
Abstract
Ovarian cancer (OC) is a fatal gynecological disease that is often diagnosed at later stages due to its asymptomatic nature and the absence of efficient early-stage biomarkers. Previous studies have identified genes with abnormal expression in OC that couldn’t be explained by methylation or mutation, indicating alternative mechanisms of gene regulation. Recent advances in human transcriptome studies have led to research on non-coding RNAs (ncRNAs) as regulators of cancer gene expression. Long non-coding RNAs (lncRNAs), a class of ncRNAs with a length greater than 200 nucleotides, have been identified as crucial regulators of physiological processes and human diseases, including cancer. Dysregulated lncRNA expression has also been found to play a crucial role in ovarian carcinogenesis, indicating their potential as novel and non-invasive biomarkers for improving OC management. However, despite the discovery of several thousand lncRNAs, only one has been approved for clinical use as a biomarker in cancer, highlighting the importance of further research in this field. In addition to their potential as biomarkers, lncRNAs have been implicated in modulating chemoresistance, a major problem in OC. Several studies have identified altered lncRNA expression upon drug treatment, further emphasizing their potential to modulate chemoresistance. In this review, we highlight the characteristics of lncRNAs, their function, and their potential to serve as tumor markers in OC. We also discuss a few databases providing detailed information on lncRNAs in various cancer types. Despite the promising potential of lncRNAs, further research is necessary to fully understand their role in cancer and develop effective strategies to combat this devastating disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ovarian cancer (OC) is a deadly gynecological malignancy accounting for 3.4% of all cancer cases (Sung et al. 2021). It ranks as the 8th most common cancer among women worldwide (Cancer today 2021), with 313,959 new cases and 207,252 deaths attributed to OC in 2020 (Sung et al. 2021). These numbers account for a significant portion of the 19.3 million new cancer cases and 10 million cancer-related deaths globally. OC is the 5th leading cause of gynecological cancer-related mortalities worldwide (Siegel et al. 2017), and by 2040, the estimated number of OC-related incident and mortality cases is expected to rise to 440,000 310,000, respectively (Cancer Tomorrow 2022). Unfortunately, OC is typically asymptomatic, and the majority of patients are diagnosed at late stages; 51% at stage III and 29% at stage IV, primarily due to the absence of reliable early-stage biomarkers (Torre et al. 2018). This, coupled with drug treatment resistance and recurrence (Norouzi-Barough et al. 2018), is a major cause of poor overall survival (Wang et al. 2019a) (Coticchia et al. 2008). The five year survival rate for early-stage OC is 89%, compared to only 20% for stage IV (Ovarian Cancer 2023). Despite advancements in OC research, fatality rates remain unchanged due to the lack of robust clinical tools for early detection (Galea et al. 2017; Chandra et al. 2019). Therefore, elucidating the process of ovarian cancer pathogenesis and identifying potential biomarkers, such as differentially expressed genes, for OC diagnosis, prognosis, and treatment is crucial (Zheng et al. 2019a).
The pathogenesis of ovarian cancer involves a series of molecular and genetic changes in healthy ovarian cells, leading to the onset and progression of cancer. However, the heterogeneity of OC complicates efforts to understand its pathogenesis (Gross et al. 2010; Kurman and Shih 2016). Various theories have been proposed to elucidate the mechanisms underlying OC pathogenesis. According to the theory of incessant ovulation, surface epithelial cells, which were previously believed to be the origin of OC, undergo physical damage that is rapidly repaired. The repetitive ovulation during a women’s life causes repeated damage to the epithelial cells, resulting in DNA damage. These cells, which are continually exposed to ovarian hormones, promote malignant growth, leading to the neoplastic transformation into cancer cells (Kurman and Shih 2010; Erickson et al. 2013; Budiana et al. 2019).
The dualistic model theory proposed by Kurman and Shih categorizes ovarian tumors into two types, namely Type-I and type-II, based on clinical, genetic, and histological observations (Kurman and Shih 2010; Koshiyama et al. 2014). Type-I ovarian carcinomas include low-grade serous, clear-cell, endometrioid, mucinous, and malignant Brenner tumors. These tumors are characterized by being low grade, indolent, and confined to the ovary at presentation, with better survival outcomes. Type-I tumors exhibit specific genetic mutations such as KRAS, PTEN, BRAF, ARID1A, PIK3CA, and ERBB2, and they are genetically stable (Koshiyama et al. 2014). The progression of Type-I cancers follows a series of sequential steps from benign precursor lesions to malignant lesions.
On the other hand, Type-II ovarian carcinomas encompass high-grade serous, high-grade endometrioid, malignant mixed, undifferentiated tumors, and mesodermal tumors. These tumors are high-grade, aggressive, and associated with unfavorable survival outcomes. They are characterized by TP53 mutations (Ledermann et al. 2013; Koshiyama et al. 2014; Chen et al. 2014; Kurman and Shih 2016; Aluloski et al. 2017; Ricciardi et al. 2018). In Type-II ovarian cancer, the precursor originates from fallopian tube, and TP53 mutations contribute to the neoplastic changes in the epithelial cells of the fallopian tube (Budiana et al. 2019). Subsequently, these cancerous cells metastasize to the ovaries and peritoneal cavities.
Previous studies in cancer have revealed aberrant gene expression that couldn’t be explained by methylation or mutation alone. Dysregulated expression of these genes could be due to their regulation by non-coding RNAs (ncRNAs), such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) (Al Saqri et al. 2022; Malgundkar et al. 2022). For many years, cancer research focused primarily on protein-coding genes. However, the discovery that protein-coding genes account for only 1.5–3% of the total genome, while the majority is transcribed to ncRNAs (15–17), has revolutionized the field. The increasing number of studies on ncRNAs suggests that they have functions beyond being an intermediary between DNA and protein. NcRNAs are a class of RNAs with no protein coding ability known to interact with DNA, RNA, or protein to modulate gene expression at the epigenetic, transcriptional, and post-transcriptional levels (Holoch and Moazed 2015).
NcRNAs, particularly lncRNAs, play a crucial role in regulating gene expression in differentiation, development, and human diseases. Given the aberrant expression of lncRNAs, they could also play a significant role in the onset and development of OC. Furthermore, the presence of lncRNAs in biological fluids makes them a promising biomarker for OC. While the list of functionally characterized lncRNAs in the cancer field is rapidly expanding, the question arises as to whether lncRNAs are solely transcriptional noise or have biological functions. Moreover, the study of lncRNAs is not as extensive as that of miRNAs, and many unknowns exist regarding their function and involvement in cancer, including OC (Salamini-Montemurri et al. 2020).
This review aims to elucidate the involvement of lncRNAs in OC, providing insights into the mechanisms by which lncRNAs contribute to the disease and their potential as diagnostic and therapeutic targets. Additionally, we will discuss several key findings on lncRNAs as biomarkers for OC.
ncRNAs and its classification
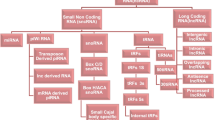
RNA is divided into two categories: messenger RNA (coding RNA) and non-coding RNA (ncRNA). Messenger RNA (mRNA) is the most well-known and studied type of RNA, constituting only 2% of the coding part of the human genome. Non-coding RNAs are further classified as structural or regulatory. House-keeping ncRNAs include transfer RNA (tRNA), ribosomal RNA (rRNA), small nucleolar RNAs (snoRNAs), and small nuclear RNAs (snRNAs). Regulatory ncRNAs are sub-classified as short or long ncRNAs based on their length. Short ncRNAs include piwi-interacting RNAs (piRNAs), microRNAs (miRNAs), and short interfering RNAs (siRNAs) (Nagano and Fraser 2011; Aliperti et al. 2021; Ahadi 2021) Fig. 1.

Classification of RNA molecule
miRNAs are short ncRNAs composed of 22 nucleotides, with a key role in post transcriptional level gene silencing. miRNAs achieve this through binding to specific sequence (3’ untranslated region) known as miRNA recognition element (MRE) on target mRNA transcripts (Ha and Kim 2014; Yang et al. 2017; Treiber et al. 2019). A previous finding revealed > 60% mRNA were regulated by miRNAs (Friedman et al. 2009). mRNAs that are perfectly complementary to the miRNA undergo cleavage via endonucleolytic activity. On the contrary, in case of partial complementarity, mRNAs are subjected to translational repression (Magri et al. 2018). Several miRNAs have been reported to be aberrantly expressed in OC, and was found to be involved in OC pathogenesis (Zaman et al. 2012; Pal et al. 2015; Mahdian-Shakib et al. 2016).
The oncogenic or tumor suppressive effects of miRNA occur through the regulation of either tumor suppressive or oncogenic protein coding genes (Svoronos et al. 2016). Overexpression of oncogenic miRNAs silences tumor suppressive genes, thereby promoting cancer. Similarly, lower levels of tumor suppressive miRNAs enhances oncogene expression (Otmani and Lewalle 2021). Aberrant miRNA expression have the ability to regulate cancer related processes such as inducing invasion, metastasis, angiogenesis, evading growth suppressors, maintaining cell growth signals (Rishabh et al. 2021).
While miRNAs acts as post-transcriptional regulators in the cytoplasm, lncRNAs occur in the cytoplasm and nucleus indicating its potential to serve as epigenetic modifiers of chromatin thereby regulating gene expression (Kapranov et al. 2010; Derrien et al. 2012; Fort et al. 2014). Furthermore, abnormal levels of miRNA have been closely associated with lncRNAs (Ye et al. 2018). The regulatory interactions between lncRNAs and miRNAs occur in two distinct ways. Firstly, lncRNAs can modulate miRNAs by serving as its precursor or can act as miRNA sponge. Secondly, miRNAs can control lncRNA expression through epigenetic mechanism. Moreover, miRNAs could trigger lncRNA degradation via argonaute (Nukala et al. 2022).
Several miRNAs have been identified as being aberrantly expressed in OC and have been implicated in its pathogenesis (Zaman et al. 2012; Pal et al. 2015; Mahdian-Shakib et al. 2016). For instance, miR-600 was found to be significantly over-expressed in OC tissue samples, leading to the downregulation of KLF9 expression and promoting OC cell proliferation and metastasis (Shan et al. 2022). Similarly, miR-149-3p facilitated EMT and cisplatin resistance in OC by reducing TIMP2 and CDKN1A levels (Wang and Liu 2021).
Another study revealed the downregulation of miR-425 in OC tissues and cell lines, which was correlated with poor overall survival. Furthermore, miR-425 was shown to inhibit OC cell migration, invasion, and proliferation by regulating PAK4 levels (Wang et al. 2022). Also, investigating the lncRNA-miRNA-mRNA network could provide insights into the mechanisms underlying OC pathogenesis, such as OIP5-AS1-miR-203a-ZEB2 axis, which regulates OC progression through EMT and metastasis (Pronina et al. 2022). Abnormal levels of miRNA have been found to be closely associated with lncRNAs (Ye et al. 2018). This study will exclusively focus on describing the role of lncRNA in OC.
