
Abstract
Up to 84% of patients with congenital pseudarthrosis of the tibia (CPT) present with neurofibromatosis type 1 (NF1) (NF1-CPT). However, the etiology of CPT not fulfilling the NIH diagnostic criteria for NF1 (non-NF1-CPT) is not well understood. Here, we collected the periosteum tissue from the pseudarthrosis (PA) site of 43 non-NF1-CPT patients and six patients with NF1-CPT, together with the blood or oral specimen of trios (probands and unaffected parents). Whole-exome plus copy number variation sequencing, multiplex ligation-dependent probe amplification (MLPA), ultra-high amplicon sequencing, and Sanger sequencing were employed to identify pathogenic variants. The result showed that nine tissues of 43 non-NF1-CPT patients (21%) had somatic mono-allelic NF1 inactivation, and five of six NF1-CPT patients (83.3%) had bi-allelic NF1 inactivation in tissues. However, previous literature involving genetic testing did not reveal somatic mosaicism in non-NF1-CPT patients so far. In NF1-CPT patients, when the results from earlier reports and the present study were combined, 66.7% of them showed somatic NF1 inactivation in PA tissues other than germline inactivation. Furthermore, no diagnostic variants from other known genes (GNAS, AKT1, PDGFRB, and NOTCH3) related to skeletal dysplasia were identified in the nine NF1 positive non-NF1-CPT patients and six NF1-CPT patients. In conclusion, we detected evident somatic mono-allelic NF1 inactivation in the non-NF1-CPT. Thus, for pediatric patients without NF1 diagnosis, somatic mutations in NF1 are important.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Congenital pseudarthrosis of the tibia (CPT) is a rare disease characterized by recalcitrant healing of the tibia followed by anterolateral bowing, pathological fractures, or pseudarthrosis in early life (Hefti et al. 2000; O'Donnell et al. 2017; Vander Have et al. 2008). Up to 84% of patients with CPT match the NIH diagnostic criteria for neurofibromatosis type 1 (NF1, MIM# 162200) (Neurofibromatosis 1988; Van Royen et al. 2016). NF1 is an autosomal-dominant genetic disease involving cutaneous, ocular, or osseous lesions, and some of the affected individuals have neurological, vascular, or cardiac manifestations. NF1 occurs in approximately 1/3000 to 1/2000 births (Evans et al. 2010; Lammert et al. 2005; Uusitalo et al. 2015), and < 5% of patients with NF1 have CPT (Friedman and Birch 1997; Schindeler and Little 2008; Young et al. 2002).
Haplo-insufficiency of the NF1 gene brought by loss-of-function (LoF) variation can cause NF1. NF1 is a tumor suppressor that encodes neuro-fibromin activating ras GTPase to control cellular proliferation, and loss of neuro-fibromin activates the RAS/MAPK signaling pathway leading to cell overgrowth (Andersen et al. 1993; Ballester et al. 1990; Basu et al. 1992; Bollag et al. 1996; Cichowski and Jacks 2001; DeClue et al. 1991; Lau et al. 2000; Xu et al. 1990a, b). Previous studies that detected the CPT pseudarthrosis (PA) tissue focused on CPT patients with NF1 (NF1-CPT) (Brekelmans et al. 2019; Lee et al. 2012; Margraf et al. 2017; Paria et al. 2014; Sakamoto et al. 2007; Sant et al. 2015; Stevenson et al. 2006; Tahaei et al. 2018), and molecular testing mainly targeted the DNA, RNA, or protein of NF1 via techniques, such as whole-exome sequencing (WES), RNA-seq, protein truncation test, multiplex ligation-dependent probe amplification (MLPA), gene-targeted microarray, quantitative PCR, and long-range PCR. However, approximately 26% of CPT cases do not meet NF1 diagnosis (non-NF1-CPT), and no genetic findings have been reported. Thus, the non-NF1-CPT etiology is still not understood.
Non-NF1-CPT patients usually have an isolated phenotype in the tibia. Generally, non-NF1-CPT has similar clinical manifestations as NF1-CPT (Zhu et al. 2019), including the time of tibia bowing or fracture onset, lateralization, abnormality of the proximal tibial epiphysis, and tibial union follow-up after surgery. Previous histological, pathological, and biological studies found no significant differences between NF1-CPT and non-NF1-CPT (Granchi et al. 2010; Hermanns-Sachweh et al. 2005), with both showing thickened periosteum with nerve cells accumulating around small arteries (Hermanns-Sachweh et al. 2005). In addition, a biological study using cultured bone marrow stromal cells from the PA site showed reduced osteogenicity in NF1-CPT and non-NF1-CPT than in controls (Granchi et al. 2010). However, our previous study of 75 patients with CPT identified no germline pathogenic NF1 variants in all 20 non-NF1-CPT patients (Zhu et al. 2019). In the reviewed 158 cases with mosaic NF1 (MNF1), two cases presented non-NF1-CPT but did not undergo genetic testing (El-Rosasy 2020; Garcia-Romero et al. 2016; Listernick et al. 2003).
Periosteum covering the outer surface of bone is essential for skeletal development and growth, and it also works as a central mediator of bone-healing after fracture (Hutmacher and Sittinger, 2003; Ozaki et al. 2000; Roberts et al. 2015). Pathologically, both NF1-CPT and non-NF1-CPT showed thicken periosteum with fibrous hyperplasia at the PA site (Ippolito et al. 2000; Sakamoto et al. 2007; Zhu et al. 2019). For NF1-CPT, some studies found bi-allelic NF1 inactivation (also known as second hit, double inactivation) in the PA tissue, which covered approximately 65% of the published cases (Lee et al. 2012; Paria et al. 2014; Sakamoto et al. 2007; Sant et al. 2015; Stevenson et al. 2006). However, for non-NF1-CPT, as no germline NF1 inactivation has been reported, we supposed that somatic NF1 inactivation could exist in the pathological periosteum at PA site related to CPT lesion. Here we obtained the DNA of the PA periosteum of 43 non-NF1-CPT patients and six patients with NF1-CPT and performed whole-exome plus copy number variation (CNV) sequencing (WES + CNV) and MLPA to analyze pathogenic variants spanning the known associated genes. We then validated the identified pathogenic variants using DNA from the trios (affected probands and unaffected parents).
