
Abstract
High glucose induces vascular smooth muscle cell (SMC) dysfunction by generating oxidative stress attributable, in part, to the up-regulated NADPH oxidases (Nox). We have attempted to elucidate the high-glucose-generated molecular signals that mediate this effect and hypothesize that products of high-glucose-induced lipid peroxidation regulate Nox by activating peroxisome proliferator-activated receptors (PPARs). Human aortic SMCs were exposed to glucose (5.5–25 mM) or 4-hydroxynonenal (1–25 μM, 4-HNE). Lucigenin assay, real-time polymerase chain reaction, western blot, and promoter analyses were employed to investigate Nox. We found that high glucose generated an increase in Nox activity and expression. It also promoted oxidative stress that consequently induced lipid peroxidation, which resulted in the production of 4-HNE. Pharmacological inhibition of Nox activity significantly reduced the formation of high-glucose-induced 4-HNE. Exposure of SMCs to non-cytotoxic concentrations (1–10 μM) of 4-HNE alone mimicked the effect of high glucose incubation, whereas scavenging of 4-HNE by N-acetyl L-cysteine completely abolished both the effects of high glucose and 4-HNE. The latter exerted its effect by activating PPARα and PPARβ/δ, but not PPARγ, as assessed pharmacologically by the inhibitory effect of selective antagonists and following the silencing of the expression of these receptors. These new data indicate that 4-HNE, generated following Nox activation, functions as an endogenous activator of PPARα and PPARβ/δ. The newly discovered “lipid peroxidation products–PPARs–Nox axis” represents a novel mechanism of Nox regulation and an additional therapeutic target for oxidative stress in diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hyperglycemia induces excess formation of reactive oxygen species (ROS) and is a major contributor to the development of diabetic complications, such as atherosclerosis, the main cause of mortality in diabetic patients (Gao and Mann 2009; Heltianu et al. 2011). The oxidative environment has been linked to the dysfunction of vascular endothelial and smooth muscle cells (SMCs), which further promotes and accelerates the development of atherosclerotic lesions. In these diseases, the phenotypic alterations of vascular SMCs represent a maladaptive response to diabetic stimuli and are considered to be partially mediated by increased levels of ROS in the cells (Csányi et al. 2009). Nevertheless, the molecular mechanisms of hyperglycemia-induced excess ROS formation in SMCs are not well defined.
Vascular NADPH oxidases (Nox) are a class of hetero-oligomeric enzymes (Nox1-5) whose exclusive function is the generation of ROS in a highly regulated manner. Increased Nox-derived ROS formation is highly detrimental in numerous pathologies, including diabetes mellitus. Typically, SMCs express the Nox1, Nox4, and Nox5 subtypes and a lower level of Nox2. The p22phox subunit is essential for Nox1 and Nox4 activities, whereas Nox5 function is calcium-dependent (Manea et al. 2008; Manea 2010). Nonetheless, the specific role and contribution of each enzyme in SMCs has not yet been elucidated. However, Nox expression and activity are reported to be significantly increased in the vasculature of diabetic subjects and these changes are associated with the development of macro- and microvascular disorders (Sedeek et al. 2012a, 2012b). Since all the members of the Nox family are major triggers of oxidative stress and might play a prominent role in diabetes-induced vasculopathies (Gao and Mann 2009; Manea et al. 2014), these enzymes and their up-stream regulators are considered major therapeutic targets (Kahles and Brandes 2012; Krause et al. 2012).
Evidence is accumulating that the activation of peroxisome proliferator-activated receptors (PPARα, −β/δ, and –γ) negatively correlates with vascular inflammation and ROS generation by mechanisms that are not yet clear. In addition, PPARs play key roles in the regulation of fatty acid metabolism and energy homeostasis. PPARs are members of a nuclear hormone receptor family comprising three major isoforms, PPARα, PPARβ/δ, and PPARγ. They dimerize with the activated retinoid X receptor (RXR) and function as ligand-activated transcription factors (Forman et al. 1997; Staels and Fruchart 2005; Das and Chakrabarti 2006; Staels 2007; Plutzky 2011).
Lipid peroxidation of polyunsaturated fatty acids (PUFAs) is initiated by ROS and hydroxyl radicals and results, following a series of non-enzymatic reactions, in the generation of 4-hydroxy-2E-hexenal (4-HHE), 4-hydroxy-2E-nonenal (4-HNE), and 4-hydroxy-2E,6Z-dodecadienal (4-HDDE). Evidence has accumulated suggesting that low and physiological levels of these molecules function as signaling messengers, most likely by activating PPARs (Riahi et al. 2010a, 2010b). Recently, 4-HNE and 4-HDDE have been reported to activate PPARβ/δ in endothelial cells and pancreatic β-cells (Riahi et al. 2010a, 2010b; Cohen et al. 2011). Whether this mechanism takes place in other cells, such as SMCs, and whether hyperglycemic conditions intensify the process remain unknown. Here, we have aimed at investigating the interactions among 4-HNE (an important lipid peroxidation end-product induced by high glucose), PPARs, and Nox-induced ROS formation in human aortic SMCs. We provide evidence that 4-HNE increases Nox expression and function by activating PPARα and PPARβ/δ. This study highlights the role of the PPAR family in redox sensing and redox control mechanisms in vascular SMCs in diabetes.
Materials and methods
Materials
Standard chemicals, antibodies, short interfering RNA (siRNA), reagents, and molecular biology kits were obtained from Sigma-Aldrich (Germany), Santa Cruz Biotechnology (USA), Invitrogen (Austria), and Qiagen (Germany). The pDR1-luc control plasmid was from Stratagene (Germany).
Cell culture
Human aortic SMCs were obtained and characterized as described in Tîrziu et al. (1999). The cell line was established in accordance with our institutional ethical guidelines. Confluent quiescent cells (at passages 7–10) cultured for 24 h in fetal bovine serum (FBS)-free Dulbecco’s modified Eagle’s Medium (DMEM, 5.5 mM glucose). To mimic hyperglycemia conditions, the cells were further exposed for up to 24 h to DMEM supplemented with normal (5.5 mM) or high (11–25 mM) glucose concentrations without or with 1–25 μM 4-HNE. Other experiments were performed under similar conditions but in the absence/presence of selective PPAR antagonists or specific siRNA for PPARα, PPARβ/δ, or PPARγ.
