
Abstract
COCH is the most abundantly expressed gene in the cochlea. Unsurprisingly, mutations in COCH underly hearing loss in mice and humans. Two forms of hearing loss are linked to mutations in COCH, the well-established autosomal dominant nonsyndromic hearing loss, with or without vestibular dysfunction (DFNA9) via a gain-of-function/dominant-negative mechanism, and more recently autosomal recessive nonsyndromic hearing loss (DFNB110) via nonsense variants. Using a combination of targeted gene panels, exome sequencing, and functional studies, we identified four novel pathogenic variants (two nonsense variants, one missense, and one inframe deletion) in COCH as the cause of autosomal recessive hearing loss in a multi-ethnic cohort. To investigate whether the non-truncating variants exert their effect via a loss-of-function mechanism, we used minigene splicing assays. Our data showed both the missense and inframe deletion variants altered RNA splicing by creating an exon splicing silencer and abolishing an exon splicing enhancer, respectively. Both variants create frameshifts and are predicted to result in a null allele. This study confirms the involvement of loss-of-function mutations in COCH in autosomal recessive nonsyndromic hearing loss, expands the mutational landscape of DFNB110 to include coding variants that alter RNA splicing, and highlights the need to investigate the effect of coding variants on RNA splicing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The harmonious interplay between many proteins is required for normal auditory function. Mutations in genes encoding these proteins give rise to hearing loss. Each of these genes has a distinct genomic and mutational signature (Azaiez et al. 2018). For some genes, variant location and variant type correlate with specific phenotypic outcomes as a consequence of the pathogenic mechanism at play; such is the case with the COCH gene (Bae et al. 2014; Janssensdevarebeke et al. 2018; Mehregan et al. 2019). Cochlin, the protein product of the COCH gene, is a large secreted extracellular protein that contains an N-terminus LCCL (Limulus factor C, Cochlin, and Late gestation lung protein Lgl1) domain and two von Willebrand factor A-like domains (vWFA1 and vWFA2). It is the most abundantly detected protein in the inner ear (Robertson et al. 1994, 1997) and plays a well-established role in post-lingual progressive autosomal dominant nonsyndromic hearing loss (ADNSHL), sometimes accompanied by vestibular dysfunction, at the DFNA9 locus (Robertson et al. 1997; Kemperman et al. 2002; Bischoff et al. 2005; Bae et al. 2014) (Fig. 1e). DFNA9 hearing loss occurs via multiple mechanisms depending on mutation location (Kemperman et al. 2002; Hildebrand et al. 2010; Bae et al. 2014; Tsukada et al. 2015). Mutations in the LCCL domain typically cause misfolding and defective multimerization of cochlin (Bae et al. 2014). Patients with these mutations tend to have later onset hearing loss that is accompanied by vestibular dysfunction (Bae et al. 2014). In contrast, mutations in the vWFA domains typically result in secretion failure and aggregate formation (Bae et al. 2014). Patients with these mutations report earlier onset hearing loss without vestibular dysfunction (Bae et al. 2014). While the toxic gain-of-function/dominant-negative effects of mutant cochlin on the auditory system and the underlying pathology that results in hearing loss have been well studied (Bae et al. 2014), little is known about the consequence and underlying pathology from loss of cochlin.

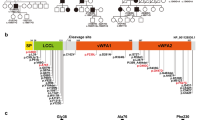
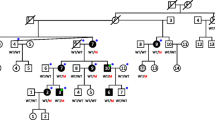
Pedigrees, audiograms, and COCH gene and protein schematic. a Segregation of p.Glu211Ter and audiograms for family PKMR266. Audiograms were obtained with air conduction with the frequency range of 250–8000 Hz. b–d Genotypes of Probands a, b, and c and their available family members. e Human COCH (NM_004086.2) has 12 exons, of which the first exon is not coding (grey). Variants identified in this study denoted in bold. The COCH protein encodes a 550 amino acid peptide consisting of a signal peptide (blue), an LCCL domain (green), and two VWFA domains (pink). Novel variants identified in this study (top) and previously reported DFNB110 variants (bottom). Square brackets represent the predicted effect of the splicing variants. f Three-dimensional modeling showing an overview of COCH protein; right panel: purple; wild-type COCH and aquamarine; mutated residues, left panel: green; targeted amino acid and golden; interacting amino acids. Upper boxes (p.Arg91Gly); on left wild-type p.Arg91 and on right mutated p.Gly91 along with interacting amino acids. Lower boxes (p.Val191Arg); on left wild-type p.Val191 and on right mutated p.Arg191 along with interacting amino acids. Yellow dotted lines represent hydrogen bonds between amino acids
Recently, two families with autosomal recessive nonsyndromic hearing loss (ARNSHL) at the DFNB110 locus have been described segregating nonsense variants in COCH (Janssensdevarebeke et al. 2018; Mehregan et al. 2019), suggesting that expression of COCH is required for proper auditory function. Here, we expand on these findings to report four novel mutations associated with DFNB110 hearing impairment, and demonstrate the involvement of missense and inframe variants in mRNA splicing. Our findings further support loss-of-function as a second pathogenic mechanism associated with COCH-related hearing loss in humans.
