
Abstract
DFNA9 sensorineural hearing loss and vestibular disorder, caused by mutations in COCH, has a unique identifying histopathology including prominent acellular deposits in cochlear and vestibular labyrinths. A recent study has shown presence of deposits also in middle ear structures of DFNA9-affected individuals (McCall et al., J Assoc Res Otolaryngol 12:141–149, 2004). To investigate the possible role of cochlin in the middle ear and in relation to aggregate formation, we evaluated middle ear histopathology in our Coch knock-in (Coch G88E/G88E) mouse model, which harbors one of the DFNA9-causative mutations. Our findings reveal accumulation of acellular deposits in the incudomalleal and incudostapedial joints in Coch G88E/G88E mice, similar to those found in human DFNA9-affected temporal bones. Aggregates are absent in negative control Coch +/+ and Coch −/− mice. Thickening of the tympanic membrane (TM) found in humans with DFNA9 was not appreciably detected in Coch G88E/G88E mice at the evaluated age. We investigated cochlin localization first in the Coch +/+mouse and in normal human middle ears, and found prominent and specific cochlin staining in the incudomalleal joint, incudostapedial joint, and the pars tensa of the TM, which are the three sites where abnormal deposits are detected in DFNA9-affected middle ears. Cochlin immunostaining of Coch G88E/G88E and DFNA9-affected middle ears showed mutant cochlin localization within areas of aggregates. Cochlin staining was heterogeneous throughout DFNA9 middle ear deposits, which appear as unorganized and overlapping mixtures of both eosinophilic and basophilic substances. Immunostaining for type II collagen colocalized with cochlin in pars tensa of the tympanic membrane. In contrast, immunostaining for type II collagen did not overlap with cochlin in interossicular joints, where type II collagen was localized in the region of the chondrocytes, but not in the thin layer of the articular surface of the ossicles nor in the eosinophilic deposits with specific cochlin staining.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
COCH (coagulation factor C homology) was initially identified and shown to be expressed at high levels in the cochlear and vestibular organs by Northern blot and tissue in situ hybridization (Robertson et al. 1994; Robertson et al. 1998). The encoded secreted protein, cochlin, is the most abundant protein detected by proteomic analyses in bovine, murine, and human cochlear and vestibular labyrinths (Ikezono et al. 2001; Robertson et al. 2006). Mutations in COCH are etiologic for mid-life onset, progressive sensorineural hearing loss, and vestibular dysfunction, DFNA9. To date, 19 missense and 2 in-frame deletion COCH mutations causing hearing loss (Fig. 1) have been reported throughout four continents. The incidence of COCH mutations is unknown, as there is currently no systematic genetic screening of COCH; rather, mutation discovery in the majority of cases has been through research studies of multigenerational pedigrees. However, reports of mutations in populations ranging from Far East Asian to Western European descent, including four distinct COCH mutations in the Netherlands alone, are suggestive of a much higher prevalence of COCH mutations than currently recognized. A query of public databases including dbSNP/1000 Genomes Project, Deafness Variation Database/OtoSCOPE, Exome Sequencing Project, and HGMD was performed for ascertainment of COCH variants, revealing 42 non-synonymous, 5 frame-shift, and 32 synonymous variants in addition to the known pathogenic mutations for a total of 99 variants within the COCH protein-coding region, as well as 11 intronic variants ±15 nucleotides from splice junctions. Future studies may elucidate the clinical significance of these variants.

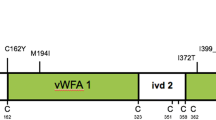
Cochlin, encoded by COCH, is a secreted protein with a signal peptide (SP) followed by a domain initially designated as FCH (factor C-homology), also known as the LCCL, (Limulus factor C, cochlin, lung gestational protein) domain, followed by an intervening domain (ivd1), and two von Willebrand factor A-like domains (vWFA1 and vWFA2) separated by an intervening domain (ivd2). To date, 19 missense mutations and two in-frame deletions causing hearing loss have been reported. Asterisk denotes the Coch G88E mutation that was replicated in our knock-in mouse model by targeted homologous recombination. The positions of all cysteine residues are shown as “C”.
Characteristic histopathological findings that are pathognomonic for DFNA9 are accumulation of cochlin-staining acellular deposits and marked loss of cellularity in the spiral ligament, limbus, and stroma underlying vestibular sensory epithelia, which are also sites of normal cochlin production (Khetarpal et al. 1991; Robertson et al. 1998). Further examination of DFNA9-affected temporal bones has revealed presence of acellular deposits also in structures within the middle ear cavity (McCall et al. 2011) (Figs. 5C, 6C, and 7C). This observation prompted us to investigate more thoroughly the nature of these deposits and the potential pathogenic role of cochlin, encoded by the gene harboring the mutations causative for DFNA9. For the study of aggregate formation, we utilized our Coch knock-in (Coch G88E/G88E) mouse model where one of the DFNA9-causing missense mutations was incorporated into the mouse genome by targeted homologous recombination (Robertson et al. 2008). Histological evaluation of middle ears in Coch G88E/G88E, in comparison to those in Coch +/+ and Coch −/−, has revealed deposit formation and recapitulation of human DFNA9 middle ear histopathology in the Coch G88E/G88E mouse model. The DFNA9-affected temporal bone in this study is from a member of a large kindred with the V66G mutation, which is in the same domain (FCH/LCCL) as the G88E mutation in our mouse model. All human DFNA9 temporal bones analyzed to date have mutations in the FCH/LCCL domain (P51S, V66G, G88E, and W117R), and all show the characteristic histopathology including eosinophilic deposits. For characterization of normal cochlin expression and localization, we performed cochlin immunostaining in Coch +/+ mouse and normal control human middle ears, showing specific and prominent localization in the same sites as those with deposits. This is the first report showing cochlin as a major component of specific structures within the middle ear. We characterized cochlin expression and localization within the deposits by performing immunohistochemistry in both Coch G88E/G88E mouse and DFN9-affected human middle ears. We also evaluated colocalization of type II collagen with cochlin, which contains vWFA domains, known to interact with collagens.