Primary cultures of adult human aortic SMCs (Lonza, Switzerland) were used to validate the data obtained with the SMC line. The cells, which were grown to confluence according to the supplier’s instructions, were treated as described above.
This study was conducted in accordance with the ethical principles for medical research involving human subjects (World Medical Association Declaration of Helsinki), and the local committee on human research approved the study protocol.
Measurement of ROS production
NADPH oxidase-dependent O2 •- production was determined in membrane fractions obtained from SMCs by lucigenin-enhanced chemiluminescence assay as previously described (Ungvari et al. 2003; Touyz et al. 2002). Employing siRNA-mediated silencing of Nox1, Nox4 or Nox5, we have shown previously that each Nox subtype contributes to the overall NADPH-dependent O2 •- formation in SMCs (Manea et al. 2012). However, the involvement of other NADPH-dependent O2 •--generating oxidases should be considered. Nox activity was calculated from the ratio of mean light units to total protein level and expressed as arbitrary units. The formation of O2 •- in intact SMCs was evaluated by the dihydroethidium (DHE) method (Manea et al. 2010a).
Real-time polymerase chain reaction
mRNA levels were quantified by the amplification of cDNA with SYBR Green I and the comparative CT method. The sequences of oligonucleotide primers were as follows: Nox1 (NM_013955) forward: 5′-CACAAGAAAAATCCTTGGGTCAA-3′, reverse: 5′-GACAGCAGATTGCGACACACA-3′; Nox4 (NM_016931) forward: 5′-TGGCTGCCCATC TGGTGAATG-3′, reverse: 5′-CAGCAGCCCTCCTGAAACATGC-3′; Nox5 (NM_024505) forward: 5′-CAGGCACCAGAAAAGAAAGCAT-3′, reverse: 5′-ATGTTGTCTTGGACACCTTCGA-3′; β-actin (NM_001101) forward: 5′- CTGGCACCCAGCACAATG -3′, reverse: 5′- GCCGATCCACACGGAGTACT -3′. Optimized amplification conditions were 0.2 μM of each primer, 2.5 mM MgCl2, annealing at 60 °C, and extension at 72 °C for 40 cycles. The gene expression levels of Nox1, Nox4, and Nox5 were normalized to β-actin and expressed as arbitrary units.
Western blot
Cell lysate preparation and western blot analysis were performed as previously described (Manea et al. 2012; Manea et al. 2013). Briefly, cultured SMCs were washed twice in ice-cold phosphate-buffered saline before lysis in 2 x Laemmli’s electrophoresis sample buffer (Serva, Germany) and boiled for 20 min. Equal amounts of protein (50 μg) were separated by 10 % SDS-polyacrylamide gel electrophoresis (SDS-PAGE; for Nox1, Nox4, 4-HNE, PPARα, PPARβ/δ, PPARγ) or 8 % SDS-PAGE (for Nox5) and electroblotted onto nitrocellulose membranes (Biorad, USA). The membranes were exposed to blocking reagent TBS Blotto A (sc-2333) and then incubated overnight at 4 °C with primary antibodies against Nox1 (rabbit polyclonal, sc-25545), Nox4 (goat polyclonal, sc-55142), Nox5 (goat polyclonal, sc-34707), 4-HNE-protein adducts (goat polyclonal, sc-130083), PPARα (rabbit polyclonal, sc-9000), PPARβ/δ (rabbit polyclonal, sc-7197), PPARγ (rabbit polyclonal, sc-7196), or β-actin (mouse monoclonal, sc-47778). After being washed with TRIS-buffered saline containing 0.05 % Tween 20, the membranes were exposed to horseradish peroxidase (HRP)-conjugated secondary antibodies for 40 min. Protein bands were detected by using chemiluminescence substrate solution (Pierce, Germany), and images were taken with a digital detection system (ImageQuant LAS 4000, Fujifilm, Japan). Quantification (TotalLab) of Nox proteins and 4-HNE-modified proteins was performed by normalization to β-actin protein and expressed as arbitrary units.
Transfection of siRNA
At 24 h prior to transfections, exponentially growing SMCs were seeded at 4 × 105 cells (≈50 % confluence) in tissue culture plates (Ø 60 mm). Control (sc-37007), PPARα (sc-36307), PPARβ/δ (sc-36305), or PPARγ (sc-29455) siRNA (20 nM; Santa Cruz Biotechnology) were transfected into SMC by using Hiperfect reagent according to the manufacturer’s protocol (Qiagen). Silencing efficiency was evaluated by western blot at 48–72 h after siRNA transfection.
Transient transfection and luciferase assay
Transient transfection was performed as in Zalba et al. (2001) by using Superfect reagent (Qiagen). Promoter activity was calculated from the ratio of firefly luciferase to β-galactosidase levels (Beta-Glo assay, Promega) and expressed as arbitrary units.
Cell impedance measurements
To evaluate the effects of high glucose and 4-HNE on SMC proliferation, the impedance-based assay for real-time monitoring of cell dynamics (xCELLigence, Roche) was employed. SMCs were seeded at 5 × 103 cells/well in 16-well E-Plates and, 24 h later, were exposed to 5.5 or 25 mM glucose in the presence or absence of 1–25 μM 4-HNE. The results were analyzed by using RTCA software. Notably, the changes in cell impedance represented an overall index of various biological processes, such as cell attachment, growth, proliferation, migration, and matrix deposition.
MTT cell proliferation assay
To investigate the involvement of Nox in mediating high-glucose-induced SMC proliferation, the Vybrant MTT cell proliferation assay [MTT = 3-(4,5-dimethyl-thiazolyl-2)-2,5-diphenol tetrazolium bromide] was used according to the manufacturer’s protocol (Molecular Probes).
Statistical analysis
Data derived from a minimum of three independent experiments were expressed as means ± standard deviation (SD). Statistical analysis was performed by one-way analysis of variance followed by Tukey’s post hoc test; P < 0.05 was considered as statistically significant.