Methods
Subjects
One Pakistani (PKMR266) family, and three probands of European (Proband A), Middle-Eastern (Proband B), and unknown ethnicity (Proband C) segregating ARNSHL were ascertained for this study. Affected individuals underwent clinical examination and pure tone audiometry to measure hearing thresholds at 0.25, 0.5, 1, 2, 3, 4, and 8 kHz. For family PKMR266, audiometry was performed in a noise free quiet room at the audiologist office, a sound proof chamber was not available near the family ascertainment area in Pakistan. After obtaining written informed consent to participate in this study, blood samples were obtained from all affected and unaffected family members and genomic DNA was extracted. The human research Institutional Review Boards approved all procedures at the University of Iowa, Iowa City, Iowa, USA, University of Maryland, School of Medicine, Baltimore, USA, and Shaheed Zulfiqar Ali Bhutto Medical University, Islamabad, Pakistan.
Next-generation sequencing and bioinformatic analysis
Probands A–C underwent comprehensive genetic testing to screen all known genes implicated in NSHL, common NSHL mimics, and common syndromic forms of hearing loss using the OtoSCOPE® panel as described (Booth et al. 2015, 2018b, c, d). Similarly, a proband from family PKMR266 underwent genetic testing as described (Zein et al. 2015; Richard et al. 2019). Bioinformatic and variant analysis were also completed as described (Booth et al. 2018b, d; Richard et al. 2019) using the GenomeAnalysisToolkit following the Broad best practices pipeline. In brief, after mapping of raw sequence reads and variant calling, variants were annotated and filtered based on quality (depth > 5 and call quality > 30), minor allele frequency (MAF) < 2% in Genome Aggregation Database (gnomAD) (Karczewski et al. 2020), and variant effect (missense, nonsense, indel, or splice-site). Retained variants were prioritized based on their conservation (GERP and PhyloP) and predicted deleteriousness [SIFT, PolyPhen2, MutationTaster, LRT, and the Combined Annotation Dependent Depletion (CADD)] (Liu et al. 2016; Rentzsch et al. 2019). For SIFT, PolyPhen2, MutationTaster, and LRT recommended thresholds for and against a deleterious effect were based on their published guidelines (Liu et al. 2011, 2016). Scores ≥ 0.95 and > 0 were considered conserved for PhyloP and GERP, respectively. Variant effect on splicing was assessed using Human Splicing Finder (HSF) (Desmet et al. 2009). HSF predicts the impact of variants on canonical splice sites and on auxiliary splicing sequences like exonic splicing enhancers (ESEs) and exonic splicing silencers (ESSs). The algorithm takes into account the global impact on both signals ESE/ESS and only variants significantly disturbing the balance between positive and negative signals are considered. Samples that were tested on OtoSCOPE also underwent copy number variant (CNV) analysis using a sliding window and read depth ratio method as described (Nord et al. 2011). Segregation analysis was carried out on available family members using Sanger sequencing.
In vitro splicing analysis
In vitro splicing assays were performed as described (Booth et al. 2018a, b, d). COCH (NM_004086) gene-specific primers were used to amplify exons 5, 7, 8, and 11 using patient genomic DNA and control DNA to obtain the mutant and wildtype allele, respectively (Supplementary Table 1). The amplicons were then ligated into the pET01 Exontrap vector (MoBiTec, Goettingen, Germany). Colonies were selected and grown, and plasmid DNA was harvested using the ZymoPure Plasmid Midiprep Kit (ZYMO Research, Irvine, California, USA). After sequence confirmation, wildtype and mutant minigenes were transfected in triplicate into COS7, HEK293, and MDCK cells, and total RNA was extracted 36 h post-transfection using the Quick-RNA MiniPrep Plus kit (ZYMO Research). Using a primer specific to the 3′ native exon of the pET01 vector, cDNA was synthesized using AMV Reverse Transcriptase (New England BioLabs). After PCR amplification, products were visualized on a 1.5% agarose gel, extracted, and then sequenced.
