with SAGEs decreased both TNF-α (1.84-fold, P ≤ 0.05) and IL-1β (1.16-fold P ≤ 0.05) in the SH- exposed animals when compared with the animals treated with SHS alone (Fig. 3b, c). To further investigate RAGE signal transduction in the placenta, we next looked at phosphorylated levels of ERK, JNK and p38. A characteristic western blot for ERK, JNK and p38 is shown in Fig. 4a. An increase in p-ERK activation (3.0-fold, P ≤ 0.05) was observed in the placenta from animals treated with SHS when compared to RA controls (Fig. 4b). When SAGEs were added to the SHS animals, activated ERK was significantly diminished (3.1-fold, P ≤ 0.05) when compared to animals treated with SHS alone (Fig. 4b). Similarly, increased activation of JNK (2.4-fold, P ≤ 0.05) occurred following SHS exposure and treatment with SAGEs was sufficient to significantly inhibit SHS-mediated p-JNK levels (2.3-fold, P ≤ 0.05) (Fig. 4c). In our analysis of p-P38, we observed a significant increase in p-P38 levels in SHS-exposed placentas compared to RA-exposed controls (2.1-fold, P ≤ 0.05) and a significant restoration of p-P38 to unexposed baseline when SHS and SAGEs were co-administered (1.8-fold, P ≤ 0.05) (Fig. 4d).

Placental TNF-α and IL-1β expression during SHS induced IUGR. A characteristic western blot is presented for each cytokine in (a). An increase in both TNF-α (1.34-fold, P ≤ 0.05) and IL-1β (1.03-fold, P ≤ 0.05) was observed in placentas from SHS-treated animals when compared to RA controls. In contrast, a significant decrease of both TNF-α (1.84-fold, P ≤ 0.05) and IL-1β (1.16-fold, P ≤ 0.05) was observed when animals were co-treated with both SHS and SAGEs when compared to animals exposed to SHS only

Placental pERK (a), pJNK (b) and pP38 (c) expression during SHS-induced IUGR. A characteristic western blot is presented for each molecule in (a). A significant increase in pERK activation (2.61-fold, P ≤ 0.05) was observed in placentas from animals treated with SHS when compared to RA controls. In contrast, when SAGEs were added to the SHS animals, activated ERK was significantly decreased (2.26-fold, P ≤ 0.05) when compared to animals treated with SHS alone (a). A significant increase in JNK activation (1.79-fold, P ≤ 0.05) occurred following SHS exposure when compared to RA controls. When SHS animals were co-treated with SAGEs, a significant decrease in pJNK levels (1.45-fold, P ≤ 0.05) was observed (b). Similarly, a significant increase in pP38 levels in SHS exposed placentas was observed compared to RA-exposed controls (2.44-fold, P ≤ 0.05) and a significant restoration of pP38 (1.55-fold, P ≤ 0.05) was observed in the SHS and SAGEs-treated animals when compared to animals treated with SHS alone (c)
Trophoblast invasion
A cellular hallmark of diminished placental weights observed in IUGR is decreased trophoblast invasion (Arroyo and Winn 2008). Previous reports from our laboratory have already demonstrated decreased cell invasion by trophoblast cells following exposure to CSE (Mejia et al. 2016). We therefore sought to quantitatively determine the plausible protective effects of SAGEs on cultured trophoblast invasion treated with CSE. As expected, CSE treatment decreased trophoblast invasion (7.4-fold, P < 0.02) in culture (Fig. 5a). Diminished invasion orchestrated by CSE was completely reversed when SAGEs and CSE were concomitantly added to the media of cultured trophoblasts (Fig. 5b).