Results
High glucose increases Nox activity and expression in SMCs
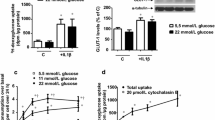
Dose-response analysis was performed to evaluate the effects of increasing concentrations of glucose on Nox expression and function in human SMCs (Fig. 1). Glucose increased Nox activity and expression of the various subtypes dose-dependently. The maximal Nox-activating effect (≈1.4-1.6-fold) was detected between 16.5 and 25 mM glucose in comparison with the 5.5 mM control (Fig. 1a). Similar patterns of increased expression of mRNA and protein levels of Nox1, Nox4, and Nox5 isotypes were detected (Fig. 1c–k). Interestingly, Nox activity and expression remained at 11 mM glucose similar to those of the 5.5 mM control. Since incubation with 25 mM glucose maximally increased Nox activity and expression, this concentration was used in further experiments. We also found similar effects of high glucose levels on O2 •- formation in SMCs by using the lucigenin-based technique and the DHE assay (Electronic Supplementary Material, Fig. S1a).

High glucose concentration increases NADPH oxidase (Nox) activity, up-regulates Nox expression, and induces smooth muscle cell (SMC) proliferation. a Quiescent SMCs were exposed to the indicated concentrations of glucose (24 h), and NADPH-dependent O2 •- production was assessed by lucigenin-enhanced chemiluminescence. b SMCs were plated at 5 × 103 cells/well on a 16-well plate and, after 24 h, were exposed to increasing concentrations of glucose in a serum-free medium. Cell impedance was monitored continuously for 48 h by using E-Plates technology. The data are representative of three independent experiments. c–h Up-regulation of Nox1, Nox4, and Nox5 mRNA (c–e) and increase in protein levels (f–h) quantified by real-time polymerase chain reaction (PCR) and western blot analyses, respectively, in cells exposed to 5.5–25 mM glucose. i–k Representative immunoblots depicting Nox1, Nox4, and Nox5 proteins following incubation of the cells with increasing concentrations of glucose. n = 4, *P < 0.05, **P < 0.01. P-values were taken in relation to the cells exposed to 5.5 mM glucose
High glucose induces SMC proliferation
Since abnormal SMC proliferation represents a key pathological manifestation in diabetes-associated vascular complications, we searched for the effect of high glucose on cultured SMCs by using the impedance-based real-time (xCELLigence) system. To monitor the effects of high glucose on SMCs proliferation, the cells were exposed to increasing glucose concentrations (5.5 to 25 mM). Figure 1b shows that exposure to increasing concentrations of glucose augmented SMC proliferation in a dose-dependent manner. Lipid peroxidation end-product 4-HNE (25 μM) was used as a negative control of cell proliferation. The quantification of xCELLigence data is presented in Electronic Supplementary Material (Fig. S2a). To validate the impedance-based measurements, MTT assays were employed to investigate the effect of high glucose on SMC proliferation. The results showed that high-glucose-induced SMC proliferation is partially mediated by ROS, possibly generated by activated Nox (Electronic Supplementary Material, Fig. S3). Figure 2 indicates that high glucose also augmented the formation of 4-HNE-modified proteins in SMCs via Nox-derived ROS. The levels of 4-HNE-modified proteins were determined for an indirect estimation of the cumulative generation of 4-HNE in cultured SMCs exposed to normal and high glucose concentrations. Figure 2a shows that the exposure of SMCs to increasing glucose levels for 24 h resulted in a steady glucose-dependent increase in 4-HNE-protein adducts formation.

High-glucose-induced accumulation of endogenous 4-HNE-protein adducts in SMCs is mediated by Nox; exogenous 4-HNE has a similar effect on cells. a Formation of 4-HNE-protein adducts as a result of cell exposure to high glucose concentrations. b Quiescent SMCs were exposed (24 h) to either normal 5.5 or 25 mM glucose in the absence or presence of 1 μM apocynin, 1 μM diphenyleneiodonium (DPI), or 2000 U/ml catalase-polyethylene glycol (PEG-CAT). c Determination of 4-HNE-protein adducts in SMCs treated with exogenous 4-HNE. The cells were exposed to increasing concentrations of 4-HNE (1–25 μM) in serum-free medium (24 h), and the formation of 4-HNE-histidine adducts was assessed by Western blot. d, e Representative immunoblots depicting 4-HNE-modified proteins (NAC N-acetyl-L-cysteine). n = 4, *P < 0.05, **P < 0.01. P-values were taken in relation to cells exposed to 5.5 mM glucose or vehicle (ethanol)
To investigate whether Nox-derived ROS mediated high-glucose-induced 4-HNE formation, quiescent SMCs were exposed to 5 or 25 mM glucose for 24 h in the absence or presence of 1 μM apocynin (an inhibitor of Nox activity with antioxidant properties), diphenyleneiodonium (DPI; a flavin-containing oxidase inhibitor), or 2000 U/ml catalase-polyethylene glycol (PEG-CAT; an intracellular scavenger of H2O2). Western blot analysis revealed that all three agents significantly reduced the level of high-glucose-induced 4-HNE-protein adducts (Fig. 2b).
Exposure of SMCs to 4-HNE induced accumulation of 4-HNE-protein adducts in a concentration-dependent manner
To determine the range of high-glucose-induced 4-HNE formation, cultured SMCs were exposed to increasing concentrations of 4-HNE for 24 h in serum-free DMEM containing 5.5 mM glucose. Whereas the reactivity was low in vehicle-treated cells, it significantly increased following exposure to increasing concentrations of 4-HNE (Fig. 2c). The polyclonal antibody against 4-HNE-histidine adducts intensely labeled protein bands of ≈ 60 and ≈ 24 kDa (Fig. 2d). In particular, the effect of 25 mM glucose on 4-HNE-modified proteins was comparable with an acute exogenous cell stimulation with 1–5 μM 4-HNE.
Since N-acetyl-L-cysteine (NAC) is an important antioxidant that neutralizes ROS and is a scavenger of α,β-unsaturated aldehydes, we used it to abolish the formation of 4-HNE adducts. SMCs were exposed to 4-HNE (50 μM) for 24 h in the absence or presence of 1 mM NAC. Under these circumstances, the formation of 4-HNE-protein adducts induced by excessive addition of exogenous 4-HNE to the cultured cells was abolished (Fig. 2e). The data indicated that NAC is a reliable 4-HNE-scavenging agent.
