
Abstract
Although Genome Wide Association Studies (GWAS) have led to many valuable insights into the genetic bases of common diseases over the past decade, the issue of missing heritability has surfaced, as the discovered main effect genetic variants found to date do not account for much of a trait’s predicted genetic component. We present a workflow, integrating epigenomics and topologically associating domain data, aimed at discovering trait-associated SNP pairs from GWAS where neither SNP achieved independent genome-wide significance. Each analyzed SNP pair consists of one SNP in a putative active enhancer and another SNP in a putative physically interacting gene promoter in a trait-relevant tissue. As a proof-of-principle case study, we used this approach to identify focused collections of SNP pairs that we analyzed in three independent Type 2 diabetes (T2D) GWAS. This approach led us to discover 35 significant SNP pairs, encompassing both novel signals and signals for which we have found orthogonal support from other sources. Nine of these pairs are consistent with eQTL results, two are consistent with our own capture C experiments, and seven involve signals supported by recent T2D literature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The majority of human GWAS efforts to date have focused on detecting main effects, i.e., individual SNPs that are associated with a given complex trait. Some would argue that this is indeed where the focus should be placed, as the majority of SNPs contribute to a given trait in an additive manner (Hill et al. 2008). However, the discovered single genetic variants do not typically account for much of a complex trait’s predicted genetic component, an issue commonly referred to as the ‘missing heritability’. There has been much discussion regarding potential sources of missing heritability (Eichler et al. 2010; Zuk et al. 2012). Based on the complexity of the biomolecular networks driving biological systems, one possible reason for this is that multiple common variants work together to affect the pathogenesis of a trait and are difficult to detect as they either act in an additive fashion, where the individual effects are too small to reach genome-wide statistical significance, or they interact. The latter would be consistent with the prevalence of epistasis in model organisms (Brem et al. 2005; Mackay 2014).
Investigating the action of multiple SNPs in GWAS is challenging. Even when focusing only on pairs of SNPs, the search space is very large, affecting computational requirements and statistical power due to the extensive multiple comparisons required. Thus, filters are typically used to reduce the number of models to analyze. One rational possibility is to apply computational filters, e.g., using methods such as ReliefF and its derivatives (Robnik-Šikonja and Kononenko 2003; Moore and White 2007; Moore 2015) or MDR (Ritchie et al. 2001), or “greedy” approaches which first identify SNPs with significant or marginally significant main effects and then investigate models involving only these SNPs (Qi et al. 2007; Verma et al. 2016). A downside of the latter is that interacting SNPs with no main effects would be missed (see Urbanowicz et al. 2014 for a discussion of pure and strict epistasis).
Another approach is to use biological filters. For example, Mitra et al. (2017) limited their SNP pair search space based on the Ras/MAPK pathway, which is known to be relevant to Autism Spectrum Disorders. Motivated by the recognition of the importance of regulatory networks in genomic studies (Cowper-Sal Iari et al. 2011; Boyle et al. 2017), we recently proposed to utilize biological filters based on enhancers and promoters (Manduchi et al. 2018). Enhancers are non-coding regions that affect the expression of possibly distant genes through chromatin looping in a tissue-specific manner. In Manduchi et al. (2018), we used interacting enhancer-promoter pairs available in EnhancerAtlas (Gao et al. 2016) for pancreas and HCT116 (a colonic carcinoma cell line) and investigated SNP pairs associated to T2D with a specific focus on epistasis, using Likelihood Ratio Tests of logistic regression models based on SNP genotypes, as implemented in PLATO (Hall et al. 2017). In the current study, we have built on this approach, extending and improving it in several ways. First of all, recognizing that our proposed filter is based on genomic interactions in cis, we conducted our tests based on 2-SNP haplotypes (using UNPHASED, Dudbridge 2008), as opposed to genotypes. Here by 2-SNP haplotype we mean the phased alleles for a SNP pair, where the SNPs in the pair are not necessarily in Linkage Disequilibrium (LD; in fact we only considered pairs with r2 < 0.6). Second, we extended our study to haplotype associations that include but are not limited to interactions, in other words we analyzed each haplotype both with a full and with an interaction model. Third, we generated enhancer-promoter pairs for tissues that are more specific to T2D, namely we considered four potential T2D relevant tissues (pancreatic islet, adipose tissue, small intestine, and liver) and we identified putative active enhancers and promoters for each tissue, based on epigenetic marks, using publicly available data sets. We then proceeded to create a putative superset of the interacting enhancer-promoter pairs for each tissue. To this end, to link each enhancer to more than just the closest promoter, while at the same time limiting the number of linked promoters, we made use of topologically associating domains (TADs). These are DNA regions within which physical interactions are believed to occur more frequently, as opposed to relatively infrequent interactions across TAD boundaries. TADs have been used to aid in the identification of candidate genes at GWAS signals (Way et al. 2017). We used these to link each putative active enhancer to all putative active promoters within the same TAD. Even if this was a potential superset of the actual interacting enhancer-promoter pairs, it was adequate to focus our search space to a manageable size. For each of the four tissues, we extracted SNP pairs consisting of one SNP in an active enhancer (at least in that given tissue) and one SNP in a linked active promoter and analyzed the 2-SNP haplotypes using three separate T2D GWAS.