Methods
Aim, design, and settings
This study aimed to investigate disease-causing variants in non-NF1-CPT patients with unknown etiologies. Toward this goal, using the PA tissues, we performed WES + CNV and MLPA in 43 non-NF1-CPT patients to first screen for known disease-causing genes. The source of mutation was then identified using trios for precise diagnosis, management, and genetic counseling. Concurrently, the six NF1-CPT patients were analyzed using the same pipeline for comparison.
Participants
After obtaining ethical approval and informed consent, we studied 43 patients with non-NF1-CPT and six patients with NF1-CPT who underwent surgery (Supp. Table S1). The patients were not diagnosed with osteofibrous dysplasia. We collected detailed clinical information, family history, and periosteum of the discarded PA tissue of each patient (Table 1). The peripheral blood or oral samples of eighteen trios (affected and unaffected parents) were also collected. According to the NF1 diagnostic criteria (NIH, 1988) (Neurofibromatosis 1988), all non-NF1-CPT cases have an isolated phenotype of tibial pseudarthrosis (HP:0009736) and do not match NF1. All six NF1-CPT patients had six or more café au lait macules (CAL, HP:0007565) > 5 mm in greatest diameter and were diagnosed with NF1 (Table 1).
Sample preparation and DNA extraction
The pathological periosteum surrounding the cortical bone of the discarded PA tissue during surgery (Supp. Fig. S1) was cut into pieces. No bone spicules were embedded. Each sample was approximately 200 mg and had a diameter of 5 mm approximately. One piece of periosteum biopsies was performed pathology detection and stained with hematoxylin and eosin (H&E) following the standard protocol. Other pieces of periosteum tissue were washed with phosphate-buffered saline (PBS), and then were frozen at − 80 °C or preserved in liquid nitrogen. Genomic DNA was extracted using the following steps: (1) the preserved periosteum was ground using the automatic sample quick grinding machine JXFSTPRP-12/16 (Tissuelyser, Shanghai, China; the ground parameter was 65 Hz for 60 s); (2) 500 μl Sodium Chloride–Tris–EDTA (STE) buffer solution (0.1 mol/L NaCl, 10 mmol/L Tris–Cl with pH = 8.0, 1 mmol/L EDTA), 10 mg/ml Proteinase K and 75 μl 10% SDS were added and digested over 24 h, (3) centrifugation and the supernatant were kept to extract DNA using the standard phenol–chloroform method, (4) the extracted DNA was then evaluated by agarose gel electrophoresis and quantified using Qubit 3.0. The genomic DNA of peripheral blood lymphocytes was extracted using the standard phenol–chloroform method. In addition, the genomic DNA of saliva or oral swabs was extracted using the Saliva DNA Purification Kit (Simgen, Hangzhou, Zhejiang, China) and Magbead Swab DNA Kit (CWBIO, Taizhou, Jiangsu, China), respectively.
Whole exome plus CNV sequencing and bioinformatics analysis
Forty-nine periosteal DNA samples and twelve blood DNA samples of unaffected parents were submitted to whole exome plus CNV sequencing (Supp. Table S1). Genomic DNA (200 ng) of each sample was fragmented, and the exome was captured using the AIExomeV1-CNV Enrichment Kit (iGeneTech, Beijing, China, capture region size: 62 Mb) for library preparation (concentration > 25 ng/ul, central peak 220–320 bp). The captured DNA was sequenced with 2 × 150 bp reads using an Illumina NovaSeq 6000 system (Illumina, San Diego, California, USA) following the manufacturer’s instructions. Each sample yielded over 15 GB of raw data with a minimum mean depth of coverage of 100 × . The average base Phred quality was over 36.1 (34.9–36.4), and the mean target coverage at 20X was 97.6% (84.4–99.4%).
The sequenced raw reads in FastQ file format were preprocessed using trim_galore (version 0.6.4, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to remove adapter-contaminated ends and low-quality bases with Phred scores < 20. Reads with > 5N bases, > 40% low-quality bases, or trimmed lengths < 30 bp were also removed. Subsequently, the clean reads were mapped to the human reference sequence (version: GRCh37) using the alignment tool Burrows–Wheeler Aligner (BWA, Version 0.7.17-r1188) (Li and Durbin 2010). SAMtools (Li et al. 2009) and SAMBLASTER (version 0.1.26) (Faust and Hall 2014) were used to remove duplicate reads. The Genome Analysis Toolkit (GATK, version 3.8) (DePristo et al. 2011) refined the alignment and call variants.
SNV and InDel analysis
The generated single-nucleotide variations (SNVs) and short insertions and deletions (InDels) from GATK with genotype quality ≥ 20, depth ≥ 5, and variant allele frequency ≥ 0.15 were kept for subsequent analysis. They were annotated using ANNOVAR (Wang et al. 2010) and InterVar (version 20,180,118) (Li and Wang 2017). We removed the SNPs and InDels with population frequency (minor allele frequency, MAF) > 0.001 in gnomAD (East Asian and popmax of continental populations using WES and whole genome sequencing) and ExAC (all populations and East Asian) databases (Karczewski et al. 2017, 2020). SNPs and InDels shared by at least two of the 12 CPT family controls or shared by less than two of our in-house 528 WES family controls (with MAF > 0.001) (Liu et al. 2021; Yang et al. 2019) were also removed. The remaining non-benign heterozygous variants annotated by InterVar or ClinVar (version 20180603) in the coding or UTR regions were then used for further analysis. The prioritized variants of the NF1 gene were screened using ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and HGMD databases (public version, http://www.hgmd.cf.ac.uk) for known pathogenic records. VarSome (Kopanos et al. 2019) was employed to classify the pathogenicity of each variant according to the American College of Medical Genetics and Genomics (ACMG) criteria (Richards et al. 2015). Protein domains and repeats and homologous super-families of neuro-fibromin were queried from InterPro (http://www.ebi.ac.uk/interpro). In addition, the known disease-causing genes GNAS, AKT1, PDGFRB, and NOTCH3, related to musculoskeletal dysplasia similar to CPT, were screened for differential diagnosis.
Detection of CNVs and structural variations (SVs)
CNVkit (version 0.9.6) (Talevich et al. 2016) was used to detect CNVs in each patient. Twelve healthy parents were used as reference, and the threshold parameter “− t = − 1.1, − 0.4, 0.3, 0.7” was used for CNV calling. We then annotated the identified CNVs using ANNOVAR against the refGene, cytoBand, wgRna, genomicSuperDups, genomicCatalog, and CNVD databases. The detected CNVs overlapping NF1, GNAS, AKT1, PDGFRB, and NOTCH3 genes were interpreted according to the ACMG and ClinGen technical standards (Riggs et al. 2020). Pathogenic or likely pathogenic CNVs were further validated using MLPA.