4-HNE increases Nox activity and expression in human SMCs
Following the above observations that 4-HNE-protein adducts were generated in SMCs exposed to high glucose (25 mM) or 1–5 μM 4-HNE (acute exposure), we asked whether 4-HNE alone could mimic the effects of long-term incubation with high glucose in these cells. Therefore, SMCs were exposed to increasing non-cytotoxic concentrations of 4-HNE (1–10 μM) for 24 h, and then the Nox activity and mRNA and protein levels were determined. Figure 3 shows that 4-HNE dose-dependently stimulated the activity, up-regulated the gene expression, and increased the protein level of each of the Nox isoforms. The maximal effect on Nox activity (≈2-fold) was observed with 5 or 10 μM 4-HNE, as assessed by lucigenin assay. A similar stimulatory effect of 4-HNE on intracellular O2 •- formation was confirmed by DHE assay (Electronic Supplementary Material, Fig. S1b). Likewise, a comparable regulatory pattern of Nox1, Nox4, and Nox5 gene and protein expression levels was detected following incubation of the cells with 5 or 10 μM 4-HNE (Fig. 3c–k).

Exposure of human aortic SMCs to exogenous 4-HNE increases Nox activity and expression. a Quiescent SMC were exposed to increasing concentrations of 4-HNE (24 h), and the NADPH-dependent O2 •- production was determined by lucigenin-enhanced chemiluminescence assay. b Analysis of SMC viability in response to 4-HNE stimulation by the E-Plates assay. c–h Concentration-dependent up-regulation of Nox1, Nox4, and Nox5 mRNA content (c-e) and protein levels (f–h) in 1–10 μM 4-HNE-treated cells. i–k Representative immunoblots depicting Nox protein regulation in response to exogenous 4-HNE. n = 4, *P < 0.05, **P < 0.01, ***P < 0.001. P-values were taken in relation to cells exposed to vehicle (ethanol)
High concentration of 4-HNE is cytotoxic to human SMCs
To determine the effects of 4-HNE on SMC viability, the impedance-based assay for real-time monitoring of cell dynamics (xCELLigence, Roche) was employed. SMCs were exposed in a single step to increasing concentrations of 4-HNE (1–25 μM), and the cell impedance was monitored continuously for 48 h. Figure 3b shows that the exposure of SMCs to 1–5 μM 4-HNE did not affect the rate of cell proliferation; this was reduced in the presence of 10 μM 4-HNE after 12 h incubation. However, high concentrations of 4-HNE (e.g., 25 μM) were cytotoxic. FBS (10 %) was employed as a positive control for SMC proliferation. The quantification of xCELLigence data is shown in the Electronic Supplementary Material (Fig. S2b). Collectively, the assay further supports the observation that the cytotoxic threshold of 4-HNE is above 10 μM.
NAC decreases high-glucose- and 4-HNE-induced augmented Nox activity and expression
NAC was employed to further investigate the idea that 4-HNE is the messenger of the high-glucose-induced up-regulation of Nox activity and expression. The cells were exposed to 5.5 mM or 25 mM glucose or 5 μM 4-HNE (5.5 mM glucose-containing medium) in the absence or presence of 1 mM NAC, and Nox activity and mRNA levels were measured. Figures S4 and S5 (Electronic supplementary material) show that NAC significantly reduced the increase of both activity and mRNA expression in cells exposed to high-glucose- or 4-HNE-induced augmented activity and expression of each Nox subtype. These findings indicate that, among other intracellular mediators triggered by high glucose, lipid peroxidation products such as 4-HNE play an important role in the regulation of the Nox complex.
High glucose activates PPARs in SMCs
Based on previous reports that link 4-hydroxyalkenals to PPAR activation (Coleman et al. 2007; Cohen et al. 2013), we asked whether such a mechanism existed in SMCs exposed to high glucose levels. To this end, we employed transactivation assays by using a pDR1-luc control plasmid carrying five repetitive and highly conserved peroxisome proliferator response elements (PPRE) cloned up-stream of the luciferase reporter gene. SMCs were transiently transfected with the plasmid in medium containing 5.5 mM glucose; at 24 h after transfection (baseline luciferase level), the cells were further exposed for 24 h to either 5.5 or 25 mM glucose, in the absence or presence of 5 μM of each PPAR antagonist (PPARα - GW6471, PPARβ/δ - GSK0660, PPARγ - GW9662) or NAC (1 mM). The results showed that high glucose alone significantly up-regulated the luciferase level directed by PPRE (≈1.8-fold) in comparison with the 5.5 mM glucose control, suggesting that various natural endogenous PPAR ligands are rapidly synthesized in response to high glucose stimulation. The pharmacological inhibition of PPARα or PPARβ/δ, but not of PPARγ, reduced significantly but also differentially the high-glucose-induced PPRE-dependent luciferase activity. Interestingly, the inhibition of PPARγ resulted in a slight reduction of high-glucose-induced DR1 activation. It is noteworthy that NAC completely abolished the luciferase induction in all experimental settings (Electronic Supplementary Material, Fig. S6).
4-HNE induces up-regulation of Nox via activation of PPARα and PPARβ/δ
The finding that high glucose activates PPARs prompted investigation into which of the various PPARs plays a role in the 4-HNE-induced up-regulation of Nox activity and expression. Quiescent SMCs were exposed (24 h) to 5 μM 4-HNE or to the vehicle (ethanol) in the absence or presence of siRNA sequences directed against each of the three PPAR isoforms. Figure 4a shows that 4-HNE-augmented Nox activity (≈two-fold) was differentially down-regulated by siRNA-mediated silencing of the PPARα (≈40 %) and PPARβ/δ (≈30 %). Likewise, pharmacological inhibition of PPARα or PPARβ/δ significantly reduced the high-glucose-induced up-regulation of NADPH oxidase-dependent O2 •- production (Electronic Supplementary Material, Fig. S7). Silencing of PPARα or PPARβ/δ led to a dissimilar reduction of 4-HNE-augmented Nox1, Nox4, and Nox5 mRNA and proteins. PPARγ silencing did not significantly affect the activity and expression of the Nox complex (Fig. 4c–k). No changes in Nox expression and activity were found in C siRNA-transfected cells. The decrease in PPAR proteins upon siRNA transfection in SMCs was confirmed by western blot (Fig. 4b). Furthermore, in other experiments, the cells were treated with specific PPAR antagonists. We observed that pharmacological inhibition of PPARα and PPARβ/δ resulted in a similar pattern of Nox regulation (data not shown). The data obtained in the SMC line were further validated in primary cultures of human aortic SMCs, by employing the same selective PPAR antagonists. The results obtained were comparable and showed that Nox activity and the mRNA levels of each Nox subtype were similarly regulated in both primary cultures and the SMC line (Electronic Supplementary Material, Fig. S8).