CSE and SAGEs treatment and trophoblast invasion. Cell invasion of cultured trophoblast cells was decreased (7.4-fold, P ≤ 0.02) during exposure to 0.5% CSE. Inhibited invasion mediated by CSE was completely reversed in trophoblasts co treated with SAGEs and CSE
Discussion
RAGE signaling has been a major focal point in inflammatory research over the past several years. Numerous early studies sought to elucidate roles for RAGE signaling in the pulmonary apparatus (Stogsdill et al. 2013; Winden et al. 2013; Barton et al. 2014); however, roles are not clearly understood in terms of systemic expression and RAGE’s relationship to non-pulmonary-related pathologies. In regards to pregnancy, recent research has demonstrated that RAGE activation may play an important role in normal placentation (Konishi et al. 2004) as RAGE is localized to the developing human trophoblast. Furthermore, current research has confirmed that increased RAGE levels are found in preeclamptic (PE) placentas (Alexander et al. 2016). Because one of the primary activators of the RAGE signaling axis is tobacco smoke exposure, we sought to examine the possible effects of SHS exposure during pregnancy. Our initial investigation revealed that the addition of SHS during embryogenesis was sufficient to cause decreased placental and fetal weights. Unsurprisingly, RAGE expression was demonstrated to be elevated by SHS. These data corroborate recent findings that the addition of SHS increases RAGE protein in a variety of tissues (Winden et al. 2014; Prasad et al. 2015).
To further confirm that RAGE signaling may be implicated in IUGR, we tested a newly developed compound that inhibits RAGE signaling. The addition of SAGEs into SHS-exposed animals during pregnancy resulted in significant protection against fetal weight loss. This positive outcome suggests that more research is needed to further evaluate the possible therapeutic benefits of RAGE targeting in the placenta. Indeed, although our data only revealed significant protection, it remains possible that a more robust study that involves a broad dose curve may reveal complete protection against fetal and placental weight loss in the context of smoke exposure.
Because our in vivo studies implicated RAGE signaling as a modulator of IUGR, we sought to characterize additional RAGE-mediated targets. ERK is a protein usually associated with proliferation. Previous studies have demonstrated that p-ERK is elevated in patients with PE and IUGR (Bahr et al. 2014). Consistent with this data, our research demonstrated increased levels of p-ERK when animals were exposed to SHS. Furthermore, as ERK is at least partially mediated through the RAGE signaling axis, when SAGEs was administered in conjunction with SHS exposure, active ERK protein levels were significantly blocked. The p38 MAPK and JNK belong to the MAPK family of intracellular signal transducers involved in mammalian growth. In the RAGE signaling pathway, these downstream mediators activate NF-kB, which then in turn influences transcriptional regulation. Studies have demonstrated that p38 and JNK protein levels remain unchanged in IUGR; however, phosphorylated p38 and JNK levels are decreased in placental disease (Laviola et al. 2005). Our own data showed increases in overall p-P38 and p-JNK protein when control and SHS groups were compared. When SHS smoke animals were given SAGEs treatment, these animals decreased both p-P38 and p-JNK. These data suggest that these potent intermediates of inflammation and their subsequent activation are at least partially enabled through the RAGE signaling pathway. Furthermore, this assertion supports previous reports that implicates RAGE signaling in the activation of these proteins in patients with PE, IUGR and gestational diabetes (Alexander et al. 2016).
Endothelial cell dysfunction is believed to largely contribute to the pathogenesis of PE/IUGR. Inflammatory cytokines such as IL-1β and TNF-α have been shown to induce functional alterations in endothelial cells (Daneva et al. 2016) and have been found elevated in PE patients (Xian et al. 2015). It has been well documented that SHS increases TNF- α and IL-1B levels. Furthermore, research has demonstrated that RAGE signaling increases both of these pro-inflammatory cytokines, thus propagating the ability of RAGE to affect systemic inflammation. Our western blot analyses demonstrated elevated levels of TNF-α and IL-1β with the addition of SHS. However, TNF-α and IL-1β were both diminished to below the control levels when SAGEs were introduced, suggesting that this potential therapeutic may be influencing IUGR by decreasing inflammation.
A hallmark of IUGR/PE is shallow invasion of trophoblast cells (Harmon et al. 2016). Diminished invasion ultimately results in a high-resistance, low-capacity perfusion system, creating large-scale nutrient and oxygen deficiencies. Using an in vitro system, we demonstrated that the addition of SHS was sufficient to drastically decrease trophoblast cell invasion and that SAGEs treatment notably restored invasion.