Our findings indicate that given the prominent and specific localization of cochlin in middle ear structures, the study of cochlin, in addition to previously known proteins in the middle ear, will enable a more comprehensive analysis of function and pathology in the middle ear. The intriguing new finding of cochlin-staining deposits in the middle ear, in a disorder previously known only as a sensorineural deficit, indicates the need for evaluation of conductive, in addition to sensorineural, hearing loss in individuals with COCH mutations. Conversely, evaluation of COCH in individuals presenting with conductive hearing loss may reveal a potential role for cochlin in pathogenesis, possibly via previously unknown mutations in this gene, or regulatory changes in its expression.
Middle ear evaluation, in addition to inner ear studies in mouse and human, provides complementary information and insight into cochlin function and DFNA9 pathology. The Coch G88E/G88E mouse model enables more thorough study of the composition of the deposits, and the onset and progression of this pathology resulting from Coch missense mutations. Future proteomic studies of aggregate contents will enable identification of potential cochlin interactors and other proteins involved in cochlin functional pathways.
METHODS
Tissues
Mouse tissues were obtained according to guidelines and protocols approved by the Harvard Medical School Standing Committee on Animals (Boston, MA, USA). Coch +/+, Coch G88E/G88E, and Coch −/− mice were evaluated in triplicate. For histology and immunohistochemistry, mice were perfused intracardially with 4 % paraformaldehyde and decapitated. Skulls, containing intact middle and inner ears, were post-fixed in 4 % paraformaldehyde for 24 h, decalcified in 120 mM EDTA for 1 week at room temperature, embedded in paraffin, serially cut at 5–8 μm thickness, and used for staining with hematoxylin and eosin (H&E). Adjacent sections were used for immunostaining.
Human temporal bones were obtained from the collection at the Massachusetts Eye and Ear Infirmary; one from a DFNA9-affected individual (age 86 years) and one from an unaffected individual (age 83 years) were utilized in this study. Previously, these temporal bones had been processed for light microscopy as described in the third edition of Schuknecht’s Pathology of the Ear (Merchant 2010). Both temporal bones were fixed in 10 % buffered formalin and decalcified in 0.27 M EDTA, embedded in celloidin, sectioned in the axial plane at 20 μm, and every tenth section stained with H&E and mounted on glass slides. Adjacent sections were used for immunostaining.
Immunohistochemistry
Immunostaining of both mouse and human tissues was performed using an anti-cochlin antibody generated against a peptide in the vWFA1 domain of cochlin corresponding to amino acid residues 163–181 of human cochlin, identical in both murine and bovine cochlin (Ikezono et al. 2004; Robertson et al. 2006). Immunostaining of mouse tissues was also performed with an anti-collagen type II antibody (Cortex Biochem, San Leandro, CA).
Immunohistochemistry of mouse paraffin-embedded temporal bones was performed as previously described (Robertson et al. 2006). Briefly, sections from 1-year-old Coch +/+, Coch G88E/G88E, and Coch −/− mice were incubated with anti-cochlin antibody at a dilution of 1:10,000 and anti-collagen type II antibody at a 1:1,000 dilution, overnight at room temperature, washed, and incubated with a secondary biotinylated anti-rabbit IgG (Vector Labs, Burlingame, CA). Immunostaining was visualized by incubation with the Vectastain ABC reagent (Vector Labs) followed by 3,3′-diaminobenzidine (DAB).
Immunohistochemistry of human celloidin-embedded temporal bones (DFNA9-affected 86-year-old female with COCH +/V66G and an unaffected 83-year-old control) was performed as previously described (O'Malley et al. 2009). Briefly, sections were adhered to slides and celloidin was removed with a sodium hydroxide methanol mixture. Sections were hydrated, rinsed in phosphate buffered saline, blocked with 5 % normal horse serum, and subsequently incubated with the same anti-cochlin antibody (as above) at a dilution of 1:2,000. The remaining steps were the same as described for the mouse.
RESULTS AND DISCUSSION
Histopathology in Mouse and Human Middle Ears
Prominent acellular deposits in DFNA9 were initially identified and characterized in the inner ear, specifically in the spiral ligament and spiral limbus of the cochlea and stroma underlying sensory epithelia of vestibular compartments (Khetarpal et al. 1991; Robertson et al. 1998). This histopathology is a unique and characteristic feature of this disorder. Examination of both DFNA9-affected and Coch G88E/G88E middle ears reveals a very similar phenomenon of deposit formation as previously characterized in cochlear and vestibular labyrinths in DFNA9 temporal bones. We first evaluated middle ear histology in Coch +/+, Coch G88E/G88E, and Coch −/− 1-year-old mice. Our findings reveal presence of acellular deposits in both the incudomalleal and incudostapedial joints in Coch G88E/G88E middle ears (Figs. 2D and G, 3D, and 4D). These deposits are present in the articular surfaces between the ossicles, as well as protruding around the perimeter of the joints. In particular, the incudomalleal joint, which has a larger articular surface than the incudostapedial joint, contains aggregates throughout the joint space, dorsally between the incus and malleal head (Figs. 2D and G), and along the length of the ossicles to more ventral processes of the ossicles (Fig. 3D). There are no aggregates present in age-matched Coch +/+ controls (Figs. 2A, 3A, and 4A) or Coch −/− mice (Figs. 2J, 3G, and 4G). Absence of this pathology in Coch −/− mice lends further support to our hypothesis of the dominant negative effect of COCH mutations in DFNA9, indicating that deposit formation is likely a consequence of a gain of function of mutant cochlin, rather than haploinsufficiency or lack of cochlin protein.