Production of 4-HNE-induced NADPH-dependent O2 •- and expression of Nox in human aortic SMCs is mediated by peroxisome proliferator-activated receptor-α (PPARα) and PPARβ/δ. Silencing of PPARα or PPARβ/δ down-regulates 4-HNE-induced Nox activity (a) and the mRNA (c–e) and protein (f–k) levels of Nox subtypes. b Representative immunoblots depicting the decrease in PPAR proteins upon short interfering RNA (siRNA) transfection in SMCs. Quiescent cells were exposed (24 h) to 5 μM 4-HNE in the absence or presence of siRNA sequences directed to silencing the expression of various PPAR subtypes. NADPH-dependent O2 •- production was determined by lucigenin-enhanced chemiluminescence. Nox gene and protein expression were assayed by real-time PCR and western blot, respectively. n = 4–5, *P < 0.05. P-values were taken in relation to siRNA-transfected cells (C siRNA)
Sequence analysis of human Nox1, Nox4, and Nox5 gene promoters
To further investigate the involvement of PPARs in the regulation of Nox subtype transcription, we performed in silico analysis (TRANSFAC) of the human Nox1, Nox4, and Nox5 proximal promoter regions. Interestingly, in silico prediction revealed the absence of typical PPRE within the promoter region of each Nox subtype. In addition to the previously indicated GAS, NF-kB, and C/EBP elements (Manea et al. 2010a, 2010b, 2012; Manea et al. 2014), the program identified the existence of highly conserved Sp1 binding sites (Electronic Supplementary Material, Fig. S9).
Discussion
Elevated oxidative stress attributable to the enhanced expression of vascular Nox enzymes has been reported in diabetic patients and in several experimental models of diabetes (Ding et al. 2007; Manea 2011; Sedeek et al. 2012a, 2012b; Gray et al. 2013). The molecular pathways of Nox regulation and the precise function of Nox-derived ROS have not yet been completely defined. Hitherto, no data have been available on the role and regulation of Nox5 in diabetes. Oxidative-stress-induced phenotypic alterations of SMCs have been reported to promote and accelerate the development of atherosclerotic lesions (Manea et al. 2008). Since oxidative stress is a common occurrence in atherosclerosis and diabetes, we have set up experiments to investigate the mediators and the signaling pathway involved in high-glucose-induced Nox and the role of glucose-derived 4-HNE in Nox function in SMCs.
The main findings of this study are: (1) high glucose conditions (mimicking diabetes) increase NADPH oxidase-dependent O2 •- production and Nox expression, in particular that of Nox1, Nox4, and Nox5 in SMCs (both the cell line and primary human cultures); (2) this is the first report showing that Nox5 is up-regulated by high glucose; (3) Nox-derived ROS contribute to 4-HNE generation; (4) in turn, 4-HNE plays a key role in the up-regulation of Nox expression and activity, and (5) high glucose up-regulates Nox expression via 4-HNE-activated PPARα and PPARβ/δ. A representation of the major findings of this study is presented in Fig. 5.

Representation of the high-glucose-induced “lipid peroxidation products–PPARs–Nox axis” leading to ROS formation in human vascular SMCs. High glucose induces the formation of 4-HNE in SMCs via redox-sensitive mechanisms as demonstrated by the inhibitory effects of DPI, apocynin, PEG-CAT, and NAC. Pharmacological inhibition/silencing of PPARα or PPARβ/δ diminishes 4-HNE-induced activity and the expression of Nox. This concept represents a novel mechanism of Nox regulation and the ensuing ROS formation in vascular SMCs exposed to hyperglycemia-mimicking conditions
Our experiments have revealed that, in SMCs, high glucose enhances NADPH oxidase-dependent O2 •- production, and that this condition is associated with significant elevations in Nox1, Nox4, and Nox5 expression levels and cell proliferation. Moreover, the data provide evidence that high-glucose-induced Nox expression is mediated by ROS, as demonstrated by the inhibitory effects of NAC (a potent antioxidant), which among its other functions, scavenges lipid peroxidation products, such as 4-HNE. Therefore, we assume that the highly reactive 4-HNE mediates, at least in part, the regulatory effects of high glucose.
The role of oxidative-stress-induced lipid peroxidation in diabetes is well-documented (Manea and Simionescu 2012). The peroxidation of hydroperoxy derivatives of n-3 and n-6 PUFAs is initiated by ROS attack and ultimately generates chemically reactive 4-hydroxyalkenals (Staels and Fruchart 2005; Cohen et al. 2012, 2013). Our study clearly shows that exposure of SMCs to high glucose concentrations induces progressive accumulation of 4-HNE-protein adducts.
Reportedly, activated Nox increases the rate of 4-HNE formation in monocytes (Kimura et al. 2005). To investigate the role of activated Nox in high-glucose-induced 4-HNE formation in SMCs, apocynin and DPI were employed. Our results showed that both compounds significantly inhibited the formation of 4-HNE. Mechanistically, apocynin blocks the assembly of Nox1- and Nox2-NADPH oxidases into active O2 •--generating complexes (Schlüter et al. 2008; Stefanska and Pawliczak 2008). Since SMCs typically express the Nox1, Nox4, and Nox5 isoforms, the inhibitory effect of apocynin is probably attributable to Nox1-containing Nox, rather than Nox4, which is constitutively active, and Nox5, which is Ca2+-dependent. Although apocynin is widely employed as a pharmacological inhibitor of Nox function, evidence exists that this compound is not a specific inhibitor of Nox activity but an antioxidant (Heumüller et al. 2008). Therefore, the involvement of other sources of ROS should also be considered when using apocynin. DPI, which binds to and inhibits flavin-containing oxidases (Ellmark et al. 2005), including all members of the Nox family, also reduces the formation of 4-HNE-protein adducts in high-glucose-exposed SMCs. In addition, the formation of 4-HNE is greatly decreased by PEG-CAT, a scavenger of intracellular H2O2 potentially generated by activated Nox4. Together, these data indicate that, in high-glucose-exposed SMCs, the generated Nox-derived ROS play a major role in lipid peroxidation.