SVs were detected using the LUMPY-sv (version v0.3.0) (Layer et al. 2014). After filtering out the SVs shared by the 12 unaffected parents, the remaining SVs overlapping NF1, GNAS, AKT1, PDGFRB, and NOTCH3 genes were subjected to for further validation. Integrative Genomics Viewer (IGV) (Thorvaldsdottir et al. 2013) was employed to view the variant-located chromosomal region using the patient’s bam file to confirm the CNVs and SVs.
Validation by Sanger sequencing
The identified pathogenic or likely pathogenic, and LoF (nonsense, frameshifting indels and canonical splice-site) SNV and InDels that overlapped with the NF1 gene were validated using Sanger sequencing in the trios [PA tissue of probands, peripheral blood, or saliva or oral swabs of probands and their parents (see Table 1 and Supp. Fig. S2 and Supp. Fig. S3 for details)]. First, we employed the Primer-BLAST program (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) to design PCR primers (primers for validating SNPs and InDels were provided in Supp. Table S2). Subsequently, all variants were validated via independent PCR amplification and bidirectional DNA sequencing suing ABI 3130 DNA analyzer (Applied Biosystems, Foster City, CA, USA).
Multiplex ligation-dependent probe amplification (MLPA)
For suspected CNVs overlapping the NF1 gene, MLPA was performed to validate deletions or duplications encompassing ≥ 1 NF1 exons or the entire gene. We used the SALSA MLPA probe P081 NF1 mix 1 and P082 NF1 mix 2 (MRC-HOLLAND, Amsterdam, Netherlands) to detect CNVs in the extracted DNA and performed dosage analysis following the manufacturer’s instructions.
Mosaic variant validation using ultra-high amplicon sequencing
NF1-related pathogenic or likely pathogenic SNVs and InDels with allele fractions ≥ 5% and ≤ 30% were considered to suspect mosaic variations. Only one variant c.334C > T with an allele fraction of 18%, was used for further validation. The Primer-BLAST program was used to design PCR primers specific for the variant. The PCR product with the target length (699 bp) using the extracted tissue DNA was submitted for ultra-high amplicon sequencing. The target-captured DNA was then sequenced with 2 × 150 bp reads using an Illumina NovaSeq 6000 system (Illumina, San Diego, California, USA) following the manufacturer’s instructions. Furthermore, 1.32 Gb of raw data was obtained with a minimum mean depth of coverage over 70,000 × and an average base quality of 36. The raw reads were polished and then aligned to the human reference sequence (version: GRCh37) using BWA MEM. Variants in the target regions were identified using GATK. IGV was deployed to confirm the variants using the generated bam file.
Literature search
The literature search in PubMed was performed using the search terms “pseudarthrosis AND (tibia OR tibial) AND (NF1 OR neurofibromatosis) AND (genetic OR molecular)”. Sixty-four articles that were published by September 30, 2021 were reviewed. Further filtering was done using keywords “somatic OR mosaic OR segmental OR localized” for non-NF1-CPT reports. We further searched for publications on NF1-CPT with somatic bi-allelic NF1 inactivation using the keywords “second hit”, “biallelic”, or “double inactivation”. We then manually screened these publications with clinical features and NF1 variants reported in patients.
Results
Non-NF1-CPT patients have pathogenic somatic NF1 variants of in the PA periosteum
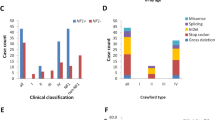
After quality and allele frequency filtration, 742 pathogenic, likely pathogenic or LoF SNPs and InDels were left in the WES data of all the 43 non-NF1-CPT and 6 NF1-CPT PA periosteal samples. They were distributed in 631 genes, and the NF1 gene covered a significantly higher proportion (14/49; 28.6%) of the sequenced cases than other genes (< 10% each). In the PA periosteum tissue of 43 non-NF1-CPT patients, 9 (21%, 9/43) were identified NF1 heterozygous variants (Fig. 1). All nine variants were classified as pathogenic based on the ACMG criteria. Further by validating the variants in family trios, seven of nine variants were found in the PA periosteum but not in the peripheral blood or oral epithelial cells of patients, or their parents (Table 1, Supp. Fig. S1). Therefore, the seven variations were confirmed to be somatic. For example, P1 was identified a heterozygous NF1 gene loss by MLPA in the PA tissue. P3 has ClinVar known pathogenic splicing variant c.4269 + 2T > C. P4 has c.484C > T (p.Gln162Ter) leading to premature translation termination. P9 was detected a mosaic c.334C > T (p.Gln112Ter) in the PA periosteum. The three variants were recorded in the ClinVar or HGMD databases as pathogenic. In addition, P6 and P7 harbor novel splicing variants c.205-1G > T and c.2002-1G > C; while P8 has a novel frameshift deletion variant c.4173_4174del (p.Arg1391SerfsTer11).

Strategy and genetic analysis overview of two groups of CPT patients in this study. het: heterozygous. hom: homozygous
In case of the other two variants, which are loss of contact and lack of family samples, it was not confirmed whether c.1185 + 1G > A in P2 and c.3897del (p.Lys1299AsnfsTer10) in P5 were germline or somatic.
Bi-allelic NF1 inactivation occurs in the PA site of patients with NF1-CPT
Using the same pipeline, all six patients with NF1 identified pathogenic NF1 variants, and three had bi-allelic NF1 inactivation in the periosteum of the PA site (Table 1, Supp. Fig. S2). Patients P11, P12, and P13 were found heterozygous NF1 pathogenic variants in the blood or oral epithelial cells (p.Gln1374ValfsTer10 for P11, p.Arg2237Ter for P12, and p.Gln11Ter for P13), whereas the three variants were homozygous in the PA periosteum. All three variants were related to premature termination of translation. Among them, the variants in P11 and P12 were not found in their unaffected parents and were confirmed to be de novo. Another variant in P13 was confirmed to be maternally inherited and the patient’s mother had NF1 findings. Notably, the InDel variant in P11’s tissue was identified as heterozygous with low-variant allele frequency (719/234; 25%) by WES, while by Sanger sequencing, this variant was homozygous in the patient’s PA periosteum and heterozygous in the patient’s blood. This variant was not detected in P11’s father and mother by Sanger sequencing. Moreover, the tibia of P11 and P13 were not un-ioned after surgery (Supp. Table S1). We supposed that one of the reasons was non-autogenous iliac bone graft applied.