H&E staining of the joint between the malleus (M) and the incus (I) (the more dorsal part) in 1-year-old (A) Coch +/+, D and G) Coch G88E/G88E, and (J) Coch −/− mice shows eosinophilic deposits (asterisk) in the Coch G88E/G88E mouse. Immunostaining with anti-cochlin antibody of (B) Coch +/+, E and H) Coch G88E/G88E, and K) Coch −/− (negative control) mice of sections adjacent to those with H&E staining shows cochlin staining in the incudomalleal joint of Coch +/+ and Coch G88E/G88E mice. Immunostaining for type II collagen in adjacent sections in C) Coch +/+, F and I) Coch G88E/G88E, and L) Coch −/− mice shows localization in the area of chondrocytes, but not in the articular joint surfaces or in eosinophilic deposits, which show cochlin staining.

H&E staining of the joint between the malleus (M) and the incus (I) (the more ventral part) of 1-year-old (A) Coch +/+, (D) Coch G88E/G88E, and (G) Coch −/− mice shows eosinophilic deposits (asterisk) in the Coch G88E/G88E mouse. Immunostaining with anti-cochlin antibody of (B) Coch +/+, (E) Coch G88E/G88E, and (H) Coch −/− (negative control) mice of sections adjacent to those with H&E staining shows cochlin staining in the incudomalleal joint of Coch +/+ and Coch G88E/G88E mice. Immunostaining for type II collagen in adjacent sections in (C) Coch +/+, (F) Coch G88E/G88E, and (I) Coch −/− mice shows localization in the area of chondrocytes, but not in the articular joint surfaces or eosinophilic deposits, which show cochlin staining.

H&E staining of the joint between the incus (I) and stapes (S) of 1-year-old A) Coch +/+, (D) Coch G88E/G88E, and (G) Coch −/− mice shows eosinophilic deposits (asterisk) in the Coch G88E/G88E mouse. Immunostaining with anti-cochlin antibody of (B) Coch +/+, (E) Coch G88E/G88E, and (H) Coch −/− (negative control) mice of sections adjacent to those with H&E staining shows cochlin staining in the incudostapedial joint of Coch +/+ and Coch G88E/G88E mice. Immunostaining for type II collagen in adjacent sections in (C) Coch +/+, (F) Coch G88E/G88E, and (I) Coch −/− mice shows localization in the area of chondrocytes, but not in the articular joint surfaces or eosinophilic deposits, which show cochlin staining.
Our findings in the Coch G88E/G88E mouse model closely parallel and recapitulate the phenotype in middle ears of human DFNA9-affected temporal bones, where aggregates are present in both the incudomalleal (Fig. 5C) and incudostapedial (Fig. 6C) joints. A study of 12 temporal bones from 7 individuals with DFNA9 shows presence of these deposits in all cases examined (McCall et al. 2011). Aggregates are also present in the middle lamina propria layer of the pars tensa (PT) of the tympanic membrane (TM) in human DFNA9 middle ears (Fig. 7C). Among all cases examined, the degree of TM thickening is variable and the area of affected TM ranges from 5 to 50 % (McCall et al. 2011). Substantial TM thickening is not consistently discernible in Coch G88E/G88E mice (Fig. 8D). It is possible that there are species differences, or that TM pathology occurs subsequent to that of the joints, as the DFNA9 temporal bones are from advanced ages (45–86 years old) and show variable degrees of thickness. No other appreciable abnormalities were detected in other structures of Coch G88E/G88E mouse middle ears or human DFNA9 middle ears, including the stapediovestibular joint, tensor tympani tendon, and incudal ligament.

H&E staining of human (A) unaffected age-matched control, and C) DFNA9-affected middle ears shows a mixture of eosinophilic and basophilic deposits (asterisk) in the DFNA9 joint between the malleus (M) and the incus (I). Immunostaining with anti-cochlin antibody of sections adjacent to those with H&E staining in human (B) unaffected and (D) DFNA9-affected middle ears shows cochlin staining in both unaffected and DFNA9 incudomalleal joints, with the latter showing an uneven pattern of cochlin localization, with some areas lacking cochlin staining.

H & E staining of human (A) unaffected age-matched control and (C) DFNA9-affected middle ears shows a mixture of eosinophilic and basophilic deposits (asterisk) in the DFNA9 joint between the incus (I) and the stapes (S). Immunostaining with anti-cochlin antibody of sections adjacent to those with H&E staining in human (B) unaffected and (D) DFNA9-affected middle ears shows cochlin staining in both unaffected and DFNA9 incudomalleal joints, with the latter showing an uneven pattern of cochlin localization, with some areas lacking cochlin staining.

H&E staining of (A) human unaffected age-matched control, and (C) DFNA9-affected middle ears shows a mixture of eosinophilic and basophilic deposits (asterisk) in the DFNA9 pars tensa of the tympanic membrane (TM). Immunostaining with anti-cochlin antibody of sections adjacent to those with H&E staining in (B) unaffected and (D) DFNA9-affected human middle ears shows cochlin staining in both unaffected and DFNA9 tympanic membranes, with the latter showing an uneven pattern of cochlin localization, with some areas lacking cochlin staining. The point of attachment of the malleus (M) to the TM is indicated.

H&E staining of 1-year-old (A) Coch +/+, (D) Coch G88E/G88E, and (G) Coch −/− mice and immunostaining with anti-cochlin antibody of (B) Coch +/+, (E) Coch G88E/G88E, and (H) Coch −/− (negative control) mice of sections adjacent to those with H&E staining shows cochlin staining in the pars tensa of the tympanic membrane (TM-PT), but not in the pars flaccida (TM-PF) of Coch +/+ and Coch G88E/G88E mice. Immunostaining for type II collagen in adjacent sections in (C) Coch +/+, (F) Coch G88E/G88E, and (I) Coch −/− mice shows localization in the same structures as detected for cochlin immunostaining. (M = malleus).