Evidence exists that, in SMCs, Nox-derived ROS are induced by oxidized PUFAs (Li et al. 2003), and 4-HNE activates Nox in macrophages by mechanisms that are not completely defined (Yun et al. 2010). Our results extend these findings to SMCs, in which we report that 4-HNE increases NADPH oxidase-dependent O2 •- production in a concentration-dependent manner. In addition, we provide novel supporting data that the gene and protein levels of each Nox isoform are up-regulated in response to 4-HNE stimulation.
Some reports have shown that, at low and non-cytotoxic concentrations, 4-HNE functions as a natural activator of PPARβ/δ (Coleman et al. 2007; Russo 2009), whereas the progressive accumulation of 4-HNE-protein adducts alters normal cell functions leading ultimately to cell death (Cohen et al. 2011). PPARs regulate gene expression following dimeriziation with RXR and binding of the heterodimer to specific DNA sequence elements, the PPRE (Gross and Staels 2007). Most of the endogenous PPAR ligands are believed to be fatty acids or their derivatives, and a variety of naturally-occurring PPAR ligands are thought to be generated during cellular metabolic activities (Gervois et al. 2004). Consequently, in order to search for a role of PPARs in mediating high glucose signaling in SMCs, we have employed transactivation assays; these have demonstrated that high glucose induces PPAR-associated transcriptional responses. In addition, we have found that PPARα, PPARβ/δ, and PPARγ are differentially activated in response to high glucose. In particular, NAC blunts the high-glucose-induced PPRE-mediated luciferase level, suggesting that the activation of PPARs is redox-sensitive.
Compelling evidence has demonstrated the beneficial effects of diverse PPAR agonists in reducing oxidative stress and in improving vascular function in several animal models (Calkin et al. 2006; Quintela et al. 2012). These studies indicate that various PPAR agonists negatively regulate Nox expression and the ensuing ROS production and oxidative stress. Nevertheless, despite the large amount of existing data, uncertainty remains as to whether Nox are direct targets of PPARs, or whether their expression and function is controlled by indirect transcriptional mechanisms. These reports might also indicate that PPAR agonists induce systemic production of negative regulators of Nox.
In contrast, the genetic ablation of PPARα has been shown to abolish hypertension and to reduce atherosclerosis in hypertensive mice (Tordjman et al. 2007). These data bring into question the precise role of PPARs and the interpretation of the biological significance of PPAR-mediated mechanisms in the vasculature (Yagil and Yagil 2007).
Moreover, an interesting mechanism whereby PPARα agonists up-regulate Nox expression and activity has been previously demonstrated in both human and murine macrophages, and the activity and expression of Nox are lower in macrophages derived from PPARα-deficient mice (Teissier et al. 2004). Other than macrophages, PRARα agonists induce Nox-derived ROS and enhance cardiomyogenesis of mouse embryonic stem cells (Sharifpanah et al. 2008). Thus far, the direct involvement of the PPARβ/δ or PPARγ isoform in the regulation of Nox enzymes has not been investigated. In this context, the precise role and the mechanisms of action of PPAR members in cardiovascular cells remain elusive and require further attention (Lalloyer et al. 2011).
These studies have thus prompted us to investigate whether members of the PPAR family play a role in mediating 4-HNE-induced Nox expression in high-glucose-exposed human vascular SMCs. In order to ascertain the role played by the PPARs in the 4-HNE effect on Nox complexes, we have used various pharmacological agents and molecular interventions. Our results show that the inhibition and silencing of either PPARα or PPARβ/δ reduces significantly but also differentially 4-HNE-induced NADPH oxidase-dependent O2 •- production and the expression of Nox1/4/5 in both the SMC line and primary cultures of human vascular SMCs in which comparable PPARα- and PPARβ/δ-dependent mechanisms mediate the 4-HNE effects on Nox gene expression. In contrast, a dissimilar regulation was detected at the protein level in the SMC line. The silencing of PPARα or PPARβ/δ significantly decreases the Nox1 protein in 4-HNE-exposed cells. Although we have detected significant down-regulation in Nox4 and Nox5 mRNA expression in cells treated with specific siRNA sequences directed against PPARα or PPARβ/δ isoforms, no significant changes in the Nox4 protein level have been determined. Conversely, the expression of Nox5 protein is regulated by 4-HNE-activated PPARβ/δ. These results suggest that, in addition to PPARs, alternative signaling pathways, transcription factors, and mRNA processing mechanisms are involved in the regulation of Nox4 and Nox5 protein expression in response to 4-HNE stimulation. Our data also suggest that PPARγ is not involved in these activities.
To further investigate the molecular basis of Nox regulation by PPARs, we searched for the existence of PPRE within Nox1, Nox4, and Nox5 proximal promoter regions. However, in silico analysis has revealed that human Nox proximal promoters do not possess typical PPRE, and that most likely PPARα and PPARβ/δ regulate Nox expression by indirect transcriptional mechanisms. Of note, in addition to prototypical ligand-dependent transactivation activity, various pathways of PPAR-induced gene regulation exist (Ricote and Glass 2007). Reportedly, all PPARs can interact under physiological and different pathophysiological conditions with the Sp1 transcription factor (Gizard et al. 2005; Okazaki et al. 2010). In good agreement with these results, the existence of Sp1-binding sites has been predicted within human Nox1, Nox4, and Nox5 gene promoters by in silico analysis. In addition, we have reported that the Nox5 gene is positively regulated by Sp1 (Manea et al. 2012). Nevertheless, the precise nature of PPAR-Nox promoter interactions needs further elucidation. In addition to the major catalytic components (e.g., Nox1, Nox4, Nox5), the potential regulation of p22phox, the cytosolic regulatory subunits, and the expression-dependent and expression-independent mechanisms affecting Nox activity should be considered.
Collectively, the present data indicate the existence of a 4-HNE–PPAR-Nox axis in human vascular SMCs exposed to hyperglycemic-like conditions. Nevertheless, the role and the biological significance of the PPAR-dependent transcriptional regulation of Nox in diabetes remains to be further characterized in vivo. Based on previous findings (Teissier et al. 2004; Riahi et al. 2010a, 2010b) and on the results reported here, we can reliably propose that the 4-HNE generated via Nox activation functions as an endogenous activator of PPARα and PPARβ/δ. Furthermore, the exploration of the novel “lipid peroxidation products–PPARs–Nox axis” might lead to the discovery of alternative molecular mechanisms of ROS regulation and ensuing novel or additional therapeutic targets for oxidative-stress-related disorders.