Molecular modeling and thermodynamic predictions
COCH protein amino acid sequence (Uniprot ID: O43405-1) was used to obtain protein predictive models from I-Tasser (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). Among the proposed structures, the model with the highest confidence score, i.e., − 2.32, was selected. This score is based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations assessed by the online tool. PyMOL Molecular Graphic System 2.3.4 was then used to analyze the effect of mutagenesis on COCH protein model in terms of amino acid size and interaction. Thermodynamic predictions were calculated using STRUM (Quan et al. 2016) server (https://zhanglab.ccmb.med.umich.edu/STRUM/).
Results
Subjects and variant identification
We ascertained a consanguineous Pakistani family and three probands of European (Proband A), Middle-Eastern (Proband B), and unknown ancestry (Proband C) with ARSNHL (Fig. 1a–d). Hearing loss in family PKMR266 is prelingual moderate-to-profound (Fig. 1a). Audiograms could not be obtained for probands A–C; however, clinical description and history were provided. Proband A is an 11-year-old male with congenital and moderate hearing loss. Proband B is a 3-year-old male with congenital downsloping mild-to-severe hearing loss. Finally, Proband C is a 15-year-old male with prelingual downsloping mild hearing loss. Families for probands B and C also reported consanguinity.
A combination of OtoSCOPE® panel and NGS was used to identify underlying genetic cause of hearing loss segregating in these affected individuals. Genetic variant filtering for MAF, quality, effect, and recessive or X-linked inheritance yielded candidate variants in the COCH gene for all probands. In Family PKMR266, a homozygous nonsense variant [c.631G > T; p.(Glu211Ter)] in exon 9 was identified, which segregated with the hearing loss phenotype in five affected individuals spanning multiple generations. Probands A–C underwent OtoSCOPE testing. In Proband A, we identified two variants in COCH: a nonsense variant (c.439A > T; p.Lys147Ter) and a dinucleotide change (c.571_572delinsAG; p.Val191Arg), in exons 7 and 8, respectively (Fig. 1e). These variants were confirmed to be in trans via visualization on Integrative Genomics Viewer (IGV: https://igv.org) and cloning. Proband B carries a homozygous missense variant (c.271C > G; p.Arg91Gly) in exon 5, and in Proband C, a homozygous inframe deletion (c.1093_1101del; p.Ser365_Asn367del) in exon 11 was identified. All identified variants are novel (absent from gnomAD) or ultra-rare (Table 1) and occur in conserved residues (Table 1). Variants p.Arg91Gly and p.Val191Arg are predicted to be deleterious by computational tools (Table 1). No CNVs were detected in patients screened by NGS. We also identified two variants in LOXHD1 [c.553G > T;(p.Gly185Cys)] and [c.4871G > C; (p.Gly1624Ala)] in Proband B (Supplementary Table 3). Both variants are rare and have conflicting scores as to conservation and predicted deleteriousness. No other candidate variants in either recessive or dominant genes were identified (Supplementary Tables 2–4).
Computational and in vitro splicing analysis
Since the previously described DFNB110-causing mutations were all truncating loss-of-function variants, we sought to explore the effects of the missense and inframe variants found in Probands A–C on RNA splicing. Variants c.271C > G, c.571_572delinsAG, and c.1093_1101del, were computationally predicted to affect splicing motifs and impact splicing by various mechanisms. The c.271C > G variant is predicted by HSF to alter an exonic splicing enhancer (ESE), create an exonic splicing silencer (ESS), and create a cryptic donor site. RT-PCR of cells transfected with either wild-type or mutant minigenes revealed no alterations in splicing between the wild type and mutant (Supplementary Fig. 1).
The loss of nucleotides c.1093–1101 in exon 11 is predicted to break the binding motifs for the serine/arginine-rich splicing factors 1 and 2 (SRSF1 and SRSF2). Visualization and sequencing of splicing products from wild-type and mutant minigenes revealed the inclusion of exon 11 in the wild type but not in the mutant (Fig. 2a). Loss of exon 11 creates a shift in the reading frame and result in a truncated protein product (p.Ala321Glufs*17).