Specifically, patient P10 was the only mono-allelic variant c.4267A > G (p.Lys1423Glu) identified in blood and PA periosteum. The variant was classified as pathogenic based on the ACMG criteria and is a known pathogenic variant in ClinVar. This variant was also identified in P10’s monozygotic twin sister, who also had NF1 but without CPT manifestations. The patient’s parents did not have the variant, suggesting that it is de novo nature.
Somatic mosaicism in PA periosteum happens in both non-NF1-CPT and NF1-CPT
In non-NF1-CPT, patient P9 was identified a mosaic c.334C > T (p.Gln112Ter) with an allelic fraction of ~ 16% in the PA tissue but not in the blood (Table 1). WES and ultra-high amplicon sequencing demonstrated similar allelic fractions of 19% (depth: 23/118) and 16% (depth: 12,288/74,922), respectively. This patient was a female and manifested CPT in the left leg at birth.
In NF1-CPT, in addition to the identified germline heterozygous NF1 inactivation, two patients (P14 and P15) were detected as carriers of a new NF1 mosaic variant in their PA periosteum. Among them, c.989C > A (p.Ala330Glu) in P14 had an allele fraction of 34%, which was not found in her oral epithelial cells. According to ACMG criteria, this variant was classified as likely pathogenic (PM1 PM2 PM5 PP2 PP3) and was recorded in ClinVar as variant of uncertain significance (VUS). Another variant c.1381C > T (p.Arg461Ter) in P15 had an allele fraction of 18% in the PA tissue but not in the oral epithelial cells. This variant was recorded as pathogenic in ClinVar. Both patients were female, and they manifested CPT near 2 years old (24 months old for P14 and 22 months old for P15).
Non-NF1-CPT patients with somatic NF1 inactivation have early and unilateral onset and similar union time to NF1-CPT
All nine non-NF1-CPT patients had unilateral onset with no family history of NF1 (Table 1). They all initially presented tibia bowing or fracture in infancy, including four infants presented at birth. The average age of onset was three months (Supp. Table S1). There was no significant bias in left or right lateral onset (5:4), but there were more male patients than females (6:3) in this cohort. H&E staining of the periosteum prevalently showed fibro-vascular tissue hyperplasia with partially hyaline degeneration, and thick-walled angiogenesis in non-NF1-CPT patients like NF1-CPT (Supp. Table S1). Compared with the six patients with NF1, the average age of tibia bowing or fracture presentation was two months, and the ratio of left to right lateralization was 2:4. Patients P11, P12, and P13 with bi-allelic NF1 inactivation in PA tissue presented tibia bowing in three months after birth. Referring to the 34 non-NF1-CPT patients without somatic NF1 inactivation, the average age of tibia bowing or fracture presentation was near nine months, which was older than the nine non-NF1-CPT patients with somatic NF1 inactivation.
As for the union time of tibia after surgery, the average union time of the nine non-NF1-CPT patients with somatic NF1 inactivation was five months. All the 43 non-NF1-CPT patients have a similar average union time (Supp. Table S1). Excluding the two patients (P11 and P13) without tibia union, the average union time of the other four NF1-CPT patients was four months. The union time of the NF1-CPT and non-NF1 CPT was quite close.
66.7% of NF1-CPT patients showed somatic NF1 inactivation in PA tissue
Based on a review of 10 articles from 2006 to 2020 (Brekelmans et al. 2019; Lee et al. 2012; Leskela et al. 2009; Margraf et al. 2017; Paria et al. 2014; Sakamoto et al. 2007; Sant et al. 2015; Stevenson et al. 2006; Tahaei et al. 2018), a total of 57 patients with NF1-CPT were reported with NF1 genetic testing in the PA tissue (Table 2). These patients were all diagnosed with NF1 based on the NIH diagnostic criteria. DNA, RNA, or protein from the PA tissue was detected using traditional molecular or cytogenetic detection methods (e.g., genotyping NF1 markers, restriction fragment length polymorphism, MS-PCR, MLPA), and NGS-based technology (e.g., WES, RNA-seq). Thirty-seven patients were identified as having a second hit in NF1 in the PA tissue other than a heterozygous germline NF1 inactivation. Combined with the five of six NF1 patients in our study, a total of 42 NF1-CPT patients (42/63, 66.7%) were found to have a somatic NF1 inactivation in addition to a germline mutation in the PA tissue, which formed bi-allelic NF1 inactivation (Table 2).
No pathogenic variants were found in the known skeletal dysplasia causing genes
For differential diagnostic purposes, in the variants called from WES + CNV sequencing of PA tissue, we screened the non-benign variants of the prioritized genes related to skeletal dysplasia with features similar to those of CPT. The known phenotype-relevant genes include GNAS related to fibrous dysplasia, AKT1-associated Proteus syndrome, PDGFRB, and NOTCH3-related autosomal-dominant infantile myofibromatosis. Based on the ACMG criteria, no likely pathogenic variants of these genes were identified in the nine non-NF1-CPT and six NF1-CPT patients with NF1 mutation. For the other 34 patients, only three missense variants with uncertain significance (VUS) were identified in the periosteum: P34 had c.166_167delGCinsAT (p.Ala56Met) in GNAS, P36 had c.881C > T (p.Thr294Met) in NOTCH3, and P47 had c.914C > T (p.Thr305Ile) in AKT1.
Discussion
This study focused on non-NF1-CPT patients that did not meet the clinical diagnostic criteria for NF1. These patients had an isolated clinical manifestation of tibial pseudarthrosis, and we primarily detected the PA periosteum using WES + CNV analysis to screen for disease-causing variants. We found that 21% of the detected non-NF1-CPT patients had somatic mono-allelic NF1 inactivation in the PA periosteum but not in blood or oral samples. Furthermore, using the same pipeline in the six NF1-CPT patients, we identified a second somatic NF1 inactivation in addition to germline mono-allelic NF1 inactivation in the PA periosteum from five of them. Our findings revealed that somatic NF1 variation exists in non-NF1-CPT and NF1-CPT patients with a significant proportion, and both have early onset.