References
Calkin AC, Cooper ME, Jandeleit-Dahm KA, Allen TJ (2006) Gemfibrozil decreases atherosclerosis in experimental diabetes in association with a reduction in oxidative stress and inflammation. Diabetologia 49:766–774
Cohen G, Riahi Y, Shamni O, Guichardant M, Chatgilialoglu C, Ferreri C, Kaiser N, Sasson S (2011) Role of lipid peroxidation and PPAR-δ in amplifying glucose-stimulated insulin secretion. Diabetes 60:2830–2842
Cohen G, Riahi Y, Sasson S (2012) Free radicals and metabolic disorders. In: Chatgilialoglu C, Studer A (eds) Handbook of radicals in chemistry and biology. Wiley, Chichester, pp 1679–1700
Cohen G, Riahi Y, Sunda V, Deplano S, Chatgilialoglu C, Ferreri C, Kaiser N, Sasson S (2013) Signaling properties of 4-hydroxyalkenals formed by lipid peroxidation in diabetes. Free Radic Biol Med 65:978–987
Coleman JD, Prabhu KS, Thompson JT, Reddy PS, Peters JM, Peterson BR, Reddy CC, Vanden Heuvel JP (2007) The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta). Free Radic Biol Med 42:1155–1164
Csányi G, Taylor WR, Pagano PJ (2009) NOX and inflammation in the vascular adventitia. Free Radic Biol Med 47:1254–1266
Das SK, Chakrabarti R (2006) Role of PPAR in cardiovascular diseases. Recent Pat Cardiovasc Drug Discov 1:193–209
Ding H, Hashem M, Triggle C (2007) Increased oxidative stress in the streptozotocin-induced diabetic apoE-deficient mouse: changes in expression of NADPH oxidase subunits and eNOS. Eur J Pharmacol 561:121–128
Ellmark SH, Dusting GJ, Fui MN, Guzzo-Pernell N, Drummond GR (2005) The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res 65:495–504
Forman BM, Chen J, Evans RM (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A 94:4312–4317
Gao L, Mann GE (2009) Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signaling. Cardiovasc Res 82:9–20
Gervois P, Fruchart JC, Staels B (2004) Inflammation, dyslipidaemia, diabetes and PPARs: pharmacological interest of dual PPARalpha and PPARgamma agonists. Int J Clin Pract Suppl 143:22–29
Gizard F, Amant C, Barbier O, Bellosta S, Robillard R, Percevault F, Sevestre H, Krimpenfort P, Corsini A, Rochette J, Glineur C, Fruchart JC, Torpier G, Staels B (2005) PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J Clin Invest 115:3228–3238
Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JP, Touyz RM, Wingler K, Cooper ME, Schmidt HH, Jandeleit-Dahm KA (2013) NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127:1888–1902
Gross B, Staels B (2007) PPAR agonists: multimodal drugs for the treatment of type-2 diabetes. Best Pract Res Clin Endocrinol Metab 21:687–710
Heltianu C, Guja C, Manea SA (2011) Genetic determinants of microvascular complications in type 1 diabetes. In: Wagner D (ed) Type 1 diabetes complications. InTech Open Access, Rijeka, pp 3–28
Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, Brandes RP (2008) Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51:211–217
Kahles T, Brandes RP (2012) NADPH oxidases as therapeutic targets in ischemic stroke. Cell Mol Life Sci 69:2345–2363
Kimura H, Liu S, Yamada S, Uchida K, Matsumoto K, Mukaida M, Yoshida K (2005) Rapid increase in serum lipid peroxide 4-hydroxynonenal (HNE) through monocyte NADPH oxidase in early endo-toxemia. Free Radic Res 39:845–851
Krause KH, Lambeth D, Krönke M (2012) NOX enzymes as drug targets. Cell Mol Life Sci 69:2279–2282
Lalloyer F, Wouters K, Baron M, Caron S, Vallez E, Vanhoutte J, Baugé E, Shiri-Sverdlov R, Hofker M, Staels B, Tailleux A (2011) Peroxisome proliferator-activated receptor-alpha gene level differently affects lipid metabolism and inflammation in apolipoprotein E2 knock-in mice. Arterioscler Thromb Vasc Biol 31:1573–1579
Li WG, Stoll LL, Rice JB, Xu SP, Miller FJ Jr, Chatterjee P, Hu L, Oberley LW, Spector AA, Weintraub NL (2003) Activation of NAD(P)H oxidase by lipid hydroperoxides: mechanism of oxidant-mediated smooth muscle cytotoxicity. Free Radic Biol Med 34:937–946
Manea A (2010) NADPH oxidase-derived reactive oxygen species: involvement in vascular physiology and pathology. Cell Tissue Res 342:325–339
Manea A (2011) Vascular biology of reactive oxygen species and NADPH oxidases: role in atherogenesis. In: Paryhasarathy S (ed) Atherogenesis. InTech Open Access, Rijeka, 425–446
Manea A, Simionescu M (2012) Nox enzymes and oxidative stress in atherosclerosis. Front Biosci (Schol Ed) 4:651–670
Manea A, Manea SA, Gafencu AV, Raicu M, Simionescu M (2008) AP-1-dependent transcriptional regulation of NADPH oxidase in human aortic smooth muscle cells: role of p22phox subunit. Arterioscler Thromb Vasc Biol 28:878–885
Manea A, Tanase LI, Raicu M, Simionescu M (2010a) Jak/STAT signaling pathway regulates Nox1 and Nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol 30:105–112
Manea A, Tanase LI, Raicu M, Simionescu M (2010b) Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem Biophys Res Commun 396:901–907
Manea A, Manea SA, Florea IC, Luca CM, Raicu M (2012) Positive regulation of NADPH oxidase 5 by proinflammatory-related mechanisms in human aortic smooth muscle cells. Free Radic Biol Med 52:1497–1507
Manea SA, Todirita A, Manea A (2013) High glucose-induced increased expression of endothelin-1 in human endothelial cells is mediated by activated CCAAT/enhancer-binding proteins. PLoS One 8:e84170
Manea SA, Todirita A, Raicu M, Manea A (2014) C/EBP transcription factors regulate NADPH oxidase in human aortic smooth muscle cells. J Cell Mol Med 18:1467–1477
Okazaki M, Iwasaki Y, Nishiyama M, Taguchi T, Tsugita M, Nakayama S, Sevestre H, Krimpenfort P, Corsini A, Rochette J, Glineur C, Fruchart JC, Torpier G, Staels B (2010) PPARbeta/delta regulates the human SIRT1 gene transcription via Sp1. Endocr J 57:403–413