Minigene splicing assays. Gel electrophoresis and sequence chromatograms of wildtype, empty pET01 vector, and the c.1093_1101del (a) and c.571_572delinsAG (b). a The c.1093_1101del variant abolishes an exonic splicing enhancer in exon 11, resulting in complete exon skipping confirmed by Sanger sequencing. b The dinucleotide c.571_572delinsAG variant produced two bands. The top band (#3) corresponds to a normally spliced product and migrates in parallel with the wildtype and the c.439A > T variant. The smaller band (#4) corresponds to the aberrant splicing of exon 8. Sequence chromatograms show the read through at each exon junction, and sequence alignment shows the deletion of exon 8 in-band #4 and the dinucleotide change in band #3. Gel images correspond to the experimental results in HEK293 cells
Finally, the c.571_572delinsAG alteration is predicted to activate two cryptic acceptor sites and create an ESS. Visualization of the spliced products for the mutant minigene carrying the c.571_572delinsAG revealed two bands at ~ 440 bp and ~ 300 bp (Fig. 2b). Sequencing showed the band corresponding to ~ 440 bp contained both exon 7 and 8, whereas the smaller band (~ 300 bp) revealed skipping of exon 8 (Fig. 2b). The loss of exon 8 in the mRNA shifts the reading frame, resulting in a premature stop codon (p.Asp161Valfs*4). Since proband A DNA was used to create the minigene, we used the in trans nonsense c.439A > T variant in exon 7 as control for splicing efficiency. Minigenes carrying the c.439A > T variant revealed a single band at ~ 440 bp, identical to the wildtype; subsequent sequencing revealed the presence of the c.439A > T variant but no alterations to wildtype splicing (Fig. 2b).
For each assay, no differences were seen between the triplicate experiments or between the different cell lines. It is noteworthy that there were no obvious differences in the ratios between wildtype and mutant bands either amongst the three cell lines or the triplicate experiments as visualized on an agarose gel.
3D protein modeling
The p.Arg91 residue is located in the LCCL domain (Fig. 1e). Three-dimensional modeling results for p.Arg91Gly showed arginine, a positively charged amino acid at position 91 of COCH protein interacting with three other amino acids through hydrogen bonding. Arg91 forms one hydrogen bond with Leu49, two with Gly126, and three with Asp47 in COCH wild-type protein. Mutagenesis at position 91 to glycine, a neutral amino acid with comparatively small size, showed loss of five hydrogen-bonding interaction points, two with Gly126, and three with Asp47 (Fig. 1f). As the native amino acid resides in the LCCL domain, this change is predicted to cause misfolding of the protein due to smaller size of glycine along with loss of hydrogen interactions. Thermodynamic simulation of the change in folding energy between the wildtype and mutant (Quan et al. 2016) revealed a difference of, − 2.77, strongly suggesting that the p.Arg91Gly has an impact on protein stability.
Molecular visualization of valine at position 191 showed interaction with Phe187, Val188, Met194, and Leu195 through hydrogen bonding in wild-type protein. Change of valine to arginine at position 191 showed effect on interactions only for Val188. The distance for the hydrogen bond between Val191 and Val188 is 2.6 Å. This distance increased to 2.7 Å for p.Val191Arg (Fig. 1f). As the variant occurs in one of the protein helix, it could affect folding pattern, thus, indirectly affecting interactions with other molecules.
Discussion
We used a combination of targeted genomic enrichment and massively parallel sequencing to implicate COCH as the causal gene in one large and three small families segregating ARNSHL. Among the identified COCH pathogenic variants, two are nonsense, one a dinucleotide change, one a missense variant, and one is an inframe deletion. Of these variants, three are novel (not reported in the Genome Aggregation Database, gnomAD; Table 1). To date, only two nonsense variants have been linked to COCH-related ARNSHL (Fig. 1e, Table 1).
In humans, COCH is the most abundantly expressed gene in the cochlea (Robertson et al. 1994, 1998). The 12-exon gene encodes the 550 amino acid Cochlin protein, consisting of a signal peptide, an LCCL domain, and two vWFA domains (Fig. 1e). Unlike most genes in the inner ear linked to hearing impairment, COCH is highly expressed in the fibrocytes of the cochlea and not in the mechanosensitive hair cells (Robertson 2001; Jones et al. 2011; Robertson et al. 2014). After translation, Cochlin is secreted from the fibrocytes. While its exact function is unknown, it has been suggested that Cochlin might play an essential structural role in organizing and stabilizing the extracellular matrix via its vWFA domains. More recently, it has been shown that the LCCL domain is critical for the immune response in the inner ear (Jung et al. 2019). Its role in hearing loss implicates Cochlin as a protein required for proper auditory function in humans.