NF1 somatic mutation found in the pathological periosteum of non-NF1-CPT with fibrous proliferation could be construed as low-level mosaicism of NF1. The age of tibia bowing or fracture onset, tibia union time after surgery, and fibrosis in the periosteum had no significant differences between the non-NF1-CPT patients with NF1 somatic mutation and the NF1-CPT patients. NF1 has high phenotypic variability without clear genotype–phenotype correlations (Gutmann et al. 2017; Sabbagh et al. 2013; Zhang et al. 2015). The function of neuro-fibromin encoded by NF1 is still not fully understood. Recently, Luo et al. and Koliou et al. reported that neuro-fibromin is involved in chromosome congression and participates in somatic cell division (Koliou et al. 2016; Luo et al. 2014). This may be related to the various onset times of different NF1 manifestations. Similar to the most common CALs, CPT as a skeletal abnormality is usually symptomatic from birth to infancy (Gutmann et al. 2017). In this study, non-NF1-CPT patients did not exhibit CALs and other symptomatic manifestations, the localized lesion in tibia with NF1 somatic haplo-insufficiency during bone development could be an importance cause. Furthermore, as periosteum serves as the source of osteoprogenitor cells producing chondrocytes and mature osteoblasts for bone growth, bone modeling and remodeling after fracture (Maes et al. 2010; Ozaki et al. 2000; Roberts et al. 2015), NF1 inactivation was found in the considerable portion of CPT’s PA periosteum with highly fibrous hyperplasia might contribute to the formation of pseudarthrosis. The dysfunction of periosteum could also impair the ossification during bone growth and bone-healing after fracture.
A high frequency of LoF variations of NF1 was observed in CPT, either germline de novo or somatic. After combining the results of previous studies and our study, the overall proportion of somatic second hits of NF1 in NF1-CPT is approximately 67% (Brekelmans et al. 2019; Lee et al. 2012; Leskela et al. 2009; Margraf et al. 2017; Paria et al. 2014; Sakamoto et al. 2007; Sant et al. 2015; Stevenson et al. 2006; Tahaei et al. 2018). Interestingly, this proportion is the same as that of the somatic inactivation in skin biopsy and blood mosaicism of NF1 of the 15 MNF1 cases subjected to genetic testing (10/15, 67%) (Garcia-Romero et al. 2016). With 21 of 63 NF1-CPT cases harboring somatic second hits in the literature and nine of 43 non-NF1-CPT cases with somatic mosaicism in this study, the overall rate of mosaic somatic NF1 inactivation was about 50% in 106 cases. This proportion is close to that of the de novo germline frequency of NF1 inactivation (Evans et al. 2010; Zhu et al. 2019). However, the cause of such a high-variation frequency of NF1 is not fully understood.
With NF1 pathogenic variants identified, non-NF1-CPT caused by somatic mosaicism and NF1-CPT caused by germline NF1 inactivation will be benefit for precise treatment, follow-up management, and gene counseling in future. Molecular testing can help establish the diagnosis of NF1 and MNF1 for CPT as early. Recently Legius et al. revised the diagnostic criteria for NF1 by adding NF1 genetic diagnosis and several more specified clinical features (Legius et al. 2021; Parrozzani et al. 2015; Tadini et al. 2014; Vagge et al. 2016). With a clear diagnosis, the follow-up surveillance of tibia union and other tissue manifestations between NF1-CPT and non-NF1-CPT would be helpful for future customized management. Furthermore, up to 13% of the reported patients with MNF1 or complete NF1 have malignancy risk (Garcia-Romero et al. 2016). Malignant peripheral nerve sheath tumors (MPNSTs) are the most common and often occur in adolescents or early adulthood in individuals with NF1 (Hagel et al. 2007; McCaughan et al. 2007; Valentin et al. 2016). Patients with complete NF1 loss are at a greater risk of developing MPNST (Kehrer-Sawatzki et al. 2017; Upadhyaya et al. 2004) than other types of variants. In this study, Patient P1 was detected the entire NF1 loss in the PA periosteum, and the follow-up surveillance for malignant risk would be needed. Other tumors, such as rhabdomyosarcomas and malignant fibrous histiocytoma, may occur since childhood (Seminog and Goldacre 2013; Varan et al. 2016). Although non-NF1-CPT with a somatic mutation has a lower risk of malignant tumors than NF1-CPT and unlikely has NF1 offspring, follow-up management would be better for individuals with NF1 inactivation.
In addition to NF1, other genes might cause musculoskeletal dysplasia features that need to be differentiated from CPT. For example, polyostotic fibrous dysplasia is caused by an early embryonic postzygotic somatic mutation of GNAS. We screened GNAS in our tissue WES + CNV analysis, and no patient had pathogenic variants of this gene. Proteus syndrome is characterized by hamartomatous overgrowth of multiple tissues, such as hyperostosis, and the somatic mosaic of AKT1 contributes to 90% of affected individuals. None of our CPT cases in this study were identified any pathogenic variants of AKT1 in the detected tissue. The variant in P47 might be upgraded to likely pathogenic if it is de novo. As the parent’s samples were unavailable, the pathogenicity of this variant would be illustrated later. We also screened PDGFRB and NOTCH3 gene related to autosomal-dominant infantile myofibromatosis, and no pathogenic variants were identified. All cases in this study confirmed that there were no pathogenic or likely pathogenic variants in these genes. Excepting these four genes and NF1, other genes were identified in patients sporadically with likely pathogenic or LoF variants. Whether other genes participated in CPT etiology needs to be further investigated.
Due to available samples and testing technology limitations, 79% of the non-NF1-CPT cases were genetically negative and only the gDNA of patients were detected using WES + CNV plus MLPA. According to previous studies, approximately 1/3 of the variants affecting splicing could not be detected by gDNA sequencing (Evans et al. 2016) and cDNA sequencing demonstrated a relatively higher diagnostic sensitivity for NF1 than solely gDNA-based analysis. The lack of cultured cells derived from fresh amorphous mesenchymal tissues prevented the assessment of mRNA expression. In addition, this study was lack of whole genome sequence analysis that did not enable identification of pathogenic NF1 mutations that could alter expression of the gene or splicing of the mRNA leading to nonsense-mediated decay. Some structural variants such as cytogenetic rearrangements were not detected, which contributed to < 1% of NF1 affected individuals (Messiaen et al. 2000). Therefore, the rate of somatic mosaicism for non-NF1-CPT could be higher than previously reported. Further studies on the remaining NF1-testing negative CPT patients to investigate other disease-causing variants are required. Other limitations included the lack of matching normal periosteum for all 49 patients, and we were unable to do the pair-wise comparison of PA-affected tissues to receive a more substantial result. In addition, some patients lacked of long-term follow-up and thorough physical examination. Thus, the clinical features presented in the collected patients might have been incomplete by the time of documentation. Moreover, some clinical manifestations are rare and of late-onset, such as subcutaneous neuro-fibromas, glioma, and MPNST, and they may be asymptomatic until adolescence or adulthood. Therefore, a complete follow-up, precise diagnosis, prognosis, or clinical course will be useful for non-NF1-CPT and NF1-CPT.