We conclude that inhibition of RAGE protects against fetal and placental weight loss during SHS-induced IUGR. This conclusion is notably supported by the discovery that hindered trophoblast invasion was reversed by SAGEs treatment. Our results further suggest that there is a direct correlation between RAGE activation and the development of IUGR during SHS exposure. These studies provide insight into tobacco-mediated IUGR progression and clarify possible avenues for alleviating placental complications during SHS exposure.
References
Akkar OB, Yildiz C, Karakus S, Akkar I, Cetin A, Yanik A, Yenicesu AG, Boztosun A (2015) Antenatal counseling against passive smoking may improve birth weight for gestational age. Clin Exp Obstet Gynecol 42:805–809
Alexander KL, Mejia CA, Jordan C, Nelson MB, Howell BM, Jones CM, Reynolds PR, Arroyo JA (2016) Differential receptor for advanced Glycation end products expression in Preeclamptic, intrauterine growth restricted, and gestational diabetic placentas. Am J Reprod Immunol 75:172–180
Allaire AD, Ballenger KA, Wells SR, McMahon MJ, Lessey BA (2000) Placental apoptosis in preeclampsia. Obstet Gynecol 96:271–276
Arffin F, Al-Bayaty FH, Hassan J (2012) Environmental tobacco smoke and stress as risk factors for miscarriage and preterm births. Arch Gynecol Obstet 286:1187–1191
Arroyo JA, Winn VD (2008) Vasculogenesis and angiogenesis in the IUGR placenta. Semin Perinatol 32:172–177
Bahr BL, Price MD, Merrill D, Mejia C, Call L, Bearss D, Arroyo J (2014) Different expression of placental pyruvate kinase in normal, preeclamptic and intrauterine growth restriction pregnancies. Placenta 35:883–890
Barker DJ (1993) Fetal origins of coronary heart disease. Br Heart J 69:195–196
Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS (1993) Fetal nutrition and cardiovascular disease in adult life. Lancet 341:938–941
Barton DB, Betteridge BC, Earley TD, Curtis CS, Robinson AB, Reynolds PR (2014) Primary alveolar macrophages exposed to diesel particulate matter increase RAGE expression and activate RAGE signaling. Cell Tissue Res 358:229–238
Belmokhtar K, Robert T, Ortillon J, Braconnier A, Vuiblet V, Boulagnon-Rombi C, Diebold MD, Pietrement C, Schmidt AM, Rieu P, Toure F (2016) Signaling of serum amyloid a through receptor for advanced glycation end products as a possible mechanism for uremia-related atherosclerosis. Arterioscler Thromb Vasc Biol 36:800–809
Bianchi R, Giambanco I, Donato R (2010) S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging 31:665–677
Bickerstaff M, Beckmann M, Gibbons K, Flenady V (2012) Recent cessation of smoking and its effect on pregnancy outcomes. Aust N Z J Obstet Gynaecol 52:54–58
Brar HS, Rutherford SE (1988) Classification of intrauterine growth retardation. Semin Perinatol 12:2–10
Buckley ST, Ehrhardt C (2010) The receptor for advanced glycation end products (RAGE) and the lung. J Biomed Biotechnol 2010:917108
Daneva AM, Hadzi-Lega M, Stefanovic M (2016) Correlation of the system of cytokines in moderate and severe preeclampsia. Clin Exp Obstet Gynecol 43:220–224
Dempsey DA, Benowitz NL (2001) Risks and benefits of nicotine to aid smoking cessation in pregnancy. Drug Saf 24:277–322
DiFederico E, Genbacev O, Fisher SJ (1999) Preeclampsia is associated with widespread apoptosis of placental cytotrophoblasts within the uterine wall. Am J Pathol 155:293–301