The ultra-rare nonsense variant c.631G > T [p.(Glu211Ter)] segregating with the moderate-to-severe hearing loss in family PKMR266 (Fig. 1a) occurs in exon 9 (Fig. 1e). Given that the created termination codon is over 50 base pairs away from the last exon-exon junction, we predict that nonsense-mediated decay (NMD) will occur (Hentze and Kulozik 1999; He and Jacobson 2015), leading to a null allele. If the allele carrying this variant was to escape NMD, the translated truncated protein product would be missing ~ 50% of the protein (Fig. 1e), rendering the protein nonfunctional. The unaffected mother (IV:4) is heterozygous for the p.(Glu211Ter) variant and has normal hearing at the age of 35. These findings show that haploinsufficiency is not a pathogenic mechanism for COCH-related hearing loss (Makishima et al. 2005; Janssensdevarebeke et al. 2018; Mehregan et al. 2019).
Novel compound heterozygous variants [(c.439A > T; p.Lys147Ter) and (c.571_572delinsAG; p.Val191Arg)] were identified in Proband A. Similar to the p.Glu211Ter variant identified in family PKMR266, p.Lys147Ter variant is predicted to produce a null allele via the NMD pathway (Hentze and Kulozik 1999; He and Jacobson 2015). The dinucleotide change c.571_572delinsAG replaces a GT with an AG and results in a single amino acid substitution valine to arginine at the highly conserved residue 191 (Fig. 1e and Supplementary Fig. 2). Molecular modeling for this variant revealed that residue 191 is buried in the core of the vWFA1 domain (Fig. 1f). The change from the neutral valine to the positively charged and bulkier arginine could disable the protein and alter folding as shown by the negative change in free energy (Table 1) predicting the mutant is less stable than the wild type. We also assessed the effect of the c.571_572delinsAG alteration on RNA splicing using a minigene that contained exon 7 and 8 and their flanking introns. We found the c.571_572delinsAG substitution which resulted in aberrant splicing of exon 8 (Fig. 2b). The loss of exon 8 in the native COCH mRNA causes a shift in the reading frame, resulting in a premature stop codon (p.Asp161Valfs*4). This mutant mRNA is expected to undergo NMD and be a null allele (Hentze and Kulozik 1999; He and Jacobson 2015). Interestingly, the spliced products of the constructs carrying the c.571_572delinsAG variant also produced a correctly spliced transcript that is identical to the control minigene (Fig. 2b), in all three cell lines tested. These results could be due to the specific cell types used that would differ from the mechanism happening in vivo where the c.571_572delinsAG alteration would cause 100% exon skipping. The mis-splicing caused by c.571_572delinsAG in combination with p.Lys147Ter could be sufficient to reduce COCH expression to levels incompatible with normal hearing function. Alternatively, c.571_572delinsAG could exert its effect via two mechanisms: aberrant splicing of exon 8, and protein misfolding.
In Proband B, we identified an ultra-rare, conserved, and predicted deleterious homozygous transversion variant c.271C > G (p.Arg91Gly). While segregation analysis could not be done, we were able to rule out the possibility of hemizygosity by CNV analysis. Although in silico prediction via HSF suggested this variant might alter RNA splicing, we did not detect any differences in splicing between the wild-type and mutant minigenes (Supplementary Fig. 1). The p.Arg91 residue is located in the LCCL domain (Fig. 1e) and the significant change in folding energy strongly suggests this variant destabilizes the protein (Quan et al. 2016). Molecular modeling revealed that the p.Arg91 interacts with p.Asp47 and p.Gly126. The substitution of a glycine abolishes this interaction (Fig. 1f). Besides the difference in charge between arginine and glycine, glycines are notoriously very flexible and can disturb the required rigidity of protein domains. This increase in flexibility fits the significant change in folding energy. Two variants in LOXHD1 (p.Gly185Cys and p.Gly1624Ala) were also identified in Proband B. Pathogenic variants in LOXHD1 are responsible for hearing loss at the DFNB77 locus (Grillet et al. 2009). DFNB77-related hearing loss shows variability in onset, severity, and progression (Mori et al. 2015; Maekawa et al. 2019; Bai et al. 2020), including prelingual downsloping mild hearing loss. Based on phenotype alone, we cannot rule out the involvement of LOXHD1 in Proband B’s hearing loss. It was not possible to perform segregation analysis to determine if these variants were in cis or trans. Given the reported consanguinity and conflicting in silico predictions for the LOXHD1 variants, the COCH variant represents the more likely cause of hearing loss in this proband; however, because disease-causing status remains uncertain, we classified the COCH c.271C > G transversion as variant of uncertain significance.