It is typically difficult to replicate statistically significant interactions at the SNP pair level. For example, small differences in Minor Allele Frequencies (MAFs) across data sets can greatly affect the results (Greene et al. 2009). To address this, we utilized the three different GWAS data sets available to us to increase power by applying a standard meta-analysis method to combine p values and identify significant SNP pairs for each tissue based on evidence across multiple studies.
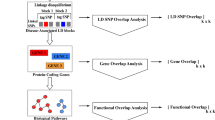
Figure 1 illustrates our workflow, which could be useful to investigate SNP pairs in GWAS in general. For any given trait, we do not expect the enhancer–promoter approach to capture all possible interesting SNP pairs; different biological filters could help explore different slices of the SNP pair space. Our aim was to take advantage of the increasing availability of tissue-specific epigenomics and ‘contactomics’ data sets to generate one biologically reasonable filter, which also has the added advantage of providing findings that could be more easily interpreted. Here by ‘contactomics’ we mean high-throughput experiments (such as HiC and capture C) aimed at detecting physical contacts between distal genomic regions, i.e., chromatin loops. Our results indicate that, whenever these types of functional genomics data are available for a tissue relevant to a GWAS trait, the ‘enhancer-promoter slice’ of the SNP pair space is worth exploring.

Analysis workflow. Epigenomics and contactomics data are used to identify putative interactive enhancer–promoter pairs in a tissue. 2-SNP haplotypes derived from these interacting regions are analyzed in multiple GWAS both in terms of a full model and in terms of an interaction model. Moreover, the individual SNPs in these pairs are also examined for main effects. For each analysis model, significant results are identified through meta-analyses, with appropriate multiple testing corrections. The results from the three models as well as information from the literature on established signals are combined to extract the most interesting SNP pairs for the full or the interaction models, e.g., SNP pairs non involving main effect SNPs. Notation: here, for each i = 1, 2, …, m, (Si1, Si2) denotes a pair of SNPs, the first in an enhancer and the second in a linked promoter
Results
In Online Resource 1 sheet 1, we report all results from our haplotype analyses (in either the full or the interaction model) having an adjusted combined p value across the three GWAS < 0.1. The p values for each of the three GWAS are indicated in Online Resource 1 sheet 1, together with the meta-analysis combined p values and the Benjamini–Hochberg (BH) adjusted combined p values. In the analyses using the full model, ‘omnibus’ association of the paired SNP haplotypes with T2D was investigated. In the analyses using the interaction model, association of the cis-phase interaction of the SNP pair with T2D was investigated. For each SNP pair we also provide the adjusted combined p values from the main effect analyses on each SNP. Moreover, SNPs associated with well-established T2D signals are marked by an asterisk. We are particularly interested in those SNP pairs in our results where neither SNP has a main effect either from our analyses or by being a proxy to an established T2D sentinel. Table 1 indicates, for each tissue, the number of significant pairs for each model and the number detected by both models; moreover, the number of full model pairs not involving main effects is reported (none of the interaction pairs involved main effects). Some pairs were present in more than one tissue SNP pair collection. The UNPHASED analyses over the 4 tissues yielded ~ 150 distinct significant 2-SNP haplotypes in the full model. A good portion of these comprised, as expected, main effect SNPs, several of which associated with well-known T2D sentinels. There were 35 distinct pairs consisting of SNPs that were not detected as main effects and were not well-known T2D signals. In addition, five distinct significant 2-SNP cis-phase interactions were also detected, none involving main effect SNPs. In Online Resource 1 sheet 2 we report, for each significant SNP pair with no main effects, the meta-analysis odd ratio results for each observed haplotype with respect to the indicated reference haplotype.