It is interesting to note that in human DFNA9 middle ears, a mixture of eosinophilic and basophilic deposits are present (Figs. 5C, 6C, and 7C), ranging from predominantly one type or the other, and gradations in between (McCall et al. 2011), whereas in the inner ears of these same DFNA9-affected temporal bones, deposits appear to have only eosinophilic staining. This may be a result of differences between cellular and extracellular compositions of the middle ear structures (interossicular joints and TM) and those of the inner ear (the spiral ligament, limbus, and vestibular stroma) where deposits are found. In the case of the Coch G88E/G88E mouse model, middle ear deposits are largely eosinophilic at the age evaluated. Some structural differences in the middle ear between the two species (including the incudomalleal joint, which is a cartilaginous synchondrosis in the mouse, as opposed to a synovial joint in the human) could account for some differences in the contents of the deposits. It is also possible that basophilic aggregates develop at more advanced ages. In addition, our previous studies of cochlear and vestibular labyrinths in 21-month-old Coch G88E/G88E mice (Robertson et al. 2008), show absence of detectable aggregates by light microscopy, although both hearing and vestibular functions are severely impaired, as shown by substantially elevated thresholds (ABR and VsEP) in all mutants tested, as well as absent ABRs, across all frequencies, in six of 11 Coch G88E/G88E and Coch G88E/+ tested. Microfibrillar deposits characteristic of end-stage human DFNA9 inner ears are not present in detectable amounts in the mouse model. However, presence of deposits in middle ears of 12-month-old Coch G88E/G88E mice, similar to findings in DFNA9 human temporal bones, provides further validation of our mouse model. Absence of substantial deposits in the membranous labyrinths of Coch G88E/G88E mice at the same age when middle ear deposits are present suggests that there may be differences in onset and progression of deposit formation between middle and inner ear structures. Mice at various ages are being evaluated for further characterization of this phenotype.
Cochlin Localization in Wild-Type Mouse and Normal Human Middle Ears
To characterize normal cochlin localization, we first performed immunohistochemistry on Coch +/+ mouse middle ears using an anti-cochlin antibody. Very prominent and specific cochlin immunostaining is detected in the incudomalleal (Figs. 2B and 3B) and incudostapedial (Fig. 4B) joints (structures corresponding to the same sites where deposits are detected in the Coch G88E/G88E and DFNA9 middle ears). In addition, intense cochlin staining is seen in the pars tensa (PT) of the TM but completely absent in the pars flaccida (PF) of the TM (Fig. 8B). This finding also corresponds to the other site of pathological deposit formation in DFNA9, namely the pars tensa of the TM. No cochlin staining was detected in negative control Coch −/− sections (Figs. 2K, 3H, 4H, and 8H), confirming specificity of the antibody.
We then evaluated cochlin localization in normal control human middle ears. Similar to findings in the mouse, very specific cochlin immunostaining was detected in the incudomalleal (Fig. 5B) and incudostapedial joints (Fig. 6B), as well as in the pars tensa of the TM (Fig. 7B). In both human and mouse, cochlin staining was quite homogeneous and intense in these structures and absent in adjacent tissues of the middle ear.
This is the first report where cochlin is identified as a major component of normally developed and functioning interossicular joints and TM of the middle ear. And furthermore, the structures where cochlin is expressed are the very sites of the pathological deposits in mouse and human middle ears carrying COCH mutations. Our previous immunohistochemistry in human cochlear and vestibular labyrinths also showed a tight correlation between sites of cochlin immunostaining and histopathological deposits, also indicating a likely direct role of mutant cochlin in pathogenesis in these structures (Robertson et al. 2006).
Cochlin Immunostaining of Mouse Coch G88E/G88E and DFNA9-Affected Middle Ears
For better characterization of aggregate formation in relation to DFNA9-causative COCH missense mutations, we performed immunohistochemistry on mouse Coch G88E/G88E and human DFNA9-affected middle ears. Immunostaining of Coch G88E/G88E middles ears (Figs. 2E and H, 3E, 4E, and 8E) reveals presence of cochlin in the same structures where normal cochlin expression is detected in wild-type mice, and within the sites of deposit formation, namely the incudomalleal and incudostapedial joints, and pars tensa of the TM. Cochlin staining is detected homogenously throughout the aggregates (which are uniformly eosinophilic) filling up the interossicular joint spaces in the Coch G88E/G88E middle ears. Because these studies are performed on Coch G88E/G88E mice, all of the staining represents mutant cochlin. This observation confirms the abundant and stable nature of the mutant protein, rather than its degradation, identifying it as one of the major components of the characteristic acellular deposits, and supporting the hypothesis of a gain of deleterious function of the mutant protein by abnormal aggregate formation.
Immunostaining in human DFNA9 middle ears shows cochlin staining throughout the same structures that contain cochlin in normal unaffected human and Coch +/+ and Coch G88E/G88E mouse middle ears, at the sites of abnormal aggregate formation. However, one interesting observation in human DFNA9 middle ears is that the pattern of cochlin staining is not homogeneous throughout the spaces containing the deposits, as was in the Coch G88E/G88E mouse, suggesting accumulation of other substances in conjunction with, as a result of, or otherwise related to presence of the mutant cochlin. These deposits appear as thick, overlapping, and intertwined mixture of eosinophilic and basophilic substances making it somewhat difficult to make an exact assessment of cochlin localization within the aggregates. However, cochlin staining corresponds predominantly to areas of eosinophilia. This finding is consistent with the fact that in the mouse middle ear, where deposits are purely eosinophilic, homogeneous cochlin staining is present. Similarly, our previous findings also show homogeneous cochlin staining of the deposits in cochlear and vestibular labyrinths (Robertson et al. 2006), which appear purely eosinophilic.