Plutzky J (2011) The PPAR-RXR transcriptional complex in the vasculature: energy in the balance. Circ Res 108:1002–1016
Quintela AM, Jiménez R, Gómez-Guzmán M, Zarzuelo MJ, Galindo P, Sánchez M, Vargas F, Cogolludo A, Tamargo J, Pérez-Vizcaíno F, Duarte J (2012) Activation of peroxisome proliferator-activated receptor-β/-δ (PPARβ/δ) prevents endothelial dysfunction in type 1 diabetic rats. Free Radic Biol Med 53:730–741
Riahi Y, Cohen G, Shamni O, Sasson S (2010a) Signaling and cytotoxic functions of 4-hydroxyalkenals. Am J Physiol Endocrinol Metab 299:E879–E886
Riahi Y, Sin-Malia Y, Cohen G, Alpert E, Gruzman A, Eckel J, Staels B, Guichardant M, Sasson S (2010b) The natural protective mechanism against hyperglycemia in vascular endothelial cells: roles of the lipid peroxidation product 4-hydroxydodecadienal and peroxisome proliferator-activated receptor delta. Diabetes 59:808–818
Ricote M, Glass CK (2007) PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta 1771:926–993
Russo GL (2009) Dietary n-6 and n-3 polyunsaturated fatty acids: from biochemistry to clinical implications in cardiovascular prevention. Biochem Pharmacol 77:937–946
Schlüter T, Steinbach AC, Steffen A, Rettig R, Grisk O (2008) Apocynin-induced vasodilation involves Rho kinase inhibition but not NADPH oxidase inhibition. Cardiovasc Res 80:271–279
Sedeek M, Callera G, Montezano A, Gutsol AF, Heitz F, Szyndralewiez C, Page P, Kennedy CR, Burns KD, Touyz RM, Hébert RL (2012a) Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Ren Physiol 299:F1348–F1358
Sedeek M, Montezano AC, Hebert RL, Gray SP, Di Marco E, Jha JC, Cooper ME, Jandeleit-Dahm K, Schiffrin EL, Wilkinson-Berka JL, Touyz RM (2012b) Oxidative stress, Nox isoforms and complications of diabetes-potential targets for novel therapies. J Cardiovasc Transl Res 5:509–518
Sharifpanah F, Wartenberg M, Hannig M, Piper HM, Sauer H (2008) Peroxisome proliferator-activated receptor alpha agonists enhance cardiomyogenesis of mouse ES cells by utilization of a reactive oxygen species-dependent mechanism. Stem Cells 26:64–71
Staels B (2007) PPAR agonists and the metabolic syndrome. Therapie 62:319–332
Staels B, Fruchart JC (2005) Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes 54:2460–2470
Stefanska J, Pawliczak R (2008) Apocynin: molecular aptitudes. Mediat Inflamm 2008:106507
Teissier E, Nohara A, Chinetti G, Paumelle R, Cariou B, Fruchart JC, Brandes RP, Shah A, Staels B (2004) Peroxisome proliferator-activated receptor alpha induces NADPH oxidase activity in macrophages, leading to the generation of LDL with PPAR-alpha activation properties. Circ Res 95:1174–1182
Tîrziu D, Jinga VV, Serban G, Simionescu M (1999) The effects of low density lipoproteins modified by incubation with chondroitin 6-sulfate on human aortic smooth muscle cells. Atherosclerosis 147:155–166
Tordjman KM, Semenkovich CF, Coleman T, Yudovich R, Bak S, Osher E, Vechoropoulos M, Stern N (2007) Absence of peroxisome proliferator-activated receptor-alpha abolishes hypertension and attenuates atherosclerosis in the Tsukuba hypertensive mouse. Hypertension 50:945–951
Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL (2002) Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res 90:1205–1213
Ungvari Z, Csiszar A, Edwards JG, Kaminski PM, Wolin MS, Kaley G, Koller A (2003) Increased superoxide production in coronary arteries in hyperhomocysteinemia: role of tumor necrosis factor-alpha, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol 23:418–424
Yagil C, Yagil Y (2007) Peroxisome proliferator-activated receptor alpha: friend or foe? Hypertension 50:847–850
Yun MR, Park HM, Seo KW, Lee SJ, Im DS, Kim CD (2010) 5-Lipoxygenase plays an essential role in 4-HNE-enhanced ROS production in murine macrophages via activation of NADPH oxidase. Free Radic Res 44:742–750
Zalba G, San José G, Beaumont FJ, Fortuño MA, Fortuño A, Díez J (2001) Polymorphisms and promoter overactivity of the p22(phox) gene in vascular smooth muscle cells from spontaneously hypertensive rats. Circ Res 88:217–222
Acknowledgments
We acknowledge the skillful assistance of Floarea Georgescu, Camelia Matei, and Constanta Stan.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by the European Foundation for the Study of Diabetes – The New Horizons Grant to A. Manea in collaboration with S. Sasson, Romanian National Authority for Scientific Research (CNCS-UEFISCDI, project numbers PN-II-ID-PCE-2011-3-0548, PN-II-RU-TE-2011-3-0142, PNII-TE 65/2010), and COST action BM1203/EU-ROS. Simona A. Manea acknowledges the support of the strategic grant POSDRU/159/1.5/S/133391 financed by the European Social Found within the Sectorial Operational Program Human Resources Development 2007–2013. S. Sasson is the Adolf D. and Horty Storch Chair in Pharmaceutical Sciences, at the Faculty of Medicine, The Hebrew University of Jerusalem, Israel. He is affiliated with the David R. Bloom Center for Pharmacy and the Dr. Adolf and Klara Brettler Center for Research of Molecular Pharmacology and Therapeutics in the Hebrew University.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 741 kb)
Rights and permissions
About this article

Cite this article
Manea, A., Manea, SA., Todirita, A. et al. High-glucose-increased expression and activation of NADPH oxidase in human vascular smooth muscle cells is mediated by 4-hydroxynonenal-activated PPARα and PPARβ/δ. Cell Tissue Res 361, 593–604 (2015). https://doi.org/10.1007/s00441-015-2120-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-015-2120-0