In what follows we focus on the significant SNP pairs with no main effects, summarized in Table 2. Some of these points to novel signals, whereas others are consistent with genes previously reported to be relevant for T2D or with GTEx results or with our own capture C data.
The only pair in pancreatic islet after main effect filtering was (enhancer SNP1 = rs7991210, promoter SNP2 = rs3742250), significant in both models. These two SNPs are in relatively high LD, having an r2 close to our threshold of 0.6 (see “Materials and methods”). The corresponding gene promoter is propionyl-CoA carboxylase alpha subunit (PCCA), whose activity was not found to be significantly different between pancreatic islets from T2D patients and controls (MacDonald et al. 2009). However, interestingly, this pair was also in the liver SNP pair collection and its enhancer SNP is in LD with rs7335993 (r2 = 0.83 from http://raggr.usc.edu/ across Europeans), which has an eQTL p value = 0.015 with PCCA in liver from GTEx.
Another pair, (rs691531, rs9433110), was present in both the small intestine and adipose tissue SNP pair collections, and was significant in the interaction analyses in small intestine as well as in the full and interaction analyses in adipose tissue. This pair is associated with the SEP15 promoter, which in turn overlaps with the HS2ST1 gene. The enhancer SNP in this pair is in LD with rs263436 (r2 = 0.89 from http://raggr.usc.edu/ across Europeans), which has an eQTL p value = 0.041 with SEP15 in adipose subcutaneous tissue from GTEx. In the small intestine, there was another pair identified in the interaction analyses involving the same promoter SNP with enhancer SNP rs691774. The latter SNP is in relatively high LD with the enhancer SNP of the previous pair (r2 = 0.86, thus marginally passed our 0.9 threshold on pairs) so they are likely to represent the same signal.
The pair (rs1563072, rs12444778) appears both in the small intestine and in the adipose tissue SNP pair data, and was significant in the full model analyses in both tissues. This pair was associated with the promoter of RP11-960L181. The enhancer SNP in this pair (which lies within an intron of GCSH) is in LD with rs1048194 (r2 = 0.89 from http://raggr.usc.edu/ across Europeans), which has an eQTL p value = 0.021 with RP11-960L18.1 in small intestine from GTEx. There was another pair involving the same promoter SNP, namely (rs12444137, rs12444778), identified by the full model both in the adipose tissue and in the liver collection. In the liver collection, there was an additional pair also involving the same promoter SNP and identified by the full model.
The pair (rs5000803, rs760294) was identified by the full model in small intestine and consists of a SNP in the promoter of ABHD16A and a SNP in a putative enhancer within an intron of HLA-DRB1.
There were two significant full model pairs in adipose tissues involving rs344954, which is both in the promoter of AC022498.1 and in a putative active enhancer in the same TAD. AC022498.1 is an uncharacterized gene which co-localizes with LPP, where a T2D signal was previously identified in American Indians (Nair et al. 2014).
The pair (rs2789686, rs74145425), identified in the adipose tissue analyses, is annotated to the promoter of ANXA11, which is one of three novel genes recently associated to T2D (Zhang et al. 2017). There are also two pairs, both involving the SNP rs2789686 and a SNP in the promoter of PLAC9 that were identified in these analyses. Besides residing in one of our putative adipose active enhancers, rs2789686 also resides within the ANXA11 region. This SNP is in LD with rs2789681 (r2 = 0.98 from http://raggr.usc.edu/ across Europeans), which has an eQTL p value = 0.023 (respectively, 0.025) with ANXA11 in adipose subcutaneous (respectively, visceral) tissue from GTEx.
Both SNP pairs (rs4631106, rs802920), associated with the REST promoter, and (rs12900028, rs8037641), associated with the ARID3B promoter, were in the adipose tissue and in the liver data and significant in the full model. This is particularly interesting for the latter pair as it is consistent with capture C data that we had generated for HEPG2 (a liver carcinoma cell line), where we found support for a cis physical interaction between the region chr15:74,832,872–74,834,488 containing rs8037641 and the region chr15:74,661,397–74,666,138 containing rs12900028 (coordinates refer to hg19), as illustrated in Online Resource 2.