Type II Collagen Immunostaining of Mouse Coch +/+, Coch G88E/G88E, and Coch −/− Middle Ears
The two tandem C-terminal vWFA (von Willebrand factor A) domains of cochlin, which show a high degree of evolutionary conservation, are present in a large number of other secreted proteins and are known to bind extracellular tissue components, including fibrillar collagens, glycoproteins, and proteoglycans (Colombatti and Bonaldo 1991; Lee et al. 1995; Whittaker and Hynes 2002). Affinity of cochlin vWFA domains for collagens has also been demonstrated in vitro (Nagy et al. 2008). Furthermore, our previous studies of cochlin localization in the cochlear and vestibular labyrinths in the spiral ligament, limbus, and stroma underlying vestibular sensory epithelia closely parallel localization of type II collagen as demonstrated in a number of reports (Yoo and Tomoda 1988; Ishibe and Yoo 1990; Slepecky et al. 1992), including our current study (data not shown). In order to characterize localization of type II collagen in the middle ears of Coch +/+, Coch G88E/G88E, and Coch −/− mouse models, and to evaluate further the content of abnormal deposits, we performed immunostaining of adjacent sections of the same middle ear structures as our cochlin immunostaining, using an anti-collagen type II antibody.
Interossicular Joints
A distinct pattern of type II collagen staining in relation to cochlin localization is observed in both the incudomalleal and incudostapedial joints (Figs. 2B–C, 3B–C, and 4B–C). Type II collagen, a known cartilage component, localizes only to the area of chondrocytes within the cartilaginous matrix. Bony areas within the ossicles that show calcification distinctly lack this staining. Cochlin staining, however, is restricted to the thin interossicular layer that forms the actual articular surface of the ossicles. Interestingly, this layer does not show any detectable type II collagen staining, but rather overlies the chondrocytes with prominent collagen staining. Therefore, areas of cochlin and type II collagen immunostaining appear to be non-overlapping, but tightly juxtaposed.
In typical joint formation, segmentation of continuous cartilaginous rods gives rise to the developing joint (for example in digit formation). This mechanism has also been demonstrated in formation of middle ear interossicular joints in the mouse (Amin and Tucker 2006). Initially, round chondrocytes flatten and become nonchondrogenic, forming the “interzone”, which will become the joint space. This process involves tight regulation of type II collagen expression by Sox9 (Bell et al. 1997), where loss of Sox9 expression in the interzone results in the absence of type II collagen specifically in this region of the developing joint, but not in the chondrocytes (Craig et al. 1987). Amin and Tucker demonstrated that the process of interossicular joint formation in the mouse is initiated between E13.5 and E14.5, where the malleus and incus become separate structures with an intervening zone devoid of type II collagen (Amin and Tucker 2006). Our data further reveal specific localization of cochlin in this interossicular space in the mouse middle ear.
In the Coch G88E/G88E mouse model, we see complete absence of type II collagen staining within interossicular eosinophilic deposits, while chondrocytes show persistent collagen staining (Figs. 2F and I, 3F, and 4F). Cochlin-staining deposits fill the joint spaces, abutting collagen-expressing chondrocytes and widening the gap between the two ossicles. In the Coch −/− mouse model, more prominent type II collagen staining is detected closer to, and in the perimeter of, joint spaces (Figs. 2L, 3I, and 4I). These data suggest a possible co-regulation of type II collagen and cochlin expression. Furthermore, even though no gross morphological abnormalities are detected in Coch −/− interossicular joints, it is possible that within the context of real environmental conditions, such as noise exposure, an altered interplay with type II collagen in the absence of cochlin could result in damage to these structures.
Tympanic Membrane
In contrast to the staining pattern in interossicular joints, type II collagen immunostaining shows remarkable colocalization with cochlin staining in the TM, namely in pars tensa, but not pars flaccida (Figs. 8B–C). In particular, the continuation of the TM to the tympanic annulus, shows intense immunostaining for both proteins in the area next to the fibrous connection to the tympanic bone. Type II collagen has been shown as a main component of the healthy pars tensa in several rodents and other mammals (Yoo and Tomoda 1988; Ishibe and Yoo 1990; Stenfeldt et al. 2006). However, an interesting study demonstrated that during healing phases after TM perforation in the rat, type II collagen levels became undetectable in the intital phase, but reappeared after full closure of the TM, and showed further proliferation and thickening in a more disarrayed form in the healed tissue (Stenfeldt et al. 2013). Another study of rat TM revealed changes in expression of the different types of fibrillar collagens during healing phases after myringotomy and infection (Stenfeldt et al. 2006). This study demonstrated that in healthy TM, type II collagen was the main constituent of the lamina propria of the pars tensa, whereas type I was predominantly in the pars flaccida, as well as in the loose connective tissue around the point of insertion of the malleus, along with type III collagen. However, after completion of healing from TM perforation and infection, the pars tensa showed substantial thickening, with scar tissue containing all three types of collagens. Type II collagen immunostaining of Coch G88E/G88E, and Coch −/− mouse TM (Fig. 8F and I) appears to have similar colocalization with cochlin in pars tensa, as for Coch +/+ (Fig. 8C). We believe that potential changes in cochlin levels, in conjunction with collagens, will be important to evaluate in the context of TM perforation, other TM structural abnormalities such as cholesteatoma, as well as in otitis media.