Overview on lncRNAs
Recent studies have revealed that a significant portion of the human genome (80%) is actively transcribed into RNA, despite having no protein-coding potential (Derrien et al. 2012; Djebali et al. 2012; de Hoon et al. 2015). A large subset of these transcripts, exceeding 200 nucleotides in length, is known as lncRNA (Nagano and Fraser 2011). LncRNAs are transcribed from the human genome by RNA Polymerase II and lack protein-coding open reading frames (Gibb et al. 2011). In a wide-scale analysis of 91,013 transcribed human genes across more than 7250 tissue samples, lncRNAs constituted around 70% (58,648 in number) of human transcripts, with 79% of these being unannotated (Iyer et al. 2015). Although over 90,000 lncRNAs have been identified in some studies, only 16,000 have been annotated in GENCODE (Harrow et al. 2012; Iyer et al. 2015). Given that the majority of the human genome is non-protein coding, and the potential of these non-coding genes to serve as biomarkers, research focus has shifted to lncRNAs, which were previously considered to be transcriptional noise (Shi et al. 2016).
Characteristics and classification of lncRNAs
LncRNAs share similarities with mRNAs, which distinguishes them from short ncRNAs that can be easily differentiated from mRNAs (Ponting et al. 2009). While lncRNAs possess a 5’ cap, exons, and poly-A tails like mRNA, they do not have functional open reading frames or protein-coding functions (Fang and Fullwood 2016). Approximately 50% of lncRNAs have a polyA tail, and nearly all are spliced (Derrien et al. 2012). Like mRNAs, lncRNA are transcribed by RNA Polymerase II (Derrien et al. 2012), but they typically have fewer exons and are longer in length (Cabili et al. 2011; Harrow et al. 2012). Despite lacking sequence conservation, LncRNAs can play a modulatory role due to the highly conserved secondary and tertiary structures of lncRNAs (Li et al. 2016b). However, their primary structure conservation is poor (Aliperti et al. 2021), as lncRNAs lack evolutionary conservation and exhibit differential expression, as revealed by the presence of epigenetic markers (Guttman et al. 2009).
In contrast to mRNAs, only 11–29% of lncRNAs are expressed in all tissues and at low levels (Derrien et al. 2012; Djebali et al. 2012). Additionally, lncRNAs exhibit high tissue specificity (~ 78%) compared to mRNAs (~ 19%) (Cabili et al. 2011). Certain lncRNAs undergo transcriptional and post-transcriptional modifications identical to protein-coding genes (Ponting et al. 2009). Furthermore, lncRNAs exhibit characteristic functions in different physiological processes, as indicated by their tissue-cell type-specific expression compared to mRNAs (Cabili et al. 2011; Derrien et al. 2012; Djebali et al. 2012).
Almost thirty years ago, the first lncRNAs, H19 and XIST, were discovered and studied for their roles in chromosome dosage compensation and genomic imprinting (Brannan et al. 1990; Brown et al. 1991). According to Brannan et al. (1990), the RNA molecule H19 was found to be distinct from classical mRNAs despite being transcribed by RNA polymerase II, undergoing splicing and polyadenylation. This distinction was due to the absence of a conserved open reading frame in the H19 RNA molecule.
Recently, lncRNAs was identified to play a crucial role in several biological processes, including RNA splicing, cell proliferation, cell differentiation, epigenetic silencing, transcription (Guttman et al. 2009), cell cycle regulation (Kitagawa et al. 2013; Heydarnezhad Asl et al. 2022), DNA methylation (Yang et al. 2022), antiviral immunity (van Solingen et al. 2022), specifying cell identity (Hu et al. 2012) and apoptosis (Jiang et al. 2021). LncRNAs achieve their functions by regulating gene expression at the epigenetic, transcriptional, and post-transcriptional levels (Kornienko et al. 2013; Fatica and Bozzoni 2014; Renganathan and Felley-Bosco 2017). The majority of lncRNAs are enriched in the nucleus, where they regulate post-transcription (Wang and Chang 2011; Quinn and Chang 2016). Additionally, lncRNAs can act as competing endogenous RNA (ceRNA) by serving as a miRNA sponge, regulating miRNA abundance, and competing with mRNA for miRNA binding (Ye et al. 2018). Furthermore, Tian et al. identified a direct association between the lncRNA WDR7-7 and the mRNA GPR30 (Tian et al. 2017), highlighting its ability to regulate gene expression through means other than the well-known ceRNA network.
Classifying lncRNAs is challenging due to their structural variations, poor sequence conservation, various molecular mechanisms of action, and differential subcellular localization (Mattick and Rinn 2015). LncRNAs can be broadly classified based on their location in the genome, mechanism of action, and function. They are categorized as intronic, intergenic, enhancer, and antisense lncRNAs based on location. Intronic lncRNAs arise from the intronic region of protein-coding genes, intergenic lncRNAs arise from the region between two protein-coding genes, enhancer lncRNAs are produced from the promoter enhancer region, and antisense lncRNAs arise from the antisense strand of the gene (Ma et al. 2013). Based on function, lncRNAs are categorized as signaling, guiding, decoy, and scaffold lncRNAs. Signaling lncRNAs are linked to specific signaling pathways, guiding lncRNAs are associated with regulatory protein complexes and direct them to particular target gene promoters, thereby regulating downstream gene expression (Wang and Chang 2011). Decoy lncRNA titrates the transcription factors away from the target gene promoters, preventing them from binding and promoting or inhibiting gene expression. Additionally, scaffold lncRNAs serve as platforms for several protein complexes, directing them to specific gene locations (Wang and Chang 2011; Ma et al. 2013).
Gene expression profiling techniques
Most of the lncRNA based research involve the use of bioinformatic analysis, RNA sequencing, microarray, functional assays for the identification and characterization of lncRNAs in cancer. Integrating multiple approaches for experimental validation and functional analysis is important to determine and characterize the lncRNAs.
Gene expression technology can be used to identify the link between genetic aberrations and OC pathogenesis, providing insight into the process of cancer onset and progression. These techniques also have the potential to reveal novel OC biomarkers (Zhang et al. 2011). The field of functional genomics has rapidly developed due to advanced computational approaches and high-throughput sequencing technologies (65, 66). Recently, sophisticated molecular biology techniques such as Next Generation Sequencing (NGS), microarrays, and Chromatin immunoprecipitation (ChIP) have been used to identify differentially expressed genes, including lncRNAs. These genes can potentially be used as biological biomarkers in epithelial ovarian cancer (EOC) (El Bairi et al. 2017).
NGS technology enables the simultaneous sequencing of millions of gene fragments (Kamps et al. 2017), providing insight into the various signaling pathways involved in OC progression (Gov et al. 2017). The most commonly employed techniques for the discovery and annotation of novel lncRNA transcripts of low abundance is RNA-seq, which also detects the lncRNA isoforms. (as reviewed in (Chowdhary et al. 2021)), however it is time consuming and costly (Lee and Kikyo 2012). Currently, RNA sequencing techniques have led to the detection of several lncRNAs, although only a few have been well characterized (Zhan et al. 2018). For instance, Abildgaard et al. identified the lncRNA SNHG12, which regulate carboplatin resistance using RNA sequencing in OC cell lines (Abildgaard et al. 2022). Similarly, RNA sequencing in OC and healthy ovarian tissues revealed 6,764 aberrantly expressed genes (Guo et al. 2020). However, this technique requires sophisticated bioinformatics techniques to interpret the data (as reviewed in Rao et al. 2019) and to predict lncRNAs.
Microarray is a widely used technique to simultaneously assess the expression of thousands of genes. It provides key data on the molecular differences in tumors, OC prognosis, mechanisms of resistance to chemotherapy, and response to chemotherapy (Spentzos et al. 2004; Konstantinopoulos et al. 2008). The use of microarray for lncRNA research is limited to only known lncRNA sequences, and it cannot detect novel lncRNAs. Microarray analysis has revealed differentially expressed lncRNA in OC samples compared to normal tissues, providing insight into their role as biomarkers (Ding et al. 2017). In a previous microarray study, 4,289 differentially expressed lncRNAs were identified in high-grade serous ovarian cancer (HGSOC) compared to healthy fallopian tissue samples (Lou et al. 2018). Similarly, another study reported 3527 aberrantly expressed lncRNAs in SKOV3 cells compared to IOSE80 cells, while 9706 dysregulated lncRNAs were identified in the cisplatin-resistant variant of SKOV3 cells (Gao et al. 2019a). Therefore, microarray analysis is a crucial tool to understand the function of lncRNAs in OC and provides a means to elaborate on the mechanisms underlying carcinogenesis, helping researchers identify significant lncRNAs for further analysis.
Many important biological processes, such as cell cycle progression, cell differentiation, transcription, DNA replication, and epigenetic silencing, rely on the interactions between DNA and proteins (Mundade et al. 2014). Chromatin immunoprecipitation (ChIP-seq) is an emerging technique that is often used in combination with DNA sequencing to identify the genomic DNA segments bound to the protein of interest, such as transcription factors, co-factors, and chromatin remodeling complexes (Wiehle and Breiling 2016; McColl et al. 2019). Therefore, ChIP is a useful technique for unraveling the molecular mechanisms of gene regulation (Mundade et al. 2014). In a previous ChIP study performed on OC cell lines, several lncRNAs that interact with E2F5, a transcription factor involved in the pathogenesis of EOC, were identified (Malgundkar et al. 2022).
To investigate the interacting partners of lncRNAs, several techniques have been developed. One commonly used technique is RNA immunoprecipitation (RNA-IP), which enables the detection of interactions between specific proteins and RNA molecules. RNA-IP is particularly useful for studying lncRNA-protein interactions and identifying lncRNAs that form complexes with specific proteins of interest (Zhang et al. 2016a). However, one disadvantage of this technique is its potential lack of specificity in identifying RNA interactions (Cao et al. 2019).
Another technique employed to determine RNA–protein interactions is cross-linking and immunoprecipitation (CLIP). This technique involves cross-linking RNA and interacting proteins, followed by immunoprecipitation to isolate RNA–protein complexes (Hafner et al. 2021).To identify the interactions between lncRNAs and chromatin, one widely used technique is chromatin isolation by RNA purification (ChIRP) (Chu and Chang 2016). For example, ChIRP was used to determine the interaction between the lncRNA NEAT 1–1 and RUNX2 promoter (Wen et al. 2020). Similarly, ChIRP was used to identify the interaction between the lncRNA GAS5 and miR-18a-5p in bladder cancer cells to elucidate its mechanism of action (Zhang et al. 2022).