The pair (rs12692585, rs3749119) in the adipose tissue collection, identified by the full model, is associated with the promoter of PLA2R1 and its enhancer SNP lies between ITGB6 and RBMS1 where T2D susceptibility loci have been previously identified (Qi et al. 2010).
There are five significant pairs from the adipose tissue collection with no main effects identified by the full model comprising rs344956. However, the main effect adjusted p value of the latter SNP was marginally significant, so these are less interesting.
The pair (rs10893517, rs112771035) from the liver data was identified by both the full and interaction models; rs112771035 maps to the promoter of ST3GAL4, a gene found to be associated with liver enzyme concentrations in plasma (Chambers et al. 2011), which are associated with increased risk of various diseases, including T2D.
Two pairs from the liver data identified by the full model involve rs12221064 in the promoter of CNNM2. This gene is among those associated with T2D (Lau et al. 2017). The enhancer SNP in one of the pairs (rs2297786) is in high LD with rs11191438 (r2 = 1 from http://raggr.usc.edu/ across Europeans), which has an eQTL p value = 0.02 with CNNM2 in liver from GTEx.
Three pairs in the liver data identified by the full model include rs927213, in the promoter of RP5-1103B4.3. This SNP is also within an intron of C8B and its three paired enhancer SNPs are all within an intron of this gene as well.
The pair (rs174358, rs62236167) identified from the liver data is associated with the promoter of CECR7 and its enhancer SNP is also proximal to the promoter of SLC25A18. The pair (rs3740392, rs4147157), also from the liver full model analyses, is associated with the WBP1L promoter and its enhancer SNP resides within an intron of AS3MT. Another pair identified in these analyses is (rs34535555, rs6440583) that is associated with the HLTF promoter and whose enhancer SNP resides within a CP intron.
Of the four full model pairs from the liver data involving the enhancer SNP rs7172774, three are associated to the SV2B promoter and one is associated to the SLCO3A1 promoter. The enhancer SNP rs7172774 is in LD with rs5814485 (r2 = 0.95 from http://raggr.usc.edu/ across Europeans), which has an eQTL p value = 0.03 with SV2B in liver from GTEx.
The pair (rs76024800, rs7577213) associated with the HECW2 promoter, was identified in the liver full model analyses. This is interesting as, similar to the pair associated with ARID3B described above, it is consistent with our HEPG2 capture C data, where we found support for a cis physical interaction between the region chr2:197,455,749–197,459,135, containing rs7577213, and the region chr2:197,486,443–197,488,380, containing three proxies (r2 > 0.8) for rs76024800: rs150785400, rs10497795, and rs10497794 (coordinates refer to hg19), as illustrated in Fig. 2.

HEPG2 capture C evidence for (rs76024800, rs7577213). The loop track indicates the HECW2 promoter bait region containing rs7577213 and the captured region containing the three proxy SNPs to rs7602480. The SNP track shows the location of the four SNPs. The location of HECW2 is indicated. Finally, the tracks with the capture C reads corresponding to the HECW2 bait in three replicate experiments are indicated. In the latter, we limited the y-axis range to [0–9] since, in capture C, peak height varies with distance from bait so including the full peaks near bait would have rendered the other peaks visually indiscernible
Online Resource 3 indicates the frequencies of the alternate alleles for each SNP involved in the pairs from Table 2 in each GWAS, together with absolute differences. The median absolute difference between the GENEVA and WTCCC frequencies (0.00722) was ~ fourfold smaller than that between GENEVA and FUSION (0.0318) and between WTCCC and FUSION (0.0334).
Discussion
We used a workflow that incorporates epigenomics data from trait-relevant tissues and TAD data to reduce the search space for 2-SNP haplotype analyses on GWAS data, reasoning that physically interacting regulatory regions may lead to plausible cooperating/interacting SNP pairs. Others have used epigenetic marks and physical chromosomal interactions to validate SNP pairs otherwise discovered (Hemani et al. 2014). In our approach, we instead used functional genomics data as our starting point to focus our searches on a manageable and reasonable SNP pair universe. This type of biological filter may miss some trait-relevant pairs, but has the advantage that pairs so identified are more easily interpretable. We have also exploited the availability of independent GWAS for a given trait (in our case T2D) to extract results which combine evidence from multiple studies.