Finally, in Proband C, we detected a homozygous 9 nucleotide inframe deletion in exon 11. It was not possible to perform segregation analysis, but we were able to rule out the possibility of hemizygosity by CNV analysis. The deletion is predicted to remove the first amino acid from the vWFA2 domain and the two proceeding amino acids. We tested the effects of the 9-nucleotide deletion on splicing using minigenes containing wild type and mutant exon 11. Spliced products showed the loss of these nine nucleotides results in a transcript lacking exon 11 (Fig. 2a). In the native mRNA, the loss of exon 11 shifts the reading frame leading to a truncated protein product (p.Ala321Glufs*17). We do not expect this allele to undergo the NMD pathway, given the newly created termination codon occurs in the last exon upstream of the native TAA. We do predict the translated protein to be nonfunctional, given that the vWFA2 domain is missing along with ~ 1/3 of the protein.
The auditory phenotype described in patients in this study is similar to the previously described hearing loss in DFNB110 patients, congenital downsloping mild/moderate-to-severe hearing loss. One subject homozygous for the p.Arg98Ter variant had vestibular dysfunction (Janssensdevarebeke et al. 2018). While not directly tested for, none of the patients in this study reported balance problems or vertigo, similar to reported DFNB110 cases. Follow-up studies are needed to see if patients with bi-allelic inactivating alleles of COCH also develop vestibular defects. Interestingly, the human phenotype is in sharp contrast to the auditory phenotype seen in the Coch knock-out mouse (Jones et al. 2011). In the mouse, loss of Coch results in very mild and slightly progressive hearing impairment that is detectable after 1 year of age in the high frequencies (Jones et al. 2011). These mice also exhibit elevated vestibular evoked potentials (VsEP) after 1 year of age, suggesting a potential vestibular defect, without any apparent menifestation of circling or twirling behavior, the hallmarks of vestibular impairment.
Our data highlight the need to exercise caution when classifying genetic variants in COCH as different pathologic mechanisms are involved, gain-of-function/dominant-negative for ADNSHL, and loss-of-function for ARNSHL (Verhoeven et al. 1998; Mustapha et al. 1999; Rehman et al. 2014; Azaiez et al. 2014; Vona et al. 2015). First, variants that result in a null allele should be considered likely pathogenic for DFNB110 and not DFNA9. Second, rare missense variants in COCH identified in patients with DFNB110 phenotype should be thoroughly investigated for their effect on RNA splicing. If functional studies validate their deleterious effect on splicing, these variants should be considered likely pathogenic. The primary emphasis on coding variant interpretation focuses on the impact at the protein level. Often, if the variant falls outside the traditional splicing window, its impact on splicing is rarely considered. However, several studies have illustrated that coding variant's effect on splicing is underappreciated (Collin et al. 2008; Aparisi et al. 2013; Booth et al. 2018b, d).
In summary, we have expanded the mutational spectrum of COCH-related hearing loss to include coding splice-altering variants. Importantly, our data further emphasize the importance of comprehensive variant interpretation irrespective of the conceived predicted translational impact as coding variants could also have a damaging impact on RNA splicing.
References
Aparisi MJ, García-García G, Aller E et al (2013) Study of USH1 splicing variants through minigenes and transcript analysis from nasal epithelial cells. PLoS One 8:e57506. https://doi.org/10.1371/journal.pone.0057506
Azaiez H, Booth KT, Bu F et al (2014) TBC1D24 mutation causes autosomal-dominant nonsyndromic hearing loss. Hum Mutat 35:819–823. https://doi.org/10.1002/humu.22557
Azaiez H, Booth KT, Ephraim SS et al (2018) Genomic landscape and mutational signatures of deafness-associated genes. Am J Hum Genet 103:484–497. https://doi.org/10.1016/j.ajhg.2018.08.006
Bae SH, Robertson NG, Cho HJ et al (2014) Identification of pathogenic mechanisms of COCH mutations, abolished cochlin secretion, and intracellular aggregate formation: genotype-phenotype correlations in DFNA9 deafness and vestibular disorder. Hum Mutat 35:1506–1513. https://doi.org/10.1002/humu.22701