To determine the function of lncRNAs, one common approach is to silence the lncRNA using siRNA and GAPmers and analyze the resulting changes in gene expression to identify the genes influenced by the lncRNA (Chowdhary et al. 2021). Computational approaches for identifying lncRNA function are more challenging due to factors such as tissue specificity, low expression levels, and interactions with multiple molecular components (Chowdhary et al. 2021).
lncRNAs in cancer
LncRNAs has been linked to the onset and progression of several human diseases, including cancer, as the aberrant expression of lncRNAs can function as oncogenes or tumor suppressor genes by regulating tumor cell migration, apoptosis, proliferation, invasion, and metastasis (Li et al. 2021b). Activation of oncogenic signaling pathways, epigenetic modifications, and interactions with other RNA molecules are known to promote cancer progression by modulating lncRNA expression. lncRNAs have been found to be differentially expressed in different types of cancer. For example, the lncRNA H19 expression levels was reported to be higher in breast cancer cell lines and tissue samples. Moreover, by regulating TNFAIP8 levels, H19 was found to promote cell migration, invasion, proliferation, and EMT, (Li et al. 2020). H19 was also found to be upregulated in gastric cancer cell lines and tissues, which resulted in activation of the Wnt/β catenin pathway by promoting nuclear translocation of β catenin. This in turn resulted in EMT and metastasis of gastric cancer cells (Liu et al. 2021). In OC, H19 was significantly higher in OC samples and cell lines, which promoted invasion, migration, proliferation and EMT through modulation of the PI3K/ AKT pathway (Xu et al. 2021). Table 1 summarizes how lncRNAs can regulate OC by functioning as scaffolds, guides, decoys, or signaling molecules.
lncRNAs reflect the physiological condition of the cell due to their specific expression in tissues and development specific expression (Chowdhary et al. 2021). High-grade serous OC is the most lethal subtype of OC, characterized by low five-year survival rates and a poor prognosis. Detecting the genes associated with OC onset and progression, as well as understanding their regulatory mechanisms, is crucial for managing OC (Lou et al. 2018). Analyzing the expression status of lncRNA has the potential to classify cancer subtypes (Flippot et al. 2016). In a previous microarray profiling study, differential expression of 663 lncRNA transcripts was observed in healthy ovarian samples, benign ovarian cysts, and EOC samples. Among these, 182 lncRNAs were overexpressed, and 481 were under-expressed in EOC samples, suggesting their function as tumor suppressor/oncogene and biomarkers (Wang et al. 2016). (Wang et al. 2016). Another microarray analysis comparing high-grade serous OC samples and healthy fallopian tube samples revealed that 1,511 lncRNAs were overexpressed, while 2,778 lncRNAs were down-regulated in OC (Lou et al. 2018). The study identified several upregulated lncRNAs, including RP11-199F11.2, FAS-AS1, AC093818.1, and AK130076, as well as the downregulation of GTSE1-AS1.
Additionally, the expression of associated mRNA, such as GSTE1, PDK1, TP53, PTEN, and FAS, was found to be altered in high-grade serous OC samples, suggesting the involvement of both mRNA and their associated lncRNAs in OC progression (Lou et al. 2018). Furthermore, dysregulated lncRNA expression was associated with lymph node metastases and OC FIGO stages in high-grade serous OC patients’ samples, indicating its potential association with ovarian carcinogenesis and tumor development (Lou et al. 2018).
The lncRNA MALAT-1 was significantly upregulated in primary OC samples compared to healthy ovarian tissues. Moreover, MALAT-1 expression was associated with the FIGO stage, indicating its involvement in OC progression (Zhou et al. 2016). On the other hand, the lncRNA MEG3, a tumor suppressor, was significantly downregulated in primary OC samples as well as metastatic tumors compared to healthy ovarian and benign ovarian cancer samples. Additionally, MEG3 expression showed a negative correlation with the FIGO stage (Xiu et al. 2017).
Regulation of gene expression by lncRNA
Regulating gene expression is a fundamental life process, and its modulation is essential for the development of an organism (Schmitt and Chang 2016). Transcription factors, cofactors, chromatin modulators, and aberrant gene regulation can result in a range of malignancies, highlighting the importance of regulating gene expression patterns (Lee and Young 2013). Critical factors in gene expression regulation include DNA methylation, histone modifications, and ncRNA-associated epigenetic mechanisms (Miller and Grant 2013). One of the most significant functions of lncRNA is epigenetic regulation, achieved through interaction with epigenetic modifiers such as Polycomb repressive complexes (PRC) and histone methyltransferases (Hanly et al. 2018). Aberrant regulation of epigenetic mechanisms is a key characteristic of cancer and contributes to its onset and progression (Shen and Laird 2013).
An example of the role of lncRNA in cancer is the lncRNA KCNQ1OT1, which was found to reduce the EIF2B5 levels by recruiting DNA methyltransferase to the EIF2B5 promoter, resulting in OC metastasis (He et al. 2022). Similarly, UNC5B-AS1 interacts with EZH2 to promote trimethylation of histone-3 at lysine 27 on the NDRG2 promoter, thereby promoting OC (Wang et al. 2020a). Only a few studies have highlighted the importance of mutations in lncRNAs as a mechanism regulating their function. Similar to proteins, mutations that cause aberrations in lncRNA expression or structure can result in a range of diseases, including cancer (Cabianca et al. 2012; Xue et al. 2015; Lin et al. 2016; Castellanos-Rubio et al. 2016). Single nucleotide polymorphisms (SNPs) in the interacting site can affect the functional role of lncRNA in terms of genomic alterations in the non-coding region. Additionally, variants can alter the secondary structure of lncRNA, thereby affecting its binding ability (Rao et al. 2017). A recent analysis reported that 495,729 SNPs are prevalent in 17,436 lncRNA genes, potentially affecting their secondary structure (Gong et al. 2015).
Competing endogenous RNA (ceRNA) network in OC
A novel mechanism of gene expression regulation involves the interaction between ncRNAs and mRNAs through shared miRNAs (Karreth and Pandolfi 2013). The participation of ncRNAs in competing endogenous regulatory interactions, also known as the ceRNA network, has been well documented. According to the ceRNA hypothesis, lncRNAs and mRNAs harbor the same miRNA response elements. Furthermore, ncRNAs have the potential to titrate miRNA away from mRNA by recognizing and binding to the miRNA recognition elements (MREs), thereby promoting target gene expression (Salmena et al. 2011; Chan and Tay 2018). Among the ceRNA network, lncRNAs have gained much attention due to their involvement in a range of biological processes. LncRNAs regulate mRNA expression at the post-transcriptional level by acting as a molecular sponge for miRNA through the miRNA-response element (Prensner and Chinnaiyan 2011; Zhou et al. 2019). Therefore, lncRNA behaves as negative regulators of miRNA and positively regulates gene expression (Fig. 2). Aberrations in the lncRNA-miRNA-mRNA network, particularly lncRNAs, have a pivotal contribution to OC pathogenesis (Yan et al. 2015; Bhan et al. 2017; Slack and Chinnaiyan 2019) Table 2.

Competing endogenous RNA network. Created with BioRender.com
Involvement of lncRNAs in drug resistance
Chemotherapy, in addition to surgical debulking, is currently used to treat ovarian tumors (Miller et al. 2019). Despite the initial response to chemotherapy, the five-year survival rate of OC patients is low due to late diagnosis and chemoresistance (Coleman et al. 2013; Siegel et al. 2016; Nasioudis et al. 2016). Resistance to chemotherapy remains a major concern in the treatment of OC. One of the significant obstacles in the clinical settings is the lack of ideal platinum resistance associated biomarkers categorizing risk and determining prognosis (Zhu et al. 2022).Elucidating the mechanisms underlying chemoresistance is crucial for developing therapeutic drugs to overcome this resistance (Helleman et al. 2006). Recently, several studies have documented the potential role of lncRNAs in determining the response to standard chemotherapy (Elsayed et al. 2020). These studies have emphasized the role of aberrant lncRNA expression in chemoresistance (Heery et al. 2017; Gao et al. 2019b).
Specific lncRNAs associated with a particular type of cancer require validation in a large cohort of patients. Identifying the cellular localization of lncRNAs is crucial for identifying strategies to inhibit their function. Furthermore, computational approaches to determine the lncRNA function are complicated due to their tissue specificity, low expression levels, and interactions with various molecular components (as reviewed by (Chowdhary et al. 2021)).
Currently, only a few lncRNAs have been thoroughly studied to understand their mechanism of action. Due to the variability of lncRNA levels between different individuals as well as tissues, determining consistent lncRNA markers is challenging. Compared to other RNA molecules, lncRNAs are less stable. Therefore, appropriate methods for sample collection, processing, storage, and assessment are crucial. Despite the therapeutic potential of lncRNAs, developing small molecule drugs to target them poses challenges (Jin et al. 2021).
Silencing of ACTA2-AS1 was found to revert cisplatin resistance by sponging miR-378-3p and regulating Wnt5a level (Lin et al. 2022). Similarly, the lncRNA UCA1 was upregulated in chemoresistant OC cell line as compared to its sensitive counterpart, and silencing UCA1 reverted the resistance to cisplatin through the ceRNA network involving miR-27a-5p. Furthermore, downregulating UCA1 resulted in lower levels of the miR-27a-5p target UBE2N, which promoted higher expression of pro-apoptotic protein BIM (Wambecke et al. 2021). Several lncRNAs have been found to regulate chemoresistance by modulating various cancer-related cascades. For example, the lncRNA HOTAIR promoted chemoresistance in multiple myeloma to dexamethasone by modulating the JAK/STAT3 pathway (Guan et al. 2019). Similarly, the Myc target lncRNA LINC00839 enhances chemoresistance in breast cancer through the PI3K/AKT signaling cascade (Chen et al. 2020). In colorectal cancer, the lncRNA LINC00689 acts as a tumor suppressor, inhibiting cell metastasis, proliferation, and chemoresistance by upregulating LATS2 expression and suppressing YAP/ β-catenin levels (Du et al. 2020).
Understanding the mechanism of action of lncRNAs on tumorigenic properties and their ability to serve as biomarkers for determining chemotherapy efficiency is essential (Peng et al. 2020). Dysregulated lncRNAs can induce chemotherapeutic resistance by inhibiting cell death, inducing protective autophagy, epithelial-mesenchymal transition, promoting cell proliferation, and dysregulating glycolysis (Li et al. 2021a). A conserved cellular mechanism through which cells degrade impaired cytoplasmic components utilizing the lysosomal compartments to uphold energy equilibrium is known as autophagy. Recent studies have revealed that autophagy contributes to the development of cisplatin resistance in cancer (Elsayed et al. 2020). In OC, the lncRNA Taurine up-regulated 1 (TUG1) promoted autophagy by sequestering miR-29b-3p, leading to chemoresistance. Silencing TUG1 reduces Beclin-1 levels and impairs the conversion of LC3B-I to LC3B-II, thereby affecting paclitaxel sensitivity (Gu et al. 2020).
Another mechanism of chemoresistance employed by lncRNAs is DNA damage repair, which involves interconnected signaling cascades coordinating the repair of DNA damage to prevent detrimental genetic changes. This network includes NF-KB, the tumor suppressor, gene TP53, and p21. However, in cancer, DNA damage repair protects cancer cells from the effects of chemotherapy, promoting cell survival (Elsayed et al. 2020). In OC, the lncRNA HOTAIR is over expressed in recurrent platinum-resistance OC samples compared to primary OC samples, and it initiates DNA damage repair following platinum treatment (Özeş et al. 2016).
Previously, the lncRNA CTSLP8 was found to be overexpressed in chemo-resistant OC samples, promoting cell proliferation and cisplatin resistance. Moreover, CTSLP8 enhances chemoresistance by facilitating the interaction of PKM2 with the c-myc promoter, thereby promoting its expression and contributing to glycolysis (Li et al. 2022).Some other examples of drug resistance mechanism involving lncRNAs is described in Table 3
One of the areas of cancer research gaining wide attention is re-sensitizing cancer cells to drug for overcoming chemoresistance. In glioblastoma, the lncRNA SNHG12 was over expressed in temozolomide resistant cells and tissues, and silencing the lncRNA reversed the resistance implying its role as a therapeutic target (Lu et al. 2020). In cisplatin resistant gastric cancer cells, the lncRNA CRAL was downregulated which led to cisplatin resistance observed to the gastric cancer cells to cisplatin. CRAL reversed the chemoresistance via the ceRNA network involving miR505/ cylindromatosis/ AKT (Wang et al. 2020c). Another lncRNA, PGM5-AS1 was downregulated in oxaliplatin resistant colorectal cancer tissues and cell lines. Further, PGM5-AS1 had tumor suppressive function as it inhibited cell migration, proliferation and acquired resistance to oxaliplatin in colon cancer cells. Engineered exosomes for simultaneous delivery of oxaliplatin and PGM5-AS1 re-sensitized the colon cancer cells to the drug (Hui et al. 2022).
lncRNAs in extracellular milieu
Recently, extracellular vesicles, such as exosomes, have been identified as messengers responsible for cell-to-cell communication (Dragomir et al. 2018). The human lncRNA database available at https://bigd.big.ac.cn/lncbook/index, indicates that a substantial number (~ 80,000) of the lncRNAs have been associated with exosomes.