Structure and Function of Cochlin and Implications in Pathology
Aggregate Formation
There is mounting evidence for the aggregative properties of cochlin, including finding of cochlin-staining deposits in middle and inner ear structures, in vitro studies, and the role of cochlin in pathology in the eye. Misfolding of the cochlin FCH/LCCL domain as a result of COCH mutations has been documented in several reports (Trexler et al. 2000; Liepinsh et al. 2001; Nagy et al. 2004). Further studies have revealed dimerization of mutant misfolded cochlin, as well as its ability to induce wild-type cochlin to also form stable oligomers, providing a possible mechanism for the dominant nature of COCH mutations (Yao et al. 2010). Proteomics in the eye has revealed cochlin deposits in trabecular meshwork of glaucomatous but not normal human eyes (Bhattacharya et al. 2005a; Bhattacharya et al. 2005b). More extensive studies have explored the role of cochlin in obstruction of aqueous humor circulation leading to elevated intraocular pressure, a hallmark of open angle glaucoma (Lee et al. 2010; Goel et al. 2012). Interestingly, in the cases of glaucoma, no mutations in the COCH protein-coding region have been reported; but rather, transcriptional upregulation of COCH leading to accumulation of cochlin has been implicated (Picciani et al. 2009).
Extracellular Matrix and Protein Interactions
Cochlin is a secreted protein detected both intracellularly and in the secreted media of mammalian cultured cells transfected with COCH (Robertson et al. 2003). In vivo, it is detected in great abundance within the tissues of middle and inner ears, suggesting the stable nature of the protein, with vWFA domains known to interact with other proteins in extracellular matrix of tissues. Therefore, cochlin may likely play a role in maintaining structural integrity and stabilizing the extracellular environment of tissues in which it is expressed, which would in turn influence proper functioning of other components and processes in these organs. Therefore, it is possible that other downstream components in cochlin functional pathways and their interactions may be altered in the presence of mutant cochlin or absence of the normal protein, particularly in the context of damaging environmental challenges.
Immune Function
The less well-characterized, but also highly evolutionarily conserved N-terminal FCH/LCCL (factor C-homology/ Limulus factor C, cochlin, lung gestational protein) domain of cochlin was initially identified (Robertson et al. 1997) based on its homology to a domain in factor C of Limulus (horseshoe crab). Factor C is a serine protease involved in innate host defense that is activated by lipopolysaccharide (LPS), the major component of the outer membrane of gram-negative bacteria, initiating a coagulation cascade for host defense (Muta et al. 1991). The FCH/LCCL domain harbors 14 of the 20 COCH mutations described to date (Fig. 1). Full-length cochlin (p60) is cleaved post-translationally during its secretion at the junction between the FCH/LCCL domain and the two more C-terminal vWFA domains, creating a smaller (~16 kD) isoform composed of the FCH/LCCL domain and larger (~40 and 44 kD) isoforms, consisting of the two tandem vWFA domains (Robertson et al. 2003; Ikezono et al. 2004). Interestingly, the p16 cochlin isoform, referred to as CTP (cochlin tomoprotein), is detected only in the perilymph of the inner ear and not within the cochlear tissues where the full-length p60 cochlin is produced, making the small isoform a potential specific marker for perilymphatic fistulae (Ikezono et al. 2009; Ikezono et al. 2010; Ikezono et al. 2011).
An interesting recent study reveals the same post-translational cleavage and secretion of the small FCH/LCCL-containing isoform of cochlin from follicular dendritic cells (FDCs) in conduits of the spleen and lymph nodes, in response to bacterial infection (Py et al. 2013). (Our initial studies had identified spleen as another predominant site of COCH expression, although not as abundant as the inner ear) (Robertson et al. 1994; Robertson et al. 1997). It was demonstrated that during inflammation in response to LPS injection or bacterial infection in wild-type mice, cochlin in FDCs is cleaved and the FCH/LCCL domain released into the blood circulation, where it amplifies cytokine responses, and promotes recruitment of immune effector cells and bacterial clearance (Py et al. 2013). The same study demonstrates that cochlin-deficient (Coch −/−) mice have defects in these immune processes, which lead to their reduced survival. The cochlin immune response is mediated by a rise in tumor necrosis factor-alpha (TNF-alpha) levels. TNF-alpha and other cytokines are known to play important roles in many inflammatory processes including those in the middle ear, such as otitis media and chronic middle ear effusion (DeMaria and Murwin 1997; Maxwell et al. 1997; Willett et al. 1998; Smirnova et al. 2002). Therefore, cochlin cleavage and release of the FCH/LLCL domain, mediated downstream of TNF-alpha release in response to infection, may have interesting implications in terms of possible involvement of cochlin in immune and inflammatory response in the middle ear. In addition, it is not currently known whether the similar cleavage and release of the FCH/LCCL-containing cochlin isoform into the perilymph has any role in immune function within the inner ear. Future studies of cochlin levels and proteolytic cleavage in response to infections in the auditory system are warranted to characterize cochlin’s possible host-defense function in this context.
CONCLUSIONS
We have identified cochlin as a component of normal mouse and human middle ear structures (pars tensa of TM and interossicular joints) as well as in COCH mouse mutant knock-in and human DFNA9 middle ears. Therefore, we believe that middle ear studies, in particular those involving the tympanic membrane and interossicular joints, would be enhanced by evaluation of cochlin as well as other traditionally studied proteins such as various types of collagens. Our studies of localization of collagen type II, showing distinct overlap with cochlin in the pars tensa, but outside of, and juxtaposed to areas of cochlin staining in the interossicular joints and deposits, will enable further thorough analyses of these extracellular proteins in the context of any structural damage or other pathology in the middle ear. Changes in cochlin level, post-translational processing, and cochlin-interacting proteins would be important to assess in the context of middle ear pathological conditions such as otitis media, structural changes in the TM, and disorders of the ossicular chain. In addition, finding of cochlin-staining characteristic histopathological deposits in middle ear structures warrants evaluation of conductive, as well as sensorineural hearing loss in individuals who present with late-onset hearing loss accompanied by balance problems, which are very indicative of DFNA9 as a result of COCH mutations. Conversely, in cases presenting with conductive hearing loss, evaluation of the COCH gene may reveal mutations or regulatory changes, different from those causative of sensorineural hearing loss. The combination of inner and middle ear findings in the human and the mouse will enable more comprehensive analyses of cochlin function and its role in DFNA9 histopathology. The Coch G88E/G88E mouse model serves as a valuable tool for the study and characterization of cochlin function in both the middle and inner ears and the role of COCH mutations in pathogenesis and progression of hearing loss.