Bai X, Zhang C, Zhang F et al (2020) Five novel mutations in LOXHD1 gene were identified to cause autosomal recessive nonsyndromic hearing loss in four chinese families. Biomed Res Int. https://doi.org/10.1155/2020/1685974
Bischoff AMLC, Huygen PLM, Kemperman MH et al (2005) Vestibular deterioration precedes hearing deterioration in the P51S COCH mutation (DFNA9): an analysis in 74 mutation carriers. Otol Neurotol 26:918–925. https://doi.org/10.1097/01.mao.0000185048.84641.e3
Booth KT, Azaiez H, Kahrizi K et al (2015) PDZD7 and hearing loss: more than just a modifier. Am J Med Genet Part A 167:2957–2965. https://doi.org/10.1002/ajmg.a.37274
Booth KT, Askew JW, Talebizadeh Z et al (2018a) Splice-altering variant in COL11A1 as a cause of nonsyndromic hearing loss DFNA37. Genet Med 21(4):948–954. https://doi.org/10.1038/s41436-018-0285-0
Booth KT, Azaiez H, Kahrizi K et al (2018b) Exonic mutations and exon skipping: lessons learned from DFNA5. Hum Mutat 39:433–440. https://doi.org/10.1002/humu.23384
Booth KT, Kahrizi K, Babanejad M et al (2018c) Variants in CIB2 cause DFNB48 and not USH1J. Clin Genet 93:812–821. https://doi.org/10.1111/cge.13170
Booth KT, Kahrizi K, Najmabadi H et al (2018d) Old gene, new phenotype: splice-altering variants in CEACAM16 cause recessive non-syndromic hearing impairment. J Med Genet 55:555–560. https://doi.org/10.1136/jmedgenet-2018-105349
Collin RWJ, de Heer A-MR, Oostrik J et al (2008) Mid-frequency DFNA8/12 hearing loss caused by a synonymous TECTA mutation that affects an exonic splice enhancer. Eur J Hum Genet 16:1430–1436. https://doi.org/10.1038/ejhg.2008.110
Desmet FO, Hamroun D, Lalande M et al (2009) Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37:1–14. https://doi.org/10.1093/nar/gkp215
Grillet N, Schwander M, Hildebrand MS et al (2009) Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am J Hum Genet 85:328–337. https://doi.org/10.1016/j.ajhg.2009.07.017
He F, Jacobson A (2015) Nonsense-mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu Rev Genet 49:339–366. https://doi.org/10.1146/annurev-genet-112414-054639
Hentze MW, Kulozik AE (1999) A perfect message: RNA surveillance and nonsense-mediated decay. Cell 96:307–310. https://doi.org/10.1016/S0092-8674(00)80542-5
Hildebrand MS, Gandolfo L, Shearer AE et al (2010) A novel mutation in COCH-implications for genotype–phenotype correlations in DFNA9 hearing loss. Laryngoscope 120:2489–2493. https://doi.org/10.1002/lary.21159
Janssensdevarebeke SPF, Van Camp G, Peeters N et al (2018) Bi-allelic inactivating variants in the COCH gene cause autosomal recessive prelingual hearing impairment. Eur J Hum Genet 26:587–591. https://doi.org/10.1038/s41431-017-0066-2
Jones SM, Robertson NG, Given S et al (2011) Hearing and vestibular deficits in the Coch−/− null mouse model: comparison to the CochG88E/G88E mouse and to DFNA9 hearing and balance disorder. Hear Res 272:42–48. https://doi.org/10.1016/j.heares.2010.11.002
Jung J, Yoo JE, Choe YH et al (2019) Cleaved cochlin sequesters pseudomonas aeruginosa and activates innate immunity in the inner ear. Cell Host Microbe 25:513–525.e6. https://doi.org/10.1016/j.chom.2019.02.001
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Neale BM, Daly MJ, MacArthur DG (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581(7809):434–443
Kemperman MH, Bom SJH, Lemaire FX et al (2002) DFNA9/COCH and its phenotype. Adv Otorhinolaryngol 61:66–72. https://doi.org/10.1159/000066806
Liu X, Jian X, Boerwinkle E (2011) dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat 32:894–899. https://doi.org/10.1002/humu.21517
Liu X, Wu C, Li C, Boerwinkle E (2016) dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-Site SNVs. Hum Mutat 37:235–241. https://doi.org/10.1002/humu.22932
Maekawa K, Nishio SY, Abe S et al (2019) Mutational spectrum and clinical features of patients with LOXHD1 variants identified in an 8074 hearing loss patient cohort. Genes (Basel). https://doi.org/10.3390/genes10100735
Makishima T, Rodriguez CI, Robertson NG et al (2005) Targeted disruption of mouse Coch provides functional evidence that DFNA9 hearing loss is not a COCH haploinsufficiency disorder. Hum Genet 118:29–34. https://doi.org/10.1007/s00439-005-0001-4
Mehregan H, Mohseni M, Akbari M et al (2019) Novel Mutations in KCNQ4, LHFPL5 and COCH Genes in Iranian families with hearing impairment. Arch Iran Med 22:189–197