The shift towards investigating the function of lncRNAs in exosomes highlights their importance in cancer onset and progression. In a study, sequencing of human plasma-derived exosomal RNA sequences revealed that miRNAs constituted the major component of exosomal RNA (42.32%), while lncRNAs accounted for only 3.36% of the total RNAs (Huang et al. 2013). However, lncRNAs were primarily encapsulated and protected within the exosomes (Li et al. 2015b). Clinical trials (NCT03738319) are underway to identify and validate lncRNAs as biomarkers in OC.
Several exosomal lncRNAs have been identified to play a crucial function in OC pathogenesis. For instance, exosomal lncRNA ATB contributed to OC development through the modulation of miR-204-3p and TGFβ2 (Yuan et al. 2022). Similarly, another study reported higher expression of SOX2-OT in the plasma exosomes derived from OC patients. Furthermore, exosomal SOX2-OT promoted proliferation and inhibited apoptosis in OC cells, implying its role as a therapeutic target (Lai et al. 2021).
Approaches to elucidate lncRNA function
Despite lncRNAs constituting the majority of RNA transcripts, their functional roles are not fully elucidated. This could be due to the lack of efficient tools for the functional characterization of lncRNAs (Awwad 2019). Currently, RNAi involving siRNA and antisense oligonucleotides (ASO), along with CRISPR/Cas9 system, are being used for the functional analysis of lncRNAs (Qian et al. 2020), with RNAi being the most commonly used strategy (Fathi Dizaji 2020). To date, only ~ 5% of lncRNAs have been functionally characterized. Gapmers, synthetic antisense oligonucleotides, have been described as one of the most useful approaches to analyzing nuclear-enriched lncRNAs (Maruyama and Yokota 2020).
lncRNAs as diagnostic and prognostic markers in OC
Currently, the majority of tumor markers are proteins, despite only 2% of the human genome coding for proteins. However, with the identification of over 102,000 lncRNAs, cancer treatment based on lncRNAs shows promise as a tool (Zhao et al. 2016; Cuykendall et al. 2017). Furthermore, the functional characterization of lncRNAs has revealed their great potential as biomarkers. Identifying circulating lncRNAs in bodily fluids could differentiate cancer patients from healthy individuals at early stages and serve as prognostic factors for cancer patients (Shi et al. 2016). Pathological examination of tissues derived from cancer is currently the standard for cancer diagnosis. However, liquid biopsies characterizing cancer-derived components in the blood and other body fluids provide a new direction for cancer research and are increasingly exploited for cancer detection (Trinidad et al. 2020). LncRNAs, with their presence in bodily fluids, tissue-specific expression, and involvement in various aspects of cancer progression, can be extracted through non-invasive means and used as diagnostic and prognostic markers (Qian et al. 2020). Currently, PCA3 is the only FDA-approved lncRNA for clinical use as a biomarker in prostate cancer (Groskopf et al. 2006).
MALAT1 has been identified as a potential biomarker for ovarian tumorigenesis and metastasis, as its higher expression promotes cell proliferation, migration, and invasion (Liu et al. 2013; Zhou et al. 2016, p. 1). Similarly, UCA1 has an oncogenic function in EOC, promoting cell proliferation, invasion, metastasis, and drug resistance (Liu et al. 2013). Elevated UCA1 expression is an independent poor prognostic marker, and its levels in EOC reflect the response to chemotherapy (Liu et al. 2013). UCA1 serves as a sponge for miR-485-5p, upregulating MMP14 expression and leading to metastasis (Yang et al. 2016).
The lncRNA Growth-arrest-specific 5 (GAS5) has a tumorigenic role in EOC, and its reduced expression indicates a poor prognosis. GAS5 suppresses tumorigenesis by inducing mitochondria-mediated apoptosis and disrupting the mitochondrial membrane potential. This increases cleaved caspase-3, cleaved caspase-9, BAX, and BAK levels in EOC patients, making GAS5 a promising therapeutic target (Gao et al. 2015; Li et al. 2016a, p. 5).
Martini et al. identified four lncRNAs (PVT1, SERTAD2-3, SOX4-1, HRCT1-1) as prognostic markers for relapse and survival (Martini et al. 2017). The network comprising PVT1 and SOX4-1 included oncogenes participating in AKT, PI3K, and MAPK pathways, promoting tumor relapse in stage-I EOC patients by regulating cell proliferation and progression through the cell cycle (Martini et al. 2017). Despite the surge in the number of functionally characterized lncRNAs, only one has been approved for clinical use as a biomarker. This suggests the need for further research in this field to identify useful lncRNAs with biomarker potential.
lncRNAs as therapeutic targets
Targeting lncRNAs is a promising approach for OC therapy due to their tissue-specific expression (Wang et al. 2019b). Previous studies have suggested modulating the expression of tumor- suppressive lncRNAs as a possible therapeutic strategy for treating cancer. This can be achieved by designing vectors to promote GAS5 expression or administering synthetic mimics of TERRA. Similarly, inhibiting oncogenic lncRNA is another suggested strategy to fight cancer (Gutschner and Diederichs 2012). Several different strategies can be used to target and regulate lncRNAs, such as using siRNAs against lncRNAs, preventing RNAs-protein associations, or blocking expression via inhibitors that bind to their promoter (Arun et al. 2018). The use of lncRNAs in precision medicine will be enhanced by the emergence of advanced techniques for identifying molecules expressed at low levels (Bonetti and Carninci 2017).
Database related to lncRNAs
The exponential increase in data generation, facilitated by sophisticated techniques such as next generation sequencing (NGS) and microarray, highlights the critical role of data management and analysis in advancing our understanding of biology and therapeutic interventions (Tao et al. 2017). This has led to the development of computational and bioinformatic approaches for analyzing biological information, particularly in cancer research (Mitra et al. 2013). The use of bioinformatics methods to identify differentially expressed genes holds promise for elucidating the underlying mechanisms of OC pathogenesis.
In recent years, there has been a rise in the number of new databases exclusively for lncRNA annotation and differential expression analysis reflecting their significant involvement in cancer. These databases offer valuable resources for investigating lncRNAs in OC and shed light on their roles and mechanisms of action in this disease. One such database is lnCAR, which compiles and integrates information on the functional roles of lncRNAs in cancer. This database collects data on the expression patterns of lncRNAs, their regulatory mechanisms, and their functions in cancer (Zheng et al. 2019b). Similarly, the UALCAN database provides a list of lncRNAs with potential prognostic biomarker roles in several cancer types, including OC (Chandrashekar et al. 2022). According to the LncExp database, the number of lncRNAs associated with normal tissue, organ development, exosomes, and virus infection exceeds that of mRNAs, highlighting their importance. The exoRBase 2.0 database is a comprehensive resource that enables the identification of lncRNAs enriched in extracellular vesicles. exoRBase provides information that aids in understanding the roles of lncRNAs in intercellular communication (Li et al. 2018). Several databases have been developed to facilitate access to the structural, functional, and expression data of lncRNAs, as well as their disease associations (Gong et al. 2021). Frequently accessed databases are listed in Table 4:
While some lncRNAs have been well studied, a significant portion of lncRNAs remains poorly understood. This highlights the need for the development of novel tools and databases to facilitate investigations on lncRNA. For instance, the lncRNAdb database provides details on lncRNAs, including their functions and associations with diseases. Furthermore, it also contains elaborate annotations for each lncRNA. However, it lacks efficient tools for data assessment, and its coverage of lncRNAs is limited compared to other databases (Quek et al. 2015).
On the other hand, the LNCipedia database offers more comprehensive details on annotated lncRNAs, such as their expression and structure, and provides the option to download data. However, it lacks sufficient tools for data analysis (Volders et al. 2019).
The TCGA database contains RNA sequencing data on lncRNA expression in various cancer types. It also allows for the integration of patient’s clinical data with the lncRNA expression. However, utilizing the database effectively requires expertise in analyzing the information and employing bioinformatic tools for detailed exploration.
The Atlas of Noncoding RNAs in Cancer (TANRIC) database provides detailed information on the lncRNA expression and survival outcomes (Li et al. 2015a). However, it focuses exclusively on lncRNAs involved in cancer.
Challenges in lncRNA research
There are several challenges that need to be overcome to incorporate lncRNAs into clinical practice. In order to explore the use of circulating lncRNAs in clinical settings for quantification, it is important to consider critical factors such as the source, primer design, quantitative Real Time PCR (qRT-PCR) procedure, timing of blood sample collection (Gonzalo-Calvo et al. 2022). Moreover standardized procedure for detection and analysis is needed for reproducibility and reliability in the results. Other challenges that need to be addressed for translating lncRNAs into the clinical realm includes comprehensive characterization of the role of lncRNAs in cellular processes, and determining lncRNAs specific for a certain cancer type, which requires validation in a large cohort of patients. Identification of the localization of lncRNA in the cell is crucial for identifying strategy to inhibit the lncRNA function. Furthermore, the identification of lncRNA function through computational approaches is complicated since lncRNAs display tissue specificity, low expression levels, and interactions with several different molecular components (as reviewed in (Chowdhary et al. 2021)). Identifying lncRNAs that are specific to particular cancer types necessitate validation in large cohorts of patients. This is a crucial process to confirm the association of specific lncRNAs with certain cancer types and understanding their potential as diagnostic or prognostic markers.
Furthermore, determining the subcellular localization of lncRNAs is vital for developing strategies to inhibit their function. The identification of lncRNA function through computational approaches is challenging due to the tissue specificity, low expression levels, and interactions with several different molecular components (as reviewed in (Chowdhary et al. 2021)).
Currently, only limited number of lncRNAs have been extensively investigated to elucidate their functional mechanisms. The variability of lncRNA levels across individuals and tissues poses challenges in identifying consistent lncRNA markers. Furthermore, lncRNAs exhibit lower stability compared to other RNA molecules, underscoring the importance of employing appropriate methods for sample collection, processing, storage, and assessment.
Despite the therapeutic potential of lncRNAs, the development of small molecule drugs targeting them presents significant challenges (Jin et al. 2021).
Conclusion and future perspectives
LncRNAs have emerged as crucial regulators in the pathogenesis of OC, holding promising potential as new diagnostic and prognostic markers. These ncRNAs regulate various cellular processes, including cell proliferation, migration, invasion, and drug resistance. Despite significant progress in understanding their roles in OC, clinical applications of lncRNAs are still limited. Future research should focus on identifying the signaling pathways regulated by lncRNAs, elaborating their mechanism of action, and identifying potential therapeutic targets in OC. Standardization of protocols for lncRNA detection and quantification, along with the use of computational approaches for lncRNA annotation, will facilitate the translation of lncRNAs into clinical practice for the diagnosis, prognosis, and treatment of OC. With continued research and development, lncRNAs hold great promise for improving the clinical management of OC and other cancers.