Conclusion
We found a significant proportion of non-NF1-CPT patients without NF1 diagnosis but with somatic mono-allelic NF1 inactivation in the PA periosteum. The high frequency of NF1 somatic mutations occurred in both non-NF1-CPT and NF1-CPT patients at an early age. During bone development and growth, NF1 haplo-insufficiency in the PA periosteum may contribute to the over proliferation of fibrous tissue leading to the formation of CPT.
WEB resources used
UCSC: https://genome.ucsc.edu/
OMIM: https://www.omim.org/
wInterVar: https://wintervar.wglab.org/
Varsome: https://varsome.com/
GnomAD: https://gnomad.broadinstitute.org/
HGMD: http://www.hgmd.cf.ac.uk/ac/validate.php
ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/
Primer-BLAST: https://www.ncbi.nlm.nih.gov/tools/primer-blast/
Availability of data and materials
The novel variants found in this study were submitted to ClinVar (accession: SCV002014754-SCV002014759). Other datasets used during the current study are available from the corresponding authors on reasonable request.
Abbreviations
- CPT:
-
Congenital pseudarthrosis of the tibia
- NF1:
-
Neurofibromatosis type 1
- NF1-CPT:
-
CPT fulfilling the NIH diagnostic criteria for NF1
- Non-NF1-CPT:
-
CPT not fulfilling the NIH diagnostic criteria for NF1
- MNF1:
-
Mosaic NF1
- MLPA:
-
Multiplex ligation-dependent probe amplification
- PA:
-
Pseudarthrosis
- WES:
-
Whole-exome sequencing
- CNV:
-
Copy number variation
- CAL:
-
Café au lait macules
- PBS:
-
Phosphate-buffered saline
- SNV:
-
Single-nucleotide variations
References
Andersen LB, Ballester R, Marchuk DA, Chang E, Gutmann DH, Saulino AM, Collins FS (1993) A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol Cell Biol 13(1):487–495
Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F (1990) The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 63(4):851–859
Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J (1992) Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 356(6371):713–715. https://doi.org/10.1038/356713a0
Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Shannon K (1996) Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 12(2):144–148. https://doi.org/10.1038/ng0296-144
Brekelmans C, Hollants S, De Groote C, Sohier N, Marechal M, Geris L, Brems H (2019) Neurofibromatosis type 1-related pseudarthrosis: Beyond the pseudarthrosis site. Hum Mutat 40(10):1760–1767. https://doi.org/10.1002/humu.23783
Cichowski K, Jacks T (2001) NF1 tumor suppressor gene function: narrowing the GAP. Cell 104(4):593–604. https://doi.org/10.1016/s0092-8674(01)00245-8
DeClue JE, Cohen BD, Lowy DR (1991) Identification and characterization of the neurofibromatosis type 1 protein product. Proc Natl Acad Sci USA 88(22):9914–9918
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. https://doi.org/10.1038/ng.806
El-Rosasy MA (2020) Congenital pseudarthrosis of the tibia: the outcome of a pathology-oriented classification system and treatment protocol. J Pediatr Orthop B 29(4):337–347. https://doi.org/10.1097/BPB.0000000000000660
Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, Lalloo F (2010) Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A 152A(2):327–332. https://doi.org/10.1002/ajmg.a.33139
Evans DG, Bowers N, Burkitt-Wright E, Miles E, Garg S, Scott-Kitching V, Huson SM (2016) Comprehensive RNA analysis of the NF1 gene in classically affected NF1 affected individuals meeting NIH criteria has high sensitivity and mutation negative testing is reassuring in isolated cases with pigmentary features only. EBioMedicine 7:212–220. https://doi.org/10.1016/j.ebiom.2016.04.005
Faust GG, Hall IM (2014) SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30(17):2503–2505. https://doi.org/10.1093/bioinformatics/btu314
Friedman JM, Birch PH (1997) Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1728 patients. Am J Med Genet 70:138–143
Garcia-Romero MT, Parkin P, Lara-Corrales I (2016) Mosaic neurofibromatosis type 1: a systematic review. Pediatr Dermatol 33(1):9–17. https://doi.org/10.1111/pde.12673
Granchi D, Devescovi V, Baglio SR, Leonardi E, Donzelli O, Magnani M, Baldini N (2010) Biological basis for the use of autologous bone marrow stromal cells in the treatment of congenital pseudarthrosis of the tibia. Bone 46(3):780–788. https://doi.org/10.1016/j.bone.2009.10.044
Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ (2017) Neurofibromatosis type 1. Nat Rev Dis Prim 3:17004. https://doi.org/10.1038/nrdp.2017.4
Hagel C, Zils U, Peiper M, Kluwe L, Gotthard S, Friedrich RE, Mautner VF (2007) Histopathology and clinical outcome of NF1-associated vs. sporadic malignant peripheral nerve sheath tumors. J Neurooncol 82(2):187–192. https://doi.org/10.1007/s11060-006-9266-2
Hefti F, Bollini G, Dungl P, Fixsen J, Grill F, Ippolito E, Wientroub S (2000) Congenital pseudarthrosis of the tibia: history, etiology, classification, and epidemiologic data. J Pediatr Orthop B 9(1):11–15
Hermanns-Sachweh B, Senderek J, Alfer J, Klosterhalfen B, Buttner R, Fuzesi L, Weber M (2005) Vascular changes in the periosteum of congenital pseudarthrosis of the tibia. Pathol Res Pract 201(4):305–312. https://doi.org/10.1016/j.prp.2004.09.013
Hutmacher DW, Sittinger M (2003) Periosteal cells in bone tissue engineering. Tissue Eng 9(Suppl 1):S45-64. https://doi.org/10.1089/10763270360696978
Ippolito E, Corsi A, Grill F, Wientroub S, Bianco P (2000) Pathology of bone lesions associated with congenital pseudarthrosis of the leg. J Pediatr Orthop B 9(1):3–10
Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, MacArthur DG (2017) The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res 45(D1):D840–D845. https://doi.org/10.1093/nar/gkw971
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, MacArthur DG (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581(7809):434–443. https://doi.org/10.1038/s41586-020-2308-7
Kehrer-Sawatzki H, Mautner VF, Cooper DN (2017) Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum Genet 136(4):349–376. https://doi.org/10.1007/s00439-017-1766-y