Mori K, Moteki H, Kobayashi Y et al (2015) Mutations in LOXHD1 gene cause various types and severities of hearing loss. Ann Otol Rhinol Laryngol 124:135S–141S. https://doi.org/10.1177/0003489415574067
Mustapha M, Weil D, Chardenoux S et al (1999) An alpha-tectorin gene defect causes a newly identified autosomal recessive form of sensorineural pre-lingual non-syndromic deafness, DFNB21. Hum Mol Genet 8:409–412
Nord AS, Lee M, King M-C, Walsh T (2011) Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genom 12:184. https://doi.org/10.1186/1471-2164-12-184
Quan L, Lv Q, Zhang Y (2016) STRUM: structure-based prediction of protein stability changes upon single-point mutation. Bioinformatics 32:2936–2946. https://doi.org/10.1093/bioinformatics/btw361
Rehman AU, Santos-Cortez RLP, Morell RJ et al (2014) Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am J Hum Genet 94:144–152. https://doi.org/10.1016/j.ajhg.2013.12.004
Rentzsch P, Witten D, Cooper GM et al (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47:D886–D894. https://doi.org/10.1093/nar/gky1016
Richard EM, Santos-Cortez RLP, Faridi R et al (2019) Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum Mutat 40:53–72. https://doi.org/10.1002/humu.23666
Robertson NG (2001) Inner ear localization of mRNA and protein products of COCH, mutated in the sensorineural deafness and vestibular disorder, DFNA9. Hum Mol Genet 10:2493–2500. https://doi.org/10.1093/hmg/10.22.2493
Robertson NG, Khetarpal U, Gutiérrez-Espeleta GA et al (1994) Isolation of novel and known genes from a human fetal cochlear cDNA library using subtractive hybridization and differential screening. Genomics 23:42–50. https://doi.org/10.1006/geno.1994.1457
Robertson NG, Skvorak AB, Yin Y et al (1997) Mapping and characterization of a novel cochlear gene in human and in mouse: a positional candidate gene for a deafness disorder, DFNA9. Genomics 46:345–354. https://doi.org/10.1006/geno.1997.5067
Robertson NG, Lu L, Heller S et al (1998) Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat Genet 20:299–303. https://doi.org/10.1038/3118
Robertson NG, O’Malley JT, Ong CA et al (2014) Cochlin in normal middle ear and abnormal middle ear deposits in DFNA9 and Coch (G88E/G88E) mice. J Assoc Res Otolaryngol 15:961–974. https://doi.org/10.1007/s10162-014-0481-9
Tsukada K, Ichinose A, Miyagawa M et al (2015) Detailed hearing and vestibular profiles in the patients with COCH mutations. Ann Otol Rhinol Laryngol 124:100S–110S. https://doi.org/10.1177/0003489415573074
Verhoeven K, Van Laer L, Kirschhofer K et al (1998) Mutations in the human alpha-tectorin gene cause autosomal dominant non-syndromic hearing impairment. Nat Genet 19:60–62. https://doi.org/10.1038/ng0598-60
Vona B, Nanda I, Hofrichter MAH et al (2015) Non-syndromic hearing loss gene identification: a brief history and glimpse into the future. Mol Cell Probes 29:260–270. https://doi.org/10.1016/j.mcp.2015.03.008
Zein WM, Falsini B, Tsilou ET et al (2015) Cone responses in usher syndrome types 1 and 2 by microvolt electroretinography. Investig Ophthalmol Vis Sci 56:107–114. https://doi.org/10.1167/iovs.14-15355
Acknowledgements
We would like to thank the patients for their participation in this study. This study was funded by NIDCDs R01s DC002842, DC012049, and DC017955 to RJS and R56DC011803 to SR.
Author information
Authors and Affiliations
Contributions
KTB, AG, MH, ZMA, RJS, HA, and SR: conception and study design; KTB, MR, LTH, MH, KF, EMR, CJN, and MS performed experiments; KTB, AG, ZMA, RJS, HA, and SR analyzed data and drafted the initial manuscript. All authors have reviewed and approved the finalized manuscript.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2020_2197_MOESM2_ESM.tif
Supplementary Figure 1. Minigene splicing assays. Gel electrophoresis and sequence chromatograms of wildtype, empty pET01 vector, and the c.271C>G mutation. Visualization and Sanger sequencing showed no difference in splicing between the wildtype and mutant (TIF 9982 kb)
439_2020_2197_MOESM3_ESM.tif
Supplementary Figure 2. Amino acid conservation analysis among different vertebral species through Clustal omega (TIF 19105 kb)
Rights and permissions
About this article

Cite this article
Booth, K.T., Ghaffar, A., Rashid, M. et al. Novel loss-of-function mutations in COCH cause autosomal recessive nonsyndromic hearing loss. Hum Genet 139, 1565–1574 (2020). https://doi.org/10.1007/s00439-020-02197-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02197-5