While molecular biology techniques such as NGS have advanced the characterization of lncRNAs, the identification and validation of these transcripts still require significant time and resources through experimental approaches. To complement existing experimental methods, computational approaches for lncRNA annotation have been proposed (Baek et al. 2018).
In conclusion, future research is necessary to elucidate the signaling pathways regulated by lncRNAs, elaborate their roles in cancer development and identify promising therapeutic targets for various cancer types. Further efforts are required to unravel the molecular mechanisms underlying the action of lncRNAs by identifying their RNA, DNA, and protein partners in cancer (Gutschner and Diederichs 2012). To utilize lncRNAs as cancer biomarkers and translate them into clinical practice, future studies should focus on standardizing protocols for sample preparation, identifying endogenous lncRNA controls in body fluids, optimizing lncRNA extraction techniques, and monitoring the quality of lncRNA (Shi et al. 2016).
The ability to detect lncRNAs in body fluids through noninvasive methods hold promising option to utilize lncRNAs in routine clinical settings. Moreover, the aberrant expression of lncRNAs upon drug treatment indicates their ability to modulate drug resistance, making them potential therapeutic targets. It is crucial to identify and functionally characterize lncRNAs as early-stage cancer biomarkers, as they could aid in patient stratification. By uncovering the lncRNA-mediated gene regulation network, key cancer-related processes can be elucidated. Future research is required in both science research and in the application of lncRNAs in the clinical realm. To translate the discovery of lncRNA biomarkers to clinical practice, the development of targeted therapies and diagnostic tests is essential. Clinical trials should be conducted to assess the potential use of lncRNAs as therapeutic markers. Further investigation of the molecular mechanisms of lncRNAs action and their interactions with other factors in OC is necessary. Advanced techniques such as CRISPR/Cas9 mediated knockdown or over expression studies are required to analyze the function of lncRNAs.
Availability of data and materials
Not applicable.
References
Abildgaard C, Canto LM, Rainho CA et al (2022) The long non-coding RNA SNHG 12 as a mediator of carboplatin resistance in ovarian cancer via epigenetic mechanisms. Cancers 14:1664. https://doi.org/10.3390/cancers14071664
Ahadi A (2021) Functional roles of lncRNAs in the pathogenesis and progression of cancer. Genes Dis 8:424–437. https://doi.org/10.1016/j.gendis.2020.04.009
Al Saqri A, Malgundkar SH, Al Kindi F et al (2022) SOCS3 gene silencing does not occur through methylation and mutations in gastric cancer. Hum Cell 35:1114–1125. https://doi.org/10.1007/s13577-022-00715-3
Aliperti V, Skonieczna J, Cerase A (2021) Long non-coding RNA (lncRNA) roles in cell biology, neurodevelopment and neurological disorders. Noncoding RNA 7:36. https://doi.org/10.3390/ncrna7020036
Aluloski I, Tanturovski M, Jovanovic R et al (2017) Survival of advanced stage high-grade serous ovarian cancer patients in the Republic of Macedonia. Open Access Maced J Med Sci 5:904–908. https://doi.org/10.3889/oamjms.2017.215
An J, Lv W, Zhang Y (2017) LncRNA NEAT1 contributes to paclitaxel resistance of ovarian cancer cells by regulating ZEB1 expression via miR-194. Onco Targets Ther 10:5377–5390. https://doi.org/10.2147/OTT.S147586
Arun G, Diermeier SD, Spector DL (2018) Therapeutic targeting of long non-coding RNAs in cancer. Trends Mol Med 24:257–277. https://doi.org/10.1016/j.molmed.2018.01.001
Awwad DA (2019) Beyond classic editing: innovative CRISPR approaches for functional studies of long non-coding RNA. Biol Methods Protocols. https://doi.org/10.1093/biomethods/bpz017
Baek J, Lee B, Kwon S, Yoon S (2018) LncRNAnet: long non-coding RNA identification using deep learning. Bioinformatics 34:3889–3897. https://doi.org/10.1093/bioinformatics/bty418
Bai Z, Wu Y, Bai S et al (2020) Long non-coding RNA SNGH7 Is activated by SP1 and exerts oncogenic properties by interacting with EZH2 in ovarian cancer. J Cell Mol Med 24:7479–7489. https://doi.org/10.1111/jcmm.15373
Bao Z, Yang Z, Huang Z et al (2019) LncRNADisease 2.0: an updated database of long non-coding RNA-associated diseases. Nucleic Acids Res 47:D1034–D1037. https://doi.org/10.1093/nar/gky905
Bhan A, Soleimani M, Mandal SS (2017) Long non-coding RNA (LncRNA) and cancer: a new paradigm. Cancer Res 77:3965–3981. https://doi.org/10.1158/0008-5472.CAN-16-2634
Bonetti A, Carninci P (2017) From bench to bedside: The long journey of long non-coding RNAs. Curr Opin Syst Biol 3:119–124. https://doi.org/10.1016/j.coisb.2017.04.016
Brannan CI, Dees EC, Ingram RS, Tilghman SM (1990) The product of the H19 gene may function as an RNA. Mol Cell Biol 10:28–36. https://doi.org/10.1128/mcb.10.1.28-36.1990
Brown CJ, Ballabio A, Rupert JL et al (1991) A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349:38–44. https://doi.org/10.1038/349038a0
Budiana ING, Angelina M, Pemayun TGA (2019) Ovarian cancer: Pathogenesis and current recommendations for prophylactic surgery. J Turk Ger Gynecol Assoc 20:47–54. https://doi.org/10.4274/jtgga.galenos.2018.2018.0119
Cabianca DS, Casa V, Bodega B et al (2012) A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 149:819–831. https://doi.org/10.1016/j.cell.2012.03.035
Cabili MN, Trapnell C, Goff L et al (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927. https://doi.org/10.1101/gad.17446611
Cancer today. http://gco.iarc.fr/today/home. Accessed 12 Sep 2021
Cancer Tomorrow. https://gco.iarc.fr/tomorrow/en/dataviz/trends?cancers=25. Accessed 2 Dec 2022
Cao M, Zhao J, Hu G (2019) Genome-wide methods for investigating long noncoding RNAs. Biomed Pharmacother 111:395–401. https://doi.org/10.1016/j.biopha.2018.12.078
Castellanos-Rubio A, Fernandez-Jimenez N, Kratchmarov R et al (2016) A long noncoding RNA associated with susceptibility to celiac disease. Science 352:91–95. https://doi.org/10.1126/science.aad0467
Chan JJ, Tay Y (2018) Noncoding RNA:RNA regulatory networks in cancer. Int J Mol Sci 19:1310. https://doi.org/10.3390/ijms19051310
Chandra A, Pius C, Nabeel M et al (2019) Ovarian cancer: current status and strategies for improving therapeutic outcomes. Cancer Med 8:7018–7031. https://doi.org/10.1002/cam4.2560
Chandrashekar DS, Karthikeyan SK, Korla PK et al (2022) UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 25:18–27. https://doi.org/10.1016/j.neo.2022.01.001
Chang L, Guo R, Yuan Z et al (2018) LncRNA HOTAIR regulates CCND1 and CCND2 expression by sponging miR-206 in ovarian cancer. Cell Physiol Biochem 49:1289–1303. https://doi.org/10.1159/000493408
Chen X-J, An N (2021) Long noncoding RNA ATB promotes ovarian cancer tumorigenesis by mediating histone H3 lysine 27 trimethylation through binding to EZH2. J Cell Mol Med 25:37–46. https://doi.org/10.1111/jcmm.15329
Chen M, Jin Y, Bi Y et al (2014) A survival analysis comparing women with ovarian low-grade serous carcinoma to those with high-grade histology. Onco Targets Ther 7:1891–1899. https://doi.org/10.2147/OTT.S67812
Chen Q, Shen H, Zhu X et al (2020) A nuclear lncRNA Linc00839 as a Myc target to promote breast cancer chemoresistance via PI3K/AKT signaling pathway. Cancer Sci 111:3279–3291. https://doi.org/10.1111/cas.14555
Chowdhary A, Satagopam V, Schneider R (2021) Long non-coding RNAs: mechanisms, experimental, and computational approaches in identification, characterization, and their biomarker potential in cancer. Front Genet. https://doi.org/10.3389/fgene.2021.649619
Chu C, Chang HY (2016) Understanding RNA-Chromatin Interactions Using Chromatin Isolation by RNA Purification (ChIRP). In: Lanzuolo C, Bodega B (eds) Polycomb Group Proteins: Methods and Protocols. Springer, New York, NY, pp 115–123
Coleman RL, Monk BJ, Sood AK, Herzog TJ (2013) Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat Rev Clin Oncol 10:211–224. https://doi.org/10.1038/nrclinonc.2013.5
Coticchia CM, Yang J, Moses MA (2008) Ovarian cancer biomarkers: current options and future promise. J Natl Compr Canc Netw 6:795–802
Cuykendall TN, Rubin MA, Khurana E (2017) Non-coding genetic variation in cancer. Curr Opin Syst Biol 1:9–15. https://doi.org/10.1016/j.coisb.2016.12.017
de Gonzalo-Calvo D, Sopić M, Devaux Y (2022) Methodological considerations for circulating long noncoding RNA quantification. Trends Mol Med 28:616–618. https://doi.org/10.1016/j.molmed.2022.05.011
de Hoon M, Shin JW, Carninci P (2015) Paradigm shifts in genomics through the FANTOM projects. Mamm Genome 26:391–402. https://doi.org/10.1007/s00335-015-9593-8
Derrien T, Johnson R, Bussotti G et al (2012) The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 22:1775–1789. https://doi.org/10.1101/gr.132159.111
Ding Y, Yang D-Z, Zhai Y-N et al (2017) Microarray expression profiling of long non-coding RNAs in epithelial ovarian cancer. Oncol Lett 14:2523–2530. https://doi.org/10.3892/ol.2017.6448
Djebali S, Davis CA, Merkel A et al (2012) Landscape of transcription in human cells. Nature 489:101–108. https://doi.org/10.1038/nature11233
Dragomir M, Chen B, Calin GA (2018) Exosomal lncRNAs as new players in cell-to-cell communication. Transl Cancer Res 7:S243–S252. https://doi.org/10.21037/tcr.2017.10.46
Du Y-L, Liang Y, Shi G-Q et al (2020) LINC00689 participates in proliferation, chemoresistance and metastasis via miR-31-5p/YAP/β-catenin axis in colorectal cancer. Exp Cell Res 395:112176. https://doi.org/10.1016/j.yexcr.2020.112176
El Bairi K, Kandhro AH, Gouri A et al (2017) Emerging diagnostic, prognostic and therapeutic biomarkers for ovarian cancer. Cell Oncol 40:105–118. https://doi.org/10.1007/s13402-016-0309-1
Elsayed AM, Amero P, Salama SA et al (2020) Back to the future: rethinking the great potential of lncRNAS for optimizing chemotherapeutic response in ovarian cancer. Cancers (Basel) 12:2406. https://doi.org/10.3390/cancers12092406