References
Amin S, Tucker AS (2006) Joint formation in the middle ear: lessons from the mouse and guinea pig. Dev Dyn 235:1326–1333
Bell DM, Leung KK, Wheatley SC, Ng LJ, Zhou S, Ling KW, Sham MH, Koopman P, Tam PP, Cheah KS (1997) SOX9 directly regulates the type-II collagen gene. Nat Genet 16:174–178
Bhattacharya SK, Annangudi SP, Salomon RG, Kuchtey RW, Peachey NS, Crabb JW (2005a) Cochlin deposits in the trabecular meshwork of the glaucomatous DBA/2J mouse. Exp Eye Res 80:741–744
Bhattacharya SK, Rockwood EJ, Smith SD, Bonilha VL, Crabb JS, Kuchtey RW, Robertson NG, Peachey NS, Morton CC, Crabb JW (2005b) Proteomics reveal Cochlin deposits associated with glaucomatous trabecular meshwork. J Biol Chem 280:6080–6084
Colombatti A, Bonaldo P (1991) The superfamily of proteins with von Willebrand factor type A-like domains: one theme common to components of extracellular matrix, hemostasis, cellular adhesion, and defense mechanisms. Blood 77:2305–2315
Craig FM, Bentley G, Archer CW (1987) The spatial and temporal pattern of collagens I and II and keratan sulphate in the developing chick metatarsophalangeal joint. Development 99:383–391
DeMaria TF, Murwin DM (1997) Tumor necrosis factor during experimental lipopolysaccharide-induced otitis media. Laryngoscope 107:369–372
Goel M, Sienkiewicz AE, Picciani R, Wang J, Lee RK, Bhattacharya SK (2012) Cochlin, intraocular pressure regulation and mechanosensing. PLoS ONE 7:e34309
Ikezono T, Omori A, Ichinose S, Pawankar R, Watanabe A, Yagi T (2001) Identification of the protein product of the Coch gene (hereditary deafness gene) as the major component of bovine inner ear protein. Biochim Biophys Acta 1535:258–265
Ikezono T, Sugizaki K, Shindo S, Sekiguchi S, Pawankar R, Baba S, Yagi T (2010) CTP (Cochlin-tomoprotein) detection in the profuse fluid leakage (gusher) from cochleostomy. Acta Otolaryngol 130:881–887
Ikezono T, Shindo S, Li L, Omori A, Ichinose S, Watanabe A, Kobayashi T, Pawankar R, Yagi T (2004) Identification of a novel cochlin isoform in the perilymph: insights to cochlin function and the pathogenesis of DFNA9. Biochem Biophys Res Commun 314:440–446
Ikezono T, Shindo S, Sekine K, Shiiba K, Matsuda H, Kusama K, Koizumi Y, Sugizaki K, Sekiguchi S, Kataoka R, Pawankar R, Baba S, Yagi T, Okubo K (2011) Cochlin-tomoprotein (CTP) detection test identifies traumatic perilymphatic fistula due to penetrating middle ear injury. Acta Otolaryngol 131:937–944
Ikezono T, Shindo S, Sekiguchi S, Hanprasertpong C, Li L, Pawankar R, Morizane T, Baba S, Koizumi Y, Sekine K, Watanabe A, Komatsuzaki A, Murakami S, Kobayashi T, Miura M, Yagi T (2009) Cochlin-tomoprotein: a novel perilymph-specific protein and a potential marker for the diagnosis of perilymphatic fistula. Audiol Neurootol 14:338–344
Ishibe T, Yoo TJ (1990) Type II collagen distribution in the monkey ear. Am J Otol 11:33–38
Khetarpal U, Schuknecht HF, Gacek RR, Holmes LB (1991) Autosomal dominant sensorineural hearing loss: pedigrees, audiologic and temporal bone findings in two kindreds. Arch Otolaryngol Head Neck Surg 117:1032–1042
Lee ES, Gabelt BT, Faralli JA, Peters DM, Brandt CR, Kaufman PL, Bhattacharya SK (2010) COCH transgene expression in cultured human trabecular meshwork cells and its effect on outflow facility in monkey organ cultured anterior segments. Invest Ophthalmol Vis Sci 51:2060–2066
Lee JO, Rieu P, Arnaout MA, Liddington R (1995) Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11b/CD18). Cell 80:631–638
Liepinsh E, Trexler M, Kaikkonen A, Weigelt J, Banyai L, Patthy L, Otting G (2001) NMR structure of the LCCL domain and implications for DFNA9 deafness disorder. Embo J 20:5347–5353
Maxwell K, Leonard G, Kreutzer DL (1997) Cytokine expression in otitis media with effusion. Tumor necrosis factor soluble receptor. Arch Otolaryngol Head Neck Surg 123:984–988
McCall AA, Linthicum FH Jr, O'Malley JT, Adams JC, Merchant SN, Bassim MK, Gellibolian R, Fayad JN (2011) Extralabyrinthine manifestations of DFNA9. J Assoc Res Otolaryngol 12:141–149
Merchant SN (2010) Methods of removal, preparation, and study. In: Merchant SN, Nadol JBJ (eds) Schuknecht’s pathology of the ear. PMPH, Shelton, CT, USA, pp 9–20
Muta T, Miyata T, Misumi Y, Tokunaga F, Nakamura T, Toh Y, Ikehara Y, Iwanaga S (1991) Limulus factor C: an endotoxin-sensitive serine protease zymogen with a mosaic structure of complement-like, epidermal growth factor-like, and lectin-like domains. J Biol Chem 266:6554–6561
Nagy I, Trexler M, Patthy L (2008) The second von Willebrand type A domain of cochlin has high affinity for type I, type II and type IV collagens. FEBS Lett 582:4003–4007