Engel SM, Scher E, Wallenstein S, Savitz DA, Alsaker ER, Trogstad L, Magnus P (2013) Maternal active and passive smoking and hypertensive disorders of pregnancy: risk with trimester-specific exposures. Epidemiology 24:379–386
Gray PH, O’Callaghan MJ, Harvey JM, Burke CJ, Payton DJ (1999) Placental pathology and neurodevelopment of the infant with intrauterine growth restriction. Dev Med Child Neurol 41:16–20
Guan J, Mao C, Xu F, Zhu L, Liu Y, Geng C, Zhang L, Xu Z (2009) Low doses of nicotine-induced fetal cardiovascular responses, hypoxia, and brain cellular activation in ovine fetuses. Neurotoxicology 30:290–297
Harmon AC, Cornelius DC, Amaral LM, Faulkner JL, Cunningham MW Jr, Wallace K, LaMarca B (2016) The role of inflammation in the pathology of preeclampsia. Clin Sci (Lond) 130:409–419
Holemans K, Van Bree R, Verhaeghe J, Aerts L, Van Assche FA (1993) In vivo glucose utilization by individual tissues in virgin and pregnant offspring of severely diabetic rats. Diabetes 42:530–536
Holmlund U, Wahamaa H, Bachmayer N, Bremme K, Sverremark-Ekstrom E, Palmblad K (2007) The novel inflammatory cytokine high mobility group box protein 1 (HMGB1) is expressed by human term placenta. Immunology 122:430–437
Hung TH, Skepper JN, Charnock-Jones DS, Burton GJ (2002) Hypoxia-reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res 90:1274–1281
Ishihara N, Matsuo H, Murakoshi H, Laoag-Fernandez JB, Samoto T, Maruo T (2002) Increased apoptosis in the syncytiotrophoblast in human term placentas complicated by either preeclampsia or intrauterine growth retardation. Am J Obstet Gynecol 186:158–166
Jorge JG, Botelho C, Silva AM, Moi GP (2016) Influence of passive smoking on learning in elementary school. J Pediatr 92:260–267
Khader YS, Al-Akour N, Alzubi IM, Lataifeh I (2011) The association between second hand smoke and low birth weight and preterm delivery. Matern Child Health J 15:453–459
Konishi H, Nakatsuka M, Chekir C, Noguchi S, Kamada Y, Sasaki A, Hiramatsu Y (2004) Advanced glycation end products induce secretion of chemokines and apoptosis in human first trimester trophoblasts. Hum Reprod 19:2156–2162
Kummet CM, Moreno LM, Wilcox AJ, Romitti PA, DeRoo LA, Munger RG, Lie RT, Wehby GL (2016) Passive smoke exposure as a risk factor for oral clefts-a large international population-based study. Am J Epidemiol 183:834–841
Laviola L, Perrini S, Belsanti G, Natalicchio A, Montrone C, Leonardini A, Vimercati A, Scioscia M, Selvaggi L, Giorgino R, Greco P, Giorgino F (2005) Intrauterine growth restriction in humans is associated with abnormalities in placental insulin-like growth factor signaling. Endocrinology 146:1498–1505
Lee NL, Samet JM, Yang G, Zhou M, Yang J, Correa A, Lees PS (2012a) Prenatal secondhand smoke exposure and infant birth weight in China. Int J Environ Res Public Health 9:3398–3420
Lee SL, Lam TH, Leung TH, Wong WH, Schooling M, Leung GM, Lau YL (2012b) Foetal exposure to maternal passive smoking is associated with childhood asthma, allergic rhinitis, and eczema. ScientificWorldJournal 2012:542983
Li D, Lei C, Zhang S, Zhang S, Liu M, Wu B (2015) Blockade of high mobility group box-1 signaling via the receptor for advanced glycation end-products ameliorates inflammatory damage after acute intracerebral hemorrhage. Neurosci Lett 609:109–119
Lubitz I, Ricny J, Atrakchi-Baranes D, Shemesh C, Kravitz E, Liraz-Zaltsman S, Maksin-Matveev A, Cooper I, Leibowitz A, Uribarri J, Schmeidler J, Cai W, Kristofikova Z, Ripova D, LeRoith D, Schnaider-Beeri M (2016) High dietary advanced glycation end products are associated with poorer spatial learning and accelerated Abeta deposition in an Alzheimer mouse model. Aging Cell 15:309–316