Erickson BK, Conner MG, Landen CN (2013) The role of the fallopian tube in the origin of ovarian cancer. Am J Obstet Gynecol 209:409–414. https://doi.org/10.1016/j.ajog.2013.04.019
Fang Y, Fullwood MJ (2016) Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genom Proteom Bioinform 14:42–54. https://doi.org/10.1016/j.gpb.2015.09.006
Fang S, Zhang L, Guo J et al (2018) NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res 46:D308–D314. https://doi.org/10.1093/nar/gkx1107
Fathi Dizaji B (2020) Strategies to target long non-coding RNAs in cancer treatment: progress and challenges. Egypt J Med Hum Genet 21:41. https://doi.org/10.1186/s43042-020-00074-4
Fatica A, Bozzoni I (2014) Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet 15:7–21. https://doi.org/10.1038/nrg3606
Flippot R, Malouf GG, Su X, et al (2016) Cancer subtypes classification using long non-coding RNA. Oncotarget 7:54082–54093. https://doi.org/10.18632/oncotarget.10213
Fort A, Hashimoto K, Yamada D et al (2014) Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat Genet 46:558–566. https://doi.org/10.1038/ng.2965
Friedman RC, Farh KK-H, Burge CB, Bartel DP (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105. https://doi.org/10.1101/gr.082701.108
Galea M, Gauci G, Calleja-Agius J, Schembri-Wismayer P (2017) Peritoneal biomarkers in the early detection of ovarian cancer. Minerva Ginecol 69:84–99. https://doi.org/10.23736/S0026-4784.16.03943-5
Gao J, Liu M, Zou Y et al (2015) Long non-coding RNA growth arrest-specific transcript 5 is involved in ovarian cancer cell apoptosis through the mitochondria-mediated apoptosis pathway. Oncol Rep 34:3212–3221. https://doi.org/10.3892/or.2015.4318
Gao C, Zhao D, Zhao Q et al (2019a) Microarray profiling and co-expression network analysis of lncRNAs and mRNAs in ovarian cancer. Cell Death Discov 5:93. https://doi.org/10.1038/s41420-019-0173-7
Gao Y, Li X, Zhi H et al (2019b) Comprehensive characterization of somatic mutations impacting lncRNA expression for pan-cancer. Mol Ther Nucleic Acids 18:66–79. https://doi.org/10.1016/j.omtn.2019.08.004
Gao Y, Wang P, Wang Y et al (2019c) Lnc2Cancer v2.0: updated database of experimentally supported long non-coding RNAs in human cancers. Nucleic Acids Res 47:D1028–D1033. https://doi.org/10.1093/nar/gky1096
Gibb EA, Brown CJ, Lam WL (2011) The functional role of long non-coding RNA in human carcinomas. Mol Cancer 10:38. https://doi.org/10.1186/1476-4598-10-38
Gong J, Liu W, Zhang J et al (2015) lncRNASNP: a database of SNPs in lncRNAs and their potential functions in human and mouse. Nucleic Acids Res 43:D181–D186. https://doi.org/10.1093/nar/gku1000
Gong Y, Zhu W, Sun M, Shi L (2021) Bioinformatics analysis of long non-coding RNA and related diseases: an overview. Front Genet 12:2575. https://doi.org/10.3389/fgene.2021.813873
Gov E, Kori M, Arga KY (2017) RNA-based ovarian cancer research from ‘a gene to systems biomedicine’ perspective. Syst Biol Reprod Med 63:219–238. https://doi.org/10.1080/19396368.2017.1330368
Groskopf J, Aubin SMJ, Deras IL et al (2006) APTIMA PCA3 molecular urine test: development of a method to aid in the diagnosis of prostate cancer. Clin Chem 52:1089–1095. https://doi.org/10.1373/clinchem.2005.063289
Gross AL, Kurman RJ, Vang R et al (2010) Precursor lesions of high-grade serous ovarian carcinoma: morphological and molecular characteristics. J Oncol. https://doi.org/10.1155/2010/126295
Gu L, Li Q, Liu H et al (2020) Long noncoding RNA TUG1 promotes autophagy-associated paclitaxel resistance by sponging miR-29b-3p in ovarian cancer cells. Onco Targets Ther 13:2007–2019. https://doi.org/10.2147/OTT.S240434
Guan R, Wang W, Fu B et al (2019) Increased lncRNA HOTAIR expression promotes the chemoresistance of multiple myeloma to dexamethasone by regulating cell viability and apoptosis by mediating the JAK2/STAT3 signaling pathway. Mol Med Rep 20:3917–3923. https://doi.org/10.3892/mmr.2019.10603
Guo Q, Wu Y, Guo X et al (2020) The RNA-binding protein CELF2 inhibits ovarian cancer progression by stabilizing FAM198B. Mol Ther Nucleic Acids 23:169–184. https://doi.org/10.1016/j.omtn.2020.10.011
Gutschner T, Diederichs S (2012) The hallmarks of cancer. RNA Biol 9:703–719. https://doi.org/10.4161/rna.20481
Guttman M, Amit I, Garber M et al (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223–227. https://doi.org/10.1038/nature07672
Ha M, Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15:509–524. https://doi.org/10.1038/nrm3838
Hafner M, Katsantoni M, Köster T et al (2021) CLIP and complementary methods. Nat Rev Methods Primers 1:1–23. https://doi.org/10.1038/s43586-021-00018-1
Hanly DJ, Esteller M, Berdasco M (2018) Interplay between long non-coding RNAs and epigenetic machinery: emerging targets in cancer? Philosophical Transactions of the Royal Society b: Biological Sciences 373:20170074. https://doi.org/10.1098/rstb.2017.0074
Harrow J, Frankish A, Gonzalez JM et al (2012) GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res 22:1760–1774. https://doi.org/10.1101/gr.135350.111
He S-L, Chen Y-L, Chen Q-H et al (2022) LncRNA KCNQ1OT1 promotes the metastasis of ovarian cancer by increasing the methylation of EIF2B5 promoter. Mol Med 28:112. https://doi.org/10.1186/s10020-022-00521-5
Heery R, Finn SP, Cuffe S, Gray SG (2017) Long non-coding RNAs: key regulators of epithelial-mesenchymal transition, tumour drug resistance and cancer stem cells. Cancers (Basel) 9:38. https://doi.org/10.3390/cancers9040038
Helleman J, Jansen MPHM, Span PN et al (2006) Molecular profiling of platinum resistant ovarian cancer. Int J Cancer 118:1963–1971. https://doi.org/10.1002/ijc.21599
Heydarnezhad Asl M, Pasban Khelejani F, Bahojb Mahdavi SZ et al (2022) The various regulatory functions of long noncoding RNAs in apoptosis, cell cycle, and cellular senescence. J Cell Biochem 123:995–1024. https://doi.org/10.1002/jcb.30221
Holoch D, Moazed D (2015) RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 16:71–84. https://doi.org/10.1038/nrg3863
Hu W, Alvarez-Dominguez JR, Lodish HF (2012) Regulation of mammalian cell differentiation by long non-coding RNAs. EMBO Rep 13:971–983. https://doi.org/10.1038/embor.2012.145
Huang X, Yuan T, Tschannen M et al (2013) Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom 14:319. https://doi.org/10.1186/1471-2164-14-319
Hui B, Lu C, Wang J et al (2022) Engineered exosomes for co-delivery of PGM5-AS1 and oxaliplatin to reverse drug resistance in colon cancer. J Cell Physiol 237:911–933. https://doi.org/10.1002/jcp.30566
Iyer MK, Niknafs YS, Malik R et al (2015) The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47:199–208. https://doi.org/10.1038/ng.3192
Jiang N, Zhang X, Gu X et al (2021) Progress in understanding the role of lncRNA in programmed cell death. Cell Death Discov 7:1–11. https://doi.org/10.1038/s41420-021-00407-1
Jin H, Du W, Huang W et al (2021) lncRNA and breast cancer: Progress from identifying mechanisms to challenges and opportunities of clinical treatment. Mol Ther Nucleic Acids 25:613–637. https://doi.org/10.1016/j.omtn.2021.08.005
Kamps R, Brandão RD, van den Bosch BJ et al (2017) Next-generation sequencing in oncology: genetic diagnosis, risk prediction and cancer classification. Int J Mol Sci 18:308. https://doi.org/10.3390/ijms18020308
Kapranov P, St Laurent G, Raz T et al (2010) The majority of total nuclear-encoded non-ribosomal RNA in a human cell is “dark matter” un-annotated RNA. BMC Biol 8:149. https://doi.org/10.1186/1741-7007-8-149
Karagkouni D, Paraskevopoulou MD, Tastsoglou S et al (2020) DIANA-LncBase v3: indexing experimentally supported miRNA targets on non-coding transcripts. Nucleic Acids Res 48:D101–D110. https://doi.org/10.1093/nar/gkz1036
Karreth FA, Pandolfi PP (2013) ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov 3:1113–1121. https://doi.org/10.1158/2159-8290.CD-13-0202
Kitagawa M, Kitagawa K, Kotake Y et al (2013) Cell cycle regulation by long non-coding RNAs. Cell Mol Life Sci 70:4785–4794. https://doi.org/10.1007/s00018-013-1423-0
Konstantinopoulos PA, Spentzos D, Cannistra SA (2008) Gene-expression profiling in epithelial ovarian cancer. Nat Clin Pract Oncol 5:577–587. https://doi.org/10.1038/ncponc1178
Kornienko AE, Guenzl PM, Barlow DP, Pauler FM (2013) Gene regulation by the act of long non-coding RNA transcription. BMC Biol 11:59. https://doi.org/10.1186/1741-7007-11-59
Koshiyama M, Matsumura N, Konishi I (2014) Recent concepts of ovarian carcinogenesis: Type I and Type II. Biomed Res Int 2014:1–11. https://doi.org/10.1155/2014/934261
Kurman RJ, Shih I-M (2010) The origin and pathogenesis of epithelial ovarian cancer- a proposed unifying theory. Am J Surg Pathol 34:433–443. https://doi.org/10.1097/PAS.0b013e3181cf3d79
Kurman RJ, Shih I-M (2016) The dualistic model of ovarian carcinogenesis. Am J Pathol 186:733–747. https://doi.org/10.1016/j.ajpath.2015.11.011
Lai Y, Dong L, Jin H, et al (2021) Exosome long non-coding RNA SOX2-OT contributes to ovarian cancer malignant progression by miR-181b-5p/SCD1 signaling. Aging (Albany NY) 13:23726–23738. https://doi.org/10.18632/aging.203645
Ledermann JA, Raja FA, Fotopoulou C et al (2013) Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 24:vi24–vi22. https://doi.org/10.1093/annonc/mdt333
Lee C, Kikyo N (2012) Strategies to identify long noncoding RNAs involved in gene regulation. Cell Biosci 2:37. https://doi.org/10.1186/2045-3701-2-37
Lee TI, Young RA (2013) Transcriptional regulation and its misregulation in disease. Cell 152:1237–1251. https://doi.org/10.1016/j.cell.2013.02.014
Li J-H, Liu S, Zhou H et al (2014) starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 42:D92–D97. https://doi.org/10.1093/nar/gkt1248
Li J, Han L, Roebuck P et al (2015a) TANRIC: An interactive open platform to explore the function of lncRNAs in cancer. Cancer Res 75:3728–3737. https://doi.org/10.1158/0008-5472.CAN-15-0273