Nagy I, Horvath M, Trexler M, Repassy G, Patthy L (2004) A novel COCH mutation, V104del, impairs folding of the LCCL domain of cochlin and causes progressive hearing loss. J Med Genet 41:e9
O'Malley JT, Merchant SN, Burgess BJ, Jones DD, Adams JC (2009) Effects of fixative and embedding medium on morphology and immunostaining of the cochlea. Audiol Neurootol 14:78–87
Picciani RG, Diaz A, Lee RK, Bhattacharya SK (2009) Potential for transcriptional upregulation of cochlin in glaucomatous trabecular meshwork: a combinatorial bioinformatic and biochemical analytical approach. Invest Ophthalmol Vis Sci 50:3106–3111
Py BF, Gonzalez SF, Long K, Kim MS, Kim YA, Zhu H, Yao J, Degauque N, Villet R, Ymele-Leki P, Gadjeva M, Pier GB, Carroll MC, Yuan J (2013) Cochlin produced by follicular dendritic cells promotes antibacterial innate immunity. Immunity 38:1063–1072
Robertson NG, Khetarpal U, Gutiérrez-Espeleta GA, Bieber FR, Morton CC (1994) Isolation of novel and known genes from a human fetal cochlear cDNA library using subtractive hybridization and differential screening. Genomics 23:42–50
Robertson NG, Hamaker SA, Patriub V, Aster JC, Morton CC (2003) Subcellular localisation, secretion, and post-translational processing of normal cochlin, and of mutants causing the sensorineural deafness and vestibular disorder, DFNA9. J Med Genet 40:479–486
Robertson NG, Skvorak AB, Yin Y, Weremowicz S, Johnson KR, Kovatch KA, Battey JF, Bieber FR, Morton CC (1997) Mapping and characterization of a novel cochlear gene in human and in mouse: a positional candidate gene for a deafness disorder, DFNA9. Genomics 46:345–354
Robertson NG, Jones SM, Sivakumaran TA, Giersch AB, Jurado SA, Call LM, Miller CE, Maison SF, Liberman MC, Morton CC (2008) A targeted Coch missense mutation: a knock-in mouse model for DFNA9 late-onset hearing loss and vestibular dysfunction. Hum Mol Genet 17:3426–3434
Robertson NG, Lu L, Heller S, Merchant SN, Eavey RD, McKenna M, Nadol JB Jr, Miyamoto RT, Linthicum FH Jr, Lubianca Neto JF, Hudspeth AJ, Seidman CE, Morton CC, Seidman JG (1998) Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat Genet 20:299–303
Robertson NG, Cremers CW, Huygen PL, Ikezono T, Krastins B, Kremer H, Kuo SF, Liberman MC, Merchant SN, Miller CE, Nadol JB Jr, Sarracino DA, Verhagen WI, Morton CC (2006) Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum Mol Genet 15:1071–1085
Slepecky NB, Savage JE, Yoo TJ (1992) Localization of type II, IX and V collagen in the inner ear. Acta Otolaryngol 112:611–617
Smirnova MG, Kiselev SL, Gnuchev NV, Birchall JP, Pearson JP (2002) Role of the pro-inflammatory cytokines tumor necrosis factor-alpha, interleukin-1 beta, interleukin-6 and interleukin-8 in the pathogenesis of the otitis media with effusion. Eur Cytokine Netw 13:161–172
Stenfeldt K, Johansson C, Hellstrom S (2006) The collagen structure of the tympanic membrane: collagen types I, II, and III in the healthy tympanic membrane, during healing of a perforation, and during infection. Arch Otolaryngol Head Neck Surg 132:293–298
Stenfeldt K, Johansson C, Eriksson PO, Hellstrom S (2013) Collagen Type II is produced in healing pars tensa of perforated tympanic membranes: an experimental study in the rat. Otol Neurotol 34:e88–e92
Trexler M, Banyai L, Patthy L (2000) The LCCL module. Eur J Biochem 267:5751–5757
Whittaker CA, Hynes RO (2002) Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell 13:3369–3387
Willett DN, Rezaee RP, Billy JM, Tighe MB, DeMaria TF (1998) Relationship of endotoxin to tumor necrosis factor-alpha and interleukin-1 beta in children with otitis media with effusion. Ann Otol Rhinol Laryngol 107:28–33
Yao J, Py BF, Zhu H, Bao J, Yuan J (2010) Role of protein misfolding in DFNA9 hearing loss. J Biol Chem 285:14909–14919
Yoo TJ, Tomoda K (1988) Type II collagen distribution in rodents. Laryngoscope 98:1255–1260
Acknowledgements
We would like to dedicate this work to the memory of our beloved friend and colleague, Dr. Saumil Merchant. We are grateful to the individuals and families who have participated in this study and for their generous donations of temporal bones. We thank Dr. John Rosowski and Melissa McKinnon for sharing their expertise in mouse middle ear. This work was supported by National Institutes of Health grants R01 DC03402 (to C.C.M.), K08 DC010419 (to K.M.S.), and the Bertarelli Foundation (to K.M.S.).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Robertson, N.G., O’Malley, J.T., Ong, C.A. et al. Cochlin in Normal Middle Ear and Abnormal Middle Ear Deposits in DFNA9 and Coch G88E/G88E Mice. JARO 15, 961–974 (2014). https://doi.org/10.1007/s10162-014-0481-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10162-014-0481-9