Our work builds on a previous pilot study (Manduchi et al. 2018) improving its workflow in several ways. Recognizing that our biological filter is based on genomic interactions in cis, we switched from genotype-based analyses to 2-SNP haplotype-based analyses. We also extended our scope from interaction only to interaction or full (‘omnibus’) model. To borrow strength from all available GWAS, we used meta-analyses, fully imputing all three GWAS. We also refined the pre-processing of the WTCCC data set as it pertains to related-individual filtering, introducing a graph theoretical approach (see “Materials and methods”). Finally, we used tissues more closely relevant to T2D, utilizing ENCODE (ENCODE Project Consortium 2012) and RoadMap (Roadmap Epigenomics Consortium et al. 2015) histone modification, promoter, and open chromatin data, in addition to EnhancerAtlas (Gao et al. 2016) enhancer data, to define tissue-specific putative active enhancers and promoters. Additional data sets of this sort, e.g., FANTOM 5 (Andersson et al. 2014; FANTOM Consortium et al. 2014) could be utilized in this workflow. A more general direction for future work is to investigate the use of 2-SNP diplotype-based analyses in our workflow, i.e., association analyses using 2-SNP haplotype pairs on homologous chromosomes.
We used TADs to link all enhancers and promoters within the same TAD. We, therefore, expect that most of the actual interacting enhancer–promoter regions would be a subset of the paired regions we analyzed, but our filtering sufficed to reduce the search space to a manageable size. TADs are believed to be relatively tissue-invariant (Dixon et al. 2012; Nora et al. 2012), thus we used available TADs from human Embryonic Stem Cells (hESC). With the advent of high-resolution capture HiC data (Mifsud et al. 2015; Javierre et al. 2016), when available, more precise tissue-specific contactomics data sets could be employed in our workflow in lieu of TADs, enabling us to narrow down the potentially interacting enhancer–promoter pairs to a collection that coincides with the actual ones for that tissue.
Our findings include both 2-SNP haplotypes associated with the disease according to the full model and 2-SNP haplotypes whose interaction is associated with the disease. After filtering out those findings involving at least one SNP with a main effect, the remaining results are expected to include pairs of SNPs whose mechanism of association with T2D could either be epistasis (when an interaction is detected) or an additive contribution of effects too small to reach genome-wide significance. In terms of effect size, UNPHASED selects one of the haplotypes as reference and outputs the odds ratio for each of the other observed haplotypes. After computing meta-analyses odd ratios (OR) across the three studies, the median undirectional OR (undirOR defined as OR when OR ≥ 1 and 1/OR when OR < 1) was 1.14 [0.99–1.27], which is comparable to effect sizes for known T2D sentinel SNPs in Europeans (DIAGRAM Consortium et al. 2014).
Our approach shows promise, in that it is able to uncover both novel and established candidate genes, such as recently identified T2D relevant loci, or loci with the support of GTEx or our own capture C data. The capture C evidence in effect supports the physical interaction between the two SNP-containing regions in a cell line close to a relevant tissue, which is important in our case especially because we started with a superset of potential enhancer-promoter regions. However, as described above, with future direct availability of capture C data for relevant tissues, these would be used in the initial workflow step to define the SNP pairs to investigate. The GTEx evidence instead supports the specific association between the enhancer SNP and the expression of the gene relative to the associated SNP’s promoter. This is relevant, although it does not support the interaction of two SNPs directly. In the future, investigation of the genetic interaction effect on the expression of the gene may provide such direct evidence.
Most of our findings were derived from our liver, adipose and small intestine enhancer-promoter collections. This may indicate that the 2-haplotype mechanisms uncovered by this type of workflow are primarily pertaining to insulin resistance, or it may be due to the fact that pancreatic islets consist of a mixed cell population, only a fraction of which (the beta cells) are most relevant to T2D. Future availability of high-quality beta cell derived epigenomics data should yield more specific T2D relevant regulatory regions potentially leading to a greater number of significant SNP pairs in the pancreatic context. Of course, statistical epistasis is different from biological epistasis (Moore and Williams 2005, 2009; Phillips 2008), and more generally, statistical associations are different from biological associations. Thus, establishing whether our findings lead to precise biological mechanisms of variant pairs which are associated to T2D, warrants further investigation. We also note that the SNP pair collections we derived were obtained after removing LD redundancies, so our SNP pair results must be assessed with not the specific SNPs in mind but rather the genomic regions in which they are harbored.