Li Q, Shao Y, Zhang X et al (2015b) Plasma long noncoding RNA protected by exosomes as a potential stable biomarker for gastric cancer. Tumour Biol 36:2007–2012. https://doi.org/10.1007/s13277-014-2807-y
Li J, Huang H, Li Y et al (2016a) Decreased expression of long non-coding RNA GAS5 promotes cell proliferation, migration and invasion, and indicates a poor prognosis in ovarian cancer. Oncol Rep 36:3241–3250. https://doi.org/10.3892/or.2016.5200
Li R, Zhu H, Luo Y (2016b) Understanding the functions of long non-coding RNAs through their higher-order structures. Int J Mol Sci 17:702. https://doi.org/10.3390/ijms17050702
Li S, Li Y, Chen B et al (2018) exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res 46:D106–D112. https://doi.org/10.1093/nar/gkx891
Li Y, Ma H-Y, Hu X-W et al (2020) LncRNA H19 promotes triple-negative breast cancer cells invasion and metastasis through the p53/TNFAIP8 pathway. Cancer Cell Int 20:200. https://doi.org/10.1186/s12935-020-01261-4
Li G, Gong J, Cao S et al (2021a) The non-coding RNAs inducing drug resistance in ovarian cancer: a new perspective for understanding drug resistance. Front Oncol. https://doi.org/10.3389/fonc.2021.742149
Li L, Wei H, Zhang YW, et al (2021b) Differential expression of long non-coding RNAs as diagnostic markers for lung cancer and other malignant tumors. Aging (Albany NY) 13:23842–23867. https://doi.org/10.18632/aging.203523
Li X, Zhang Y, Wang X et al (2022) Long non-coding RNA CTSLP8 mediates ovarian cancer progression and chemotherapy resistance by modulating cellular glycolysis and regulating c-Myc expression through PKM2. Cell Biol Toxicol 38:1027–1045. https://doi.org/10.1007/s10565-021-09650-9
Lin A, Li C, Xing Z et al (2016) The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat Cell Biol 18:213–224. https://doi.org/10.1038/ncb3295
Lin X, Yang F, Qi X et al (2019) LncRNA DANCR promotes tumor growth and angiogenesis in ovarian cancer through direct targeting of miR-145. Mol Carcinog 58:2286–2296. https://doi.org/10.1002/mc.23117
Lin H, Shen L, Lin Q et al (2020) SNHG5 enhances Paclitaxel sensitivity of ovarian cancer cells through sponging miR-23a. Biomed Pharmacother 123:109711. https://doi.org/10.1016/j.biopha.2019.109711
Lin C, Zheng M, Yang Y et al (2022) Knockdown of lncRNA ACTA2-AS1 reverses cisplatin resistance of ovarian cancer cells via inhibition of miR-378a-3p-regulated Wnt5a. Bioengineered 13:9829–9838. https://doi.org/10.1080/21655979.2022.2061181
Liu S-P, Yang J-X, Cao D-Y, Shen K (2013) Identification of differentially expressed long non-coding RNAs in human ovarian cancer cells with different metastatic potentials. Cancer Biol Med 10:138–141. https://doi.org/10.7497/j.issn.2095-3941.2013.03.003
Liu J, Wang G, Zhao J et al (2021) LncRNA H19 promoted the epithelial to mesenchymal transition and metastasis in gastric cancer via activating Wnt/β-catenin signaling. Dig Dis 40:436–447. https://doi.org/10.1159/000518627
Lou Y, Jiang H, Cui Z et al (2017) Linc-ROR induces epithelial-to-mesenchymal transition in ovarian cancer by increasing Wnt/β-catenin signaling. Oncotarget 8:69983–69994. https://doi.org/10.18632/oncotarget.19545
Lou Y, Jiang H, Cui Z et al (2018) Gene microarray analysis of lncRNA and mRNA expression profiles in patients with high-grade ovarian serous cancer. Int J Mol Med 42:91–104. https://doi.org/10.3892/ijmm.2018.3588
Lu C, Wei Y, Wang X et al (2020) DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma. Mol Cancer 19:28. https://doi.org/10.1186/s12943-020-1137-5
Ma L, Bajic VB, Zhang Z (2013) On the classification of long non-coding RNAs. RNA Biol 10:925–933. https://doi.org/10.4161/rna.24604
Magri F, Vanoli F, Corti S (2018) miRNA in spinal muscular atrophy pathogenesis and therapy. J Cell Mol Med 22:755–767. https://doi.org/10.1111/jcmm.13450
Mahdian-Shakib A, Dorostkar R, Tat M et al (2016) Differential role of microRNAs in prognosis, diagnosis, and therapy of ovarian cancer. Biomed Pharmacother 84:592–600. https://doi.org/10.1016/j.biopha.2016.09.087
Malgundkar SH, Hassan NA, Al Badi H et al (2022) Identification and validation of a novel long non-coding RNA (LINC01465) in ovarian cancer. Hum Cell. https://doi.org/10.1007/s13577-022-00842-x
Martini P, Paracchini L, Caratti G et al (2017) lncRNAs as novel indicators of patients’ prognosis in stage I epithelial ovarian cancer: a retrospective and multicentric study. Clin Cancer Res 23:2356–2366
Maruyama R, Yokota T (2020) Knocking Down Long Noncoding RNAs Using Antisense Oligonucleotide Gapmers. In: Yokota T, Maruyama R (eds) Gapmers: Methods and Protocols. Springer, US, New York, pp 49–56
Mattick JS, Rinn JL (2015) Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol 22:5–7. https://doi.org/10.1038/nsmb.2942
McColl H, Zagozewski JL, Eisenstat DD (2019) Chromatin Immunoprecipitation (ChIP) protocols for the cancer and developmental biology laboratory. Methods Mol Biol 1869:143–153. https://doi.org/10.1007/978-1-4939-8805-1_13
Miao J-T, Gao J-H, Chen Y-Q et al (2019) LncRNA ANRIL affects the sensitivity of ovarian cancer to cisplatin via regulation of let-7a/HMGA2 axis. Biosci Rep. https://doi.org/10.1042/BSR20182101
Miller JL, Grant PA (2013) The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell Biochem 61:289–317. https://doi.org/10.1007/978-94-007-4525-4_13
Miller KD, Nogueira L, Mariotto AB et al (2019) Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin 69:363–385. https://doi.org/10.3322/caac.21565
Mitra S, Das S, Chakrabarti J (2013) Systems biology of cancer biomarker detection. Cancer Biomark 13:201–213. https://doi.org/10.3233/CBM-130363
Mundade R, Ozer HG, Wei H et al (2014) Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond. Cell Cycle 13:2847–2852. https://doi.org/10.4161/15384101.2014.949201
Nagano T, Fraser P (2011) No-nonsense functions for long noncoding RNAs. Cell 145:178–181. https://doi.org/10.1016/j.cell.2011.03.014
Nasioudis D, Sisti G, Kanninen TT et al (2016) Epidemiology and outcomes of squamous ovarian carcinoma; a population-based study. Gynecol Oncol 141:128–133. https://doi.org/10.1016/j.ygyno.2016.02.004
Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol https://www.annalsofoncology.org/article/S0923-7534(19)31561-3/fulltext. Accessed 17 Jun 2023d
Norouzi-Barough L, Sarookhani MR, Sharifi M et al (2018) Molecular mechanisms of drug resistance in ovarian cancer. J Cell Physiol 233:4546–4562. https://doi.org/10.1002/jcp.26289
Nukala SB, Jousma J, Cho Y et al (2022) Long non-coding RNAs and microRNAs as crucial regulators in cardio-oncology. Cell Biosci 12:24. https://doi.org/10.1186/s13578-022-00757-y
Otmani K, Lewalle P (2021) Tumor suppressor miRNA in cancer cells and the tumor microenvironment: mechanism of deregulation and clinical implications. Front Oncol 11:708765. https://doi.org/10.3389/fonc.2021.708765
Ovarian Cancer Stages, Survival Rate and Prognosis. In: OCRA. https://ocrahope.org/get-the-facts/staging/. Accessed 18 Feb 2023
Özeş AR, Miller DF, Özeş ON et al (2016) NF-κB-HOTAIR axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer. Oncogene 35:5350–5361. https://doi.org/10.1038/onc.2016.75
Pronina IV, Uroshlev LA, Moskovtsev AA, Zaichenko DM, Filippova EA, Fridman MV et al (2022) Dysregulation of lncRNA– miRNA–mRNA interactome as a marker of metastatic process in ovarian cancer. Biomedicines 10(4):824
Pal MK, Jaiswar SP, Dwivedi VN et al (2015) MicroRNA: a new and promising potential biomarker for diagnosis and prognosis of ovarian cancer. Cancer Biol Med 12:328–341. https://doi.org/10.7497/j.issn.2095-3941.2015.0024
Peng Y, Tang D, Zhao M et al (2020) Long non-coding RNA: a recently accentuated molecule in chemoresistance in cancer. Cancer Metastasis Rev 39:825–835. https://doi.org/10.1007/s10555-020-09910-w
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136:629–641. https://doi.org/10.1016/j.cell.2009.02.006
Prensner JR, Chinnaiyan AM (2011) The emergence of lncRNAs in cancer biology. Cancer Discov 1:391–407. https://doi.org/10.1158/2159-8290.CD-11-0209
Qian Y, Shi L, Luo Z (2020) Long non-coding RNAs in cancer: implications for diagnosis, prognosis, and therapy. Front Med (Lausanne) 7:612393. https://doi.org/10.3389/fmed.2020.612393
Quek XC, Thomson DW, Maag JLV et al (2015) lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res 43:D168–D173. https://doi.org/10.1093/nar/gku988
Quinn JJ, Chang HY (2016) Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17:47–62. https://doi.org/10.1038/nrg.2015.10
Rao AKDM, Rajkumar T, Mani S (2017) Perspectives of long non-coding RNAs in cancer. Mol Biol Rep 44:203–218. https://doi.org/10.1007/s11033-017-4103-6
Rao MS, Van Vleet TR, Ciurlionis R et al (2019) Comparison of RNA-Seq and microarray gene expression platforms for the toxicogenomic evaluation of liver from short-term rat toxicity studies. Front Genet. https://doi.org/10.3389/fgene.2018.00636
Renganathan A, Felley-Bosco E (2017) Long noncoding RNAs in cancer and therapeutic potential. In: Rao MRS (ed) Long Non Coding RNA Biology. Springer, Singapore, pp 199–222
Ricciardi E, Baert T, Ataseven B et al (2018) Low-grade serous ovarian carcinoma. Geburtshilfe Frauenheilkd 78:972–976. https://doi.org/10.1055/a-0717-5411
Rishabh K, Khadilkar S, Kumar A et al (2021) MicroRNAs as modulators of oral tumorigenesis—a focused review. Int J Mol Sci 22:2561. https://doi.org/10.3390/ijms22052561
Salamini-Montemurri M, Lamas-Maceiras M, Barreiro-Alonso A et al (2020) The challenges and opportunities of LncRNAs in ovarian cancer research and clinical use. Cancers (Basel) 12:1020. https://doi.org/10.3390/cancers12041020
Salmena L, Poliseno L, Tay Y et al (2011) A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell 146:353–358. https://doi.org/10.1016/j.cell.2011.07.014
Schmitt AM, Chang HY (2016) Long noncoding RNAs in cancer pathways. Cancer Cell 29:452–463. https://doi.org/10.1016/j.ccell.2016.03.010