Koliou X, Fedonidis C, Kalpachidou T, Mangoura D (2016) Nuclear import mechanism of neurofibromin for localization on the spindle and function in chromosome congression. J Neurochem 136(1):78–91. https://doi.org/10.1111/jnc.13401
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, Massouras A (2019) VarSome: the human genomic variant search engine. Bioinformatics 35(11):1978–1980. https://doi.org/10.1093/bioinformatics/bty897
Lammert M, Friedman JM, Kluwe L, Mautner VF (2005) Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol 141(1):71–74. https://doi.org/10.1001/archderm.141.1.71
Lau N, Feldkamp MM, Roncari L, Loehr AH, Shannon P, Gutmann DH, Guha A (2000) Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J Neuropathol Exp Neurol 59(9):759–767. https://doi.org/10.1093/jnen/59.9.759
Layer RM, Chiang C, Quinlan AR, Hall IM (2014) LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15(6):R84. https://doi.org/10.1186/gb-2014-15-6-r84
Lee SM, Choi IH, Lee DY, Lee HR, Park MS, Yoo WJ, Cho TJ (2012) Is double inactivation of the Nf1 gene responsible for the development of congenital pseudarthrosis of the tibia associated with NF1? J Orthop Res 30(10):1535–1540. https://doi.org/10.1002/jor.22121
Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, Plotkin SR (2021) Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med 23(8):1506–1513. https://doi.org/10.1038/s41436-021-01170-5
Leskela HV, Kuorilehto T, Risteli J, Koivunen J, Nissinen M, Peltonen S, Peltonen J (2009) Congenital pseudarthrosis of neurofibromatosis type 1: impaired osteoblast differentiation and function and altered NF1 gene expression. Bone 44(2):243–250. https://doi.org/10.1016/j.bone.2008.10.050
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595. https://doi.org/10.1093/bioinformatics/btp698
Li Q, Wang K (2017) InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet 100(2):267–280. https://doi.org/10.1016/j.ajhg.2017.01.004
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Genome Project Data Processing, S (2009) The sequence alignment/map format and SAM tools. Bioinformatics 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Listernick R, Mancini AJ, Charrow J (2003) Segmental neurofibromatosis in childhood. Am J Med Genet A 121A(2):132–135. https://doi.org/10.1002/ajmg.a.20183
Liu J, Zheng Y, Huang J, Zhu D, Zang P, Luo Z, Lu X (2021) Expanding the genotypes and phenotypes for 19 rare diseases by exome sequencing performed in pediatric intensive care unit. Hum Mutat 42(11):1443–1460. https://doi.org/10.1002/humu.24266
Luo G, Kim J, Song K (2014) The C-terminal domains of human neurofibromin and its budding yeast homologs Ira1 and Ira2 regulate the metaphase to anaphase transition. Cell Cycle 13(17):2780–2789. https://doi.org/10.4161/15384101.2015.945870
Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Kronenberg HM (2010) Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell 19(2):329–344. https://doi.org/10.1016/j.devcel.2010.07.010
Margraf RL, VanSant-Webb C, Sant D, Carey J, Hanson H, D’Astous J, Mao R (2017) Utilization of whole-exome next-generation sequencing variant read frequency for detection of lesion-specific, somatic loss of heterozygosity in a neurofibromatosis type 1 cohort with tibial pseudarthrosis. J Mol Diagn 19(3):468–474. https://doi.org/10.1016/j.jmoldx.2017.01.008
McCaughan JA, Holloway SM, Davidson R, Lam WW (2007) Further evidence of the increased risk for malignant peripheral nerve sheath tumour from a Scottish cohort of patients with neurofibromatosis type 1. J Med Genet 44(7):463–466. https://doi.org/10.1136/jmg.2006.048140
Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Paepe AD (2000) Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat 15(6):541–555. https://doi.org/10.1002/1098-1004(200006)15:6%3c541::AID-HUMU6%3e3.0.CO;2-N
Neurofibromatosis (1988) Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 45(5):575–578
O’Donnell C, Foster J, Mooney R, Beebe C, Donaldson N, Heare T (2017) Congenital Pseudarthrosis of the Tibia. JBJS Rev 5(4):e3. https://doi.org/10.2106/JBJS.RVW.16.00068
Ozaki A, Tsunoda M, Kinoshita S, Saura R (2000) Role of fracture hematoma and periosteum during fracture healing in rats: interaction of fracture hematoma and the periosteum in the initial step of the healing process. J Orthop Sci 5(1):64–70. https://doi.org/10.1007/s007760050010
Paria N, Cho TJ, Choi IH, Kamiya N, Kayembe K, Mao R, Rios JJ (2014) Neurofibromin deficiency-associated transcriptional dysregulation suggests a novel therapy for tibial pseudoarthrosis in NF1. J Bone Miner Res 29(12):2636–2642. https://doi.org/10.1002/jbmr.2298
Parrozzani R, Clementi M, Frizziero L, Miglionico G, Perrini P, Cavarzeran F, Midena E (2015) In vivo detection of choroidal abnormalities related to NF1: feasibility and comparison with standard NIH diagnostic criteria in pediatric patients. Invest Ophthalmol vis Sci 56(10):6036–6042. https://doi.org/10.1167/iovs.14-16053
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Martin CL (2020) Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 22(2):245–257. https://doi.org/10.1038/s41436-019-0686-8
Roberts SJ, van Gastel N, Carmeliet G, Luyten FP (2015) Uncovering the periosteum for skeletal regeneration: the stem cell that lies beneath. Bone 70:10–18. https://doi.org/10.1016/j.bone.2014.08.007
Sabbagh A, Pasmant E, Imbard A, Luscan A, Soares M, Blanche H, Wolkenstein P (2013) NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: the French experience. Hum Mutat 34(11):1510–1518. https://doi.org/10.1002/humu.22392
Sakamoto A, Yoshida T, Yamamoto H, Oda Y, Tsuneyoshi M, Iwamoto Y (2007) Congenital pseudarthrosis of the tibia: analysis of the histology and the NF1 gene. J Orthop Sci 12(4):361–365. https://doi.org/10.1007/s00776-007-1142-1
Sant DW, Margraf RL, Stevenson DA, Grossmann AH, Viskochil DH, Hanson H, Mao R (2015) Evaluation of somatic mutations in tibial pseudarthrosis samples in neurofibromatosis type 1. J Med Genet 52(4):256–261. https://doi.org/10.1136/jmedgenet-2014-102815