Mayhew TM, Ohadike C, Baker PN, Crocker IP, Mitchell C, Ong SS (2003) Stereological investigation of placental morphology in pregnancies complicated by pre-eclampsia with and without intrauterine growth restriction. Placenta 24:219–226
Mejia C, Lewis J, Jordan C, Mejia J, Ogden C, Monson T, Winden D, Watson M, Reynolds PR, Arroyo JA (2016) Decreased activation of placental mTOR family members is associated with the induction of intrauterine growth restriction by secondhand smoke in the mouse. Cell Tissue Res 367:387–395
Morbini P, Villa C, Campo I, Zorzetto M, Inghilleri S, Luisetti M (2006) The receptor for advanced glycation end products and its ligands: a new inflammatory pathway in lung disease? Mod Pathol 19:1437–1445
Nelson MB, Swensen AC, Winden DR, Bodine JS, Bikman BT, Reynolds PR (2015) Cardiomyocyte mitochondrial respiration is reduced by receptor for advanced glycation end-product signaling in a ceramide-dependent manner. Am J Phys Heart Circ Phys 309:H63–H69
Phipps K, Barker DJ, Hales CN, Fall CH, Osmond C, Clark PM (1993) Fetal growth and impaired glucose tolerance in men and women. Diabetologia 36:225–228
Pijnenborg R, Anthony J, Davey DA, Rees A, Tiltman A, Vercruysse L, van Assche A (1991) Placental bed spiral arteries in the hypertensive disorders of pregnancy. Br J Obstet Gynaecol 98:648–655
Pollack RN, Divon MY (1992) Intrauterine growth retardation: definition, classification, and etiology. Clin Obstet Gynecol 35:99–107
Prasad K, Dhar I, Caspar-Bell G (2015) Role of advanced Glycation end products and its receptors in the pathogenesis of cigarette smoke-induced cardiovascular disease. Int J Angiol 24:75–80
Reusens-Billen B, Remacle C, Hoet JJ (1989) The development of the fetal rat intestine and its reaction to maternal diabetes. II. Effect of mild and severe maternal diabetes. Diabetes Res Clin Pract 6:213–219
Reynolds PR, Cosio MG, Hoidal JR (2006) Cigarette smoke-induced Egr-1 upregulates proinflammatory cytokines in pulmonary epithelial cells. Am J Respir Cell Mol Biol 35:314–319
Reynolds PR, Kasteler SD, Cosio MG, Sturrock A, Huecksteadt T, Hoidal JR (2008) RAGE: developmental expression and positive feedback regulation by Egr-1 during cigarette smoke exposure in pulmonary epithelial cells. Am J Physiol Lung Cell Molec Physiol 294:L1094–L1101
Reynolds PR, Kasteler SD, Schmitt RE, Hoidal JR (2011) Receptor for advanced glycation end-products signals through Ras during tobacco smoke-induced pulmonary inflammation. Am J Respir Cell Mol Biol 45:411–418
Robinson AB, Stogsdill JA, Lewis JB, Wood TT, Reynolds PR (2012) RAGE and tobacco smoke: insights into modeling chronic obstructive pulmonary disease. Front Physiol 3:301
Roos S, Jansson N, Palmberg I, Saljo K, Powell TL, Jansson T (2007) Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J Physiol 582:449–459
Smith CJ, Ryckman KK, Barnabei VM, Howard BV, Isasi CR, Sarto GE, Tom SE, Van Horn LV, Wallace RB, Robinson JG (2016) The impact of birth weight on cardiovascular disease risk in the Women’s Health Initiative. Nutr Metab Cardiovasc Dis 26:239–245
Stogsdill MP, Stogsdill JA, Bodine BG, Fredrickson AC, Sefcik TL, Wood TT, Kasteler SD, Reynolds PR (2013) Conditional overexpression of receptors for advanced glycation end-products in the adult murine lung causes airspace enlargement and induces inflammation. Am J Respir Cell Mol Biol 49:128–134