Shan L, Song P, Zhao Y, An N, Xia Y, Qi Y et al (2022) miR-600 promotes ovarian cancer cells stemness, proliferation and metastasis via targeting KLF9. J Ovarian Res 15:52
Shen H, Laird PW (2013) Interplay between the cancer genome and epigenome. Cell 153:38–55. https://doi.org/10.1016/j.cell.2013.03.008
Shi T, Gao G, Cao Y (2016) Long noncoding RNAs as novel biomarkers have a promising future in cancer diagnostics. Dis Markers. https://doi.org/10.1155/2016/9085195
Siegel RL, Miller KD, Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66:7–30. https://doi.org/10.3322/caac.21332
Siegel RL, Miller KD, Jemal A (2017) Cancer statistics. CA Cancer J Clin 67:7–30. https://doi.org/10.3322/caac.21387
Slack FJ, Chinnaiyan AM (2019) The role of non-coding RNAs in oncology. Cell 179:1033–1055. https://doi.org/10.1016/j.cell.2019.10.017
Spentzos D, Levine DA, Ramoni MF et al (2004) Gene expression signature with independent prognostic significance in epithelial ovarian cancer. J Clin Oncol 22:4700–4710. https://doi.org/10.1200/JCO.2004.04.070
Sung H, Ferlay J, Siegel RL et al (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 Countries. CA A Cancer J Clin. https://doi.org/10.3322/caac.21660
Svoronos AA, Engelman DM, Slack FJ (2016) OncomiR or tumor suppressor? The duplicity of MicroRNAs in cancer. Can Res 76:3666–3670. https://doi.org/10.1158/0008-5472.CAN-16-0359
Tao Z, Shi A, Li R et al (2017) Microarray bioinformatics in cancer- a review. J BUON 22:838–843
Tian J, Wang Y, Zhang X et al (2017) Calycosin inhibits the in vitro and in vivo growth of breast cancer cells through WDR7-7-GPR30 signaling. J Exp Clin Cancer Res 36:153. https://doi.org/10.1186/s13046-017-0625-y
Torre LA, Trabert B, DeSantis CE et al (2018) Ovarian cancer statistics. CA Cancer J Clin 68:284–296. https://doi.org/10.3322/caac.21456
Treiber T, Treiber N, Meister G (2019) Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol 20:5–20. https://doi.org/10.1038/s41580-018-0059-1
Trinidad CV, Tetlow AL, Bantis LE, Godwin AK (2020) Reducing ovarian cancer mortality through early detection: approaches using circulating biomarkers. Cancer Prev Res (Phila) 13:241–252. https://doi.org/10.1158/1940-6207.CAPR-19-0184
van Solingen C, Cyr Y, Scacalossi KR et al (2022) Long noncoding RNA CHROMR regulates antiviral immunity in humans. Proc Natl Acad Sci 119:e2210321119. https://doi.org/10.1073/pnas.2210321119
Volders P-J, Anckaert J, Verheggen K et al (2019) LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic Acids Res 47:D135–D139. https://doi.org/10.1093/nar/gky1031
Wambecke A, Ahmad M, Morice P et al (2021) The lncRNA ‘UCA1’ modulates the response to chemotherapy of ovarian cancer through direct binding to miR-27a-5p and control of UBE2N levels. Mol Oncol 15:3659–3678. https://doi.org/10.1002/1878-0261.13045
Wang KC, Chang HY (2011) Molecular mechanisms of long noncoding RNAs. Mol Cell 43:904–914. https://doi.org/10.1016/j.molcel.2011.08.018
Wang F, Zhou J, Xie X et al (2015) Involvement of SRPK1 in cisplatin resistance related to long non-coding RNA UCA1 in human ovarian cancer cells. Neoplasma 62:432–438
Wang H, Fu Z, Dai C et al (2016) LncRNAs expression profiling in normal ovary, benign ovarian cyst and malignant epithelial ovarian cancer. Sci Rep 6:38983. https://doi.org/10.1038/srep38983
Wang J, Ye C, Liu J, Hu Y (2018) UCA1 confers paclitaxel resistance to ovarian cancer through miR-129/ABCB1 axis. Biochem Biophys Res Commun 501:1034–1040. https://doi.org/10.1016/j.bbrc.2018.05.104
Wang J, Dean DC, Hornicek FJ et al (2019a) RNA sequencing (RNA-Seq) and its application in ovarian cancer. Gynecol Oncol 152:194–201. https://doi.org/10.1016/j.ygyno.2018.10.002
Wang W-T, Han C, Sun Y-M et al (2019b) Noncoding RNAs in cancer therapy resistance and targeted drug development. J Hematol Oncol 12:55. https://doi.org/10.1186/s13045-019-0748-z
Wang H, Su H, Tan Y (2020a) UNC5B-AS1 promoted ovarian cancer progression by regulating the H3K27me on NDRG2 via EZH2. Cell Biol Int 44:1028–1036. https://doi.org/10.1002/cbin.11300
Wang P, Li X, Gao Y et al (2020b) LnCeVar: a comprehensive database of genomic variations that disturb ceRNA network regulation. Nucleic Acids Res 48:D111–D117. https://doi.org/10.1093/nar/gkz887
Wang Z, Wang Q, Xu G et al (2020c) The long noncoding RNA CRAL reverses cisplatin resistance via the miR-505/CYLD/AKT axis in human gastric cancer cells. RNA Biol 17:1576–1589. https://doi.org/10.1080/15476286.2019.1709296
Wang Y, Shao F, Chen L (2022) miR-425 regulates ovarian cancer proliferation, metastasis, and apoptosis by repressing PAK4 expression. Asia Pac J Clin Oncol 18(1):76–83
Wang J, Liu L (2021) MiR-149-3p promotes the cisplatin resistance and EMT in ovarian cancer through downregulating TIMP2 and CDKN1A. J Ovarian Res 14:165
Wen S, Wei Y, Zen C et al (2020) Long non-coding RNA NEAT1 promotes bone metastasis of prostate cancer through N6-methyladenosine. Mol Cancer 19:171. https://doi.org/10.1186/s12943-020-01293-4
Wiehle L, Breiling A (2016) Chromatin Immunoprecipitation. In: Lanzuolo C, Bodega B (eds) Polycomb group proteins: methods and protocols. Springer, New York, pp 7–21
Xiu Y, Sun K, Chen X, et al (2017) Upregulation of the lncRNA Meg3 induces autophagy to inhibit tumorigenesis and progression of epithelial ovarian carcinoma by regulating activity of ATG3. Oncotarget 8:31714–31725. https://doi.org/10.18632/oncotarget.15955
Xu H, Ding Y, Yang X (2021) Overexpression of long noncoding RNA H19 downregulates miR-140-5p and activates PI3K/AKT signaling pathway to promote invasion, migration and epithelial-mesenchymal transition of ovarian cancer cells. Biomed Res Int. https://doi.org/10.1155/2021/6619730
Xue Y, Ma G, Gu D et al (2015) Genome-wide analysis of long noncoding RNA signature in human colorectal cancer. Gene 556:227–234. https://doi.org/10.1016/j.gene.2014.11.060
Yan X, Hu Z, Feng Y et al (2015) Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell 28:529–540. https://doi.org/10.1016/j.ccell.2015.09.006
Yang Y, Jiang Y, Wan Y et al (2016) UCA1 functions as a competing endogenous RNA to suppress epithelial ovarian cancer metastasis. Tumour Biol 37:10633–10641. https://doi.org/10.1007/s13277-016-4917-1
Yang F, Ning Z, Ma L et al (2017) Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol Cancer 16:148. https://doi.org/10.1186/s12943-017-0718-4
Yang Z, Xu F, Teschendorff AE et al (2022) Insights into the role of long non-coding RNAs in DNA methylation mediated transcriptional regulation. Front Mol Biosci. https://doi.org/10.3389/fmolb.2022.1067406
Ye Y, Li S-L, Wang S-Y (2018) Construction and analysis of mRNA, miRNA, lncRNA, and TF regulatory networks reveal the key genes associated with prostate cancer. PLoS One. https://doi.org/10.1371/journal.pone.0198055
Yuan D, Guo T, Zhu D et al (2022) Exosomal lncRNA ATB derived from ovarian cancer cells promotes angiogenesis via regulating miR-204-3p/TGFβR2 axis. Cancer Manag Res 14:327–337. https://doi.org/10.2147/CMAR.S330368
Zaman MS, Maher DM, Khan S et al (2012) Current status and implications of microRNAs in ovarian cancer diagnosis and therapy. J Ovarian Res 5:44. https://doi.org/10.1186/1757-2215-5-44
Zhan L, Li J, Wei B (2018) Long non-coding RNAs in ovarian cancer. J Exp Clin Cancer Res 37:120. https://doi.org/10.1186/s13046-018-0793-4
Zhang B, Cai FF, Zhong XY (2011) An overview of biomarkers for the ovarian cancer diagnosis. Eur J Obstet Gynecol Reprod Biol 158:119–123. https://doi.org/10.1016/j.ejogrb.2011.04.023
Zhang Y, Feng Y, Hu Z et al (2016a) Characterization of long non-coding RNA associated proteins by RNA-immunoprecipitation. Methods Mol Biol 1402:19–26. https://doi.org/10.1007/978-1-4939-3378-5_3
Zhang Z, Cheng J, Wu Y et al (2016b) LncRNA HOTAIR controls the expression of Rab22a by sponging miR-373 in ovarian cancer. Mol Med Rep 14:2465–2472. https://doi.org/10.3892/mmr.2016.5572
Zhang Z, Liu T, Cheng C et al (2022) LncRNA GAS5 regulates the Wnt/β-catenin pathway through the miR-18a-5p/AXIN2/GSK3β axis to inhibit the proliferation and migration of bladder cancer cells. Carcinogenesis 43:1176–1189. https://doi.org/10.1093/carcin/bgac087
Zhao Y, Li H, Fang S et al (2016) NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res 44:D203–D208. https://doi.org/10.1093/nar/gkv1252
Zheng M-J, Li X, Hu Y-X et al (2019a) Identification of molecular marker associated with ovarian cancer prognosis using bioinformatics analysis and experiments. J Cell Physiol 234:11023–11036. https://doi.org/10.1002/jcp.27926
Zheng Y, Xu Q, Liu M et al (2019b) lnCAR: a comprehensive resource for lncRNAs from cancer arrays. Cancer Res 79:2076–2083. https://doi.org/10.1158/0008-5472.CAN-18-2169
Zhou Y, Xu X, Lv H et al (2016) The long noncoding RNA MALAT-1 is highly expressed in ovarian cancer and induces cell growth and migration. PLoS One 11:e0155250. https://doi.org/10.1371/journal.pone.0155250
Zhou R-S, Zhang E-X, Sun Q-F et al (2019) Integrated analysis of lncRNA-miRNA-mRNA ceRNA network in squamous cell carcinoma of tongue. BMC Cancer 19:779. https://doi.org/10.1186/s12885-019-5983-8
Zhu Y, Chen J, Zhou L et al (2022) A platinum resistance-related lncRNA signature for risk classification and prognosis prediction in patients with serous ovarian cancer. J Oncol. https://doi.org/10.1155/2022/7625138
Acknowledgements
We would like to acknowledge Biorender, through which we created Fig. 2.
Funding
SQU internal Grant: Grant number: IG/MED/BIOC/22/01.
Author information
Authors and Affiliations
Contributions
SH drafted the review article. YT contributed to the concept and reviewing of the article. All the authors approved the final version of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Malgundkar, S.H., Tamimi, Y. The pivotal role of long non-coding RNAs as potential biomarkers and modulators of chemoresistance in ovarian cancer (OC). Hum. Genet. 143, 107–124 (2024). https://doi.org/10.1007/s00439-023-02635-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-023-02635-0