Schindeler A, Little DG (2008) Recent insights into bone development, homeostasis, and repair in type 1 neurofibromatosis (NF1). Bone 42(4):616–622. https://doi.org/10.1016/j.bone.2007.11.006
Seminog OO, Goldacre MJ (2013) Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: population-based record-linkage study. Br J Cancer 108(1):193–198. https://doi.org/10.1038/bjc.2012.535
Stevenson DA, Zhou H, Ashrafi S, Messiaen LM, Carey JC, D’Astous JL, Viskochil DH (2006) Double inactivation of NF1 in tibial pseudarthrosis. Am J Hum Genet 79(1):143–148. https://doi.org/10.1086/504441
Tadini G, Milani D, Menni F, Pezzani L, Sabatini C, Esposito S (2014) Is it time to change the neurofibromatosis 1 diagnostic criteria? Eur J Intern Med 25(6):506–510. https://doi.org/10.1016/j.ejim.2014.04.004
Tahaei SE, Couasnay G, Ma Y, Paria N, Gu J, Lemoine BF, Elefteriou F (2018) The reduced osteogenic potential of Nf1-deficient osteoprogenitors is EGFR-independent. Bone 106:103–111. https://doi.org/10.1016/j.bone.2017.10.012
Talevich E, Shain AH, Botton T, Bastian BC (2016) CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 12(4):e1004873. https://doi.org/10.1371/journal.pcbi.1004873
Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2):178–192. https://doi.org/10.1093/bib/bbs017
Upadhyaya M, Han S, Consoli C, Majounie E, Horan M, Thomas NS, Cooper DN (2004) Characterization of the somatic mutational spectrum of the neurofibromatosis type 1 (NF1) gene in neurofibromatosis patients with benign and malignant tumors. Hum Mutat 23(2):134–146. https://doi.org/10.1002/humu.10305
Uusitalo E, Leppavirta J, Koffert A, Suominen S, Vahtera J, Vahlberg T, Peltonen S (2015) Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol 135(3):904–906. https://doi.org/10.1038/jid.2014.465
Vagge A, Nelson LB, Capris P, Traverso CE (2016) Choroidal freckling in pediatric patients affected by neurofibromatosis type 1. J Pediatr Ophthalmol Strabismus 53(5):271–274. https://doi.org/10.3928/01913913-20160719-05
Valentin T, Le Cesne A, Ray-Coquard I, Italiano A, Decanter G, Bompas E, Chevreau C (2016) Management and prognosis of malignant peripheral nerve sheath tumors: the experience of the French Sarcoma Group (GSF-GETO). Eur J Cancer 56:77–84. https://doi.org/10.1016/j.ejca.2015.12.015 (1879-0852 (Electronic))
Van Royen K, Brems H, Legius E, Lammens J, Laumen A (2016) Prevalence of neurofibromatosis type 1 in congenital pseudarthrosis of the tibia. Eur J Pediatr 175(9):1193–1198. https://doi.org/10.1007/s00431-016-2757-z
Vander Have KL, Hensinger RN, Caird M, Johnston C, Farley FA (2008) Congenital pseudarthrosis of the tibia. J Am Acad Orthop Surg 16(4):228–236
Varan A, Sen H, Aydin B, Yalcin B, Kutluk T, Akyuz C (2016) Neurofibromatosis type 1 and malignancy in childhood. Clin Genet 89(3):341–345. https://doi.org/10.1111/cge.12625
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. https://doi.org/10.1093/nar/gkq603
Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, Tamanoi F (1990a) The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 63(4):835–841. https://doi.org/10.1016/0092-8674(90)90149-9
Xu GF, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M et al (1990b) The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62(3):599–608. https://doi.org/10.1016/0092-8674(90)90024-9
Yang Y, Zheng Y, Li W, Li L, Tu M, Zhao L, Zhu Y (2019) SMAD6 is frequently mutated in nonsyndromic radioulnar synostosis. Genet Med 21(11):2577–2585. https://doi.org/10.1038/s41436-019-0552-8
Young H, Hyman S, North K (2002) Neurofibromatosis 1: clinical review and exceptions to the rules. J Child Neurol 17:613–621
Zhang J, Tong H, Fu X, Zhang Y, Liu J, Cheng R, Yao Z (2015) Molecular characterization of NF1 and neurofibromatosis type 1 genotype-phenotype correlations in a Chinese population. Sci Rep 5:2045–2322. https://doi.org/10.1038/srep11291
Zhu G, Zheng Y, Liu Y, Yan A, Hu Z, Yang Y, Mei H (2019) Identification and characterization of NF1 and non-NF1 congenital pseudarthrosis of the tibia based on germline NF1 variants: genetic and clinical analysis of 75 patients. Orphanet J Rare Dis 14(1):221. https://doi.org/10.1186/s13023-019-1196-0
Acknowledgements
We thank all the participated patients and their families for supporting our research. We also would like to express our sincere gratitude to all those who helped us in this study.
Funding
This work was supported by the Hunan Province Natural Science Foundation of China (Grant number: 2020JJ8005), the Health Commission of Hunan Province of China (Grant number: B2019020), Key Research and Development Program of Hunan Province (Grant number: 2020SK2113), and Clinical Research Center for Limb Deformity of Children in Hunan Province (Grant number: 2019SK4006).
Author information
Authors and Affiliations
Contributions
Conceptualization and investigation: ZH and HM, Funding acquisition, investigation, supervision, resources acquisition, methodology, and validation: HM, ZH, and YZ. Original draft writing and data analysis: YZ. Resources collection, idea refining, and data validation: GZ, YL, WZ, YY, ZL, and YF. Manuscript review and editing: ZH and HM. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval and consent to participate
This study was approved by the Ethics Committee of Hunan Children's Hospital (Approval No. HCHLL-2020-106). The samples were obtained appropriate informed consent from all participants.
Consent for publication
All authors read and approved the final manuscript for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zheng, Y., Zhu, G., Liu, Y. et al. Case series of congenital pseudarthrosis of the tibia unfulfilling neurofibromatosis type 1 diagnosis: 21% with somatic NF1 haploinsufficiency in the periosteum. Hum Genet 141, 1371–1383 (2022). https://doi.org/10.1007/s00439-021-02429-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02429-2