Subramoney S, d’Espaignet ET, Gupta PC (2010) Higher risk of stillbirth among lower and middle income women who do not use tobacco, but live with smokers. Acta Obstet Gynecol Scand 89:572–577
Vardavas CI, Hohmann C, Patelarou E, Martinez D, Henderson AJ, Granell R, Sunyer J, Torrent M, Fantini MP, Gori D, Annesi-Maesano I, Slama R, Duijts L, de Jongste JC, Aurrekoetxea JJ, Basterrechea M, Morales E, Ballester F, Murcia M, Thijs C, Mommers M, Kuehni CE, Gaillard EA, Tischer C, Heinrich J, Pizzi C, Zugna D, Gehring U, Wijga A, Chatzi L, Vassilaki M, Bergstrom A, Eller E, Lau S, Keil T, Nieuwenhuijsen M, Kogevinas M (2016) The independent role of prenatal and postnatal exposure to active and passive smoking on the development of early wheeze in children. Eur Respir J 48:115–124
Wahabi HA, Alzeidan RA, Fayed AA, Mandil A, Al-Shaikh G, Esmaeil SA (2013) Effects of secondhand smoke on the birth weight of term infants and the demographic profile of Saudi exposed women. BMC Public Health 13:341
Winden DR, Ferguson NT, Bukey BR, Geyer AJ, Wright AJ, Jergensen ZR, Robinson AB, Stogsdill JA, Reynolds PR (2013) Conditional over-expression of RAGE by embryonic alveolar epithelium compromises the respiratory membrane and impairs endothelial cell differentiation. Respir Res 14:108
Winden DR, Barton DB, Betteridge BC, Bodine JS, Jones CM, Rogers GD, Chavarria M, Wright AJ, Jergensen ZR, Jimenez FR, Reynolds PR (2014) Antenatal exposure of maternal secondhand smoke (SHS) increases fetal lung expression of RAGE and induces RAGE-mediated pulmonary inflammation. Respir Res 15:129
Wright JL, Cosio M, Churg A (2008) Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Molec Physiol 295:L1–15
Wu Q, Zhong ZM, Zhu SY, Liao CR, Pan Y, Zeng JH, Zheng S, Ding RT, Lin QS, Ye Q, Ye WB, Li W, Chen JT (2016) Advanced oxidation protein products induce chondrocyte apoptosis via receptor for advanced glycation end products-mediated, redox-dependent intrinsic apoptosis pathway. Apoptosis 21:36–50
Xian N, Chen W, Zhang Y, Li J, Zhang N, Ye Y (2015) Correlation of the expressions of advanced glycation end products and its receptor in serum and placenta with the pathogenesis of preeclampsia. Zhonghua fu chan ke za zhi 50:493–499
Zhang J, Xu X, Rao NV, Argyle B, McCoard L, Rusho WJ, Kennedy TP, Prestwich GD, Krueger G (2011) Novel sulfated polysaccharides disrupt cathelicidins, inhibit RAGE and reduce cutaneous inflammation in a mouse model of rosacea. PLoS ONE 6:e16658
Acknowledgements
The authors would like to thank Dr. Glenn D. Prestwich from the University of Utah Medical Center for his donation of SAGEs. Additionally, we would like to thank the many undergraduate students from the combined laboratories of Drs. Juan A. Arroyo and Paul R. Reynolds for their vital support.
Funding
This study was supported in part by grant 7R00HD055054-06, a grant from the Flight Attendant’s Medical Research Institute (FAMRI) and by BYU Mentoring Environment Grants.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors report no conflict of interest.
Rights and permissions
About this article

Cite this article
Lewis, J.B., Mejia, C., Jordan, C. et al. Inhibition of the receptor for advanced glycation end-products (RAGE) protects from secondhand smoke (SHS)-induced intrauterine growth restriction IUGR in mice. Cell Tissue Res 370, 513–521 (2017). https://doi.org/10.1007/s00441-017-2691-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-017-2691-z