Overall, this approach represents an opportunity to implicate additional loci contributing to the pathogenesis of complex traits. After all, much of the missing heritability has still to be resolved for common diseases (Manolio et al. 2009), including T2D, so it is clear that many more loci, or combinations thereof, remain to be characterized. The additional advantage of our approach is that the constraint of one SNP being located in a promoter leads to less ambiguity about what the effector gene could be; although one can still not be certain given other complex mechanisms that could still be at play. By reducing the degree of multiple testing by limiting testing on known genomic features, we have been successful in identifying a number of loci that yield a degree of replication support, most obviously AC022498.1, REST, ARID3B, ST3GAL4, RP5-1103B4.3, CECR7, WBP1L and SLCO3A1. Interestingly, none of these genes have strong literature support for a role in T2D, so are revealing new biology underpinning this common disease; in addition, the fact that some of these genes are non-coding in nature highlights the need to understand their mechanism of action further in the context of complex traits.
In conclusion, we have utilized available T2D GWAS and functional genomics data to propose a workflow for 2-SNP haplotype investigation which appears promising and is extensible to other traits when, in addition to GWAS data sets, epigenomics and contactomics data sets are also available for tissues that are relevant to the traits.
Materials and methods
Definition of putative active enhancer and promoter pairs
The tissue-specific data we used to define putative active enhancers and promoters are listed in Online Resource 4 and were obtained from ENCODE (http://www.encodeproject.org; ENCODE Project Consortium 2012) and EnhancerAtlas (Gao et al. 2016). We used hg19 coordinates in all cases, applying LiftOver (Hinrichs et al. 2006) when needed. Region operations were performed with BEDOPS v2.4.25 (Neph et al. 2012).
Putative active enhancers For each of the four tissues, the results from steps (1)–(4) are what we refer to as ‘putative active enhancers’:
-
1.
Intersect broad peaks from H3K4me1 and H3K27ac.
-
2.
Flank open chromatin regions by 150 bp on each side, merging any overlapping regions which result from this extension. Open chromatin regions were obtained by merging available DNase-Seq or ATAC-Seq peaks and available combined open chromatin files.
-
3.
Take all regions from (2) which overlap with a region from (1).
-
4.
Merge results from (3) with the available annotated enhancer regions for the tissue from EnhancerAtlas or ENCODE, if any (see Online Resource 4).
Putative active promoters For each of the four tissues, the results from steps (1)–(6) are what we refer to as ‘putative active promoters’:
-
1.
Take H3K4me3 broad peaks.
-
2.
Flank open chromatin regions by 150 bp on each side, merging any overlapping regions which result from this extension. Open chromatin regions were obtained by merging available DNase-Seq or ATAC-Seq peaks and available combined open chromatin files.
-
3.
Take all regions from (2) which overlap with a region from (1).
-
4.
Merge results from (3) with the available annotated promoter regions for the tissue from ENCODE, if any (see Online Resource 4).
-
5.
Take all regions from 1000 bp upstream to 500 bp downstream of GeneCode v19 Transcription Start Sites (TSS), downloaded from the UCSC Genome Browser (Hinrichs et al. 2006).
-
6.
Take any region from (5) which intersects any of the regions in (4).
Pairing We utilized TADs to narrow down enhancer–promoter pairs which may interact. TADs are believed to be relatively tissue-invariant (Dixon et al. 2012; Nora et al. 2012). We used the hESC TADs, derived from HiC data, downloaded from http://promoter.bx.psu.edu/hi-c/download.html (Dixon et al. 2012) to link our putative tissue-specific enhancers to promoters, by pairing each putative active enhancer with every putative active promoter in the same TAD. For each tissue, SNPs harbored in each putative active enhancer or promoter were identified with Biofilter v2.4.0 (https://ritchielab.psu.edu/software/biofilter-download-1; Bush et al. 2009; Pendergrass et al. 2013). All SNP pairs with one SNP in an enhancer and one SNP in a linked promoter were formed. These pairs were then used in the post-imputation quality control (QC) described below.
GWAS data set processing
The three GWAS data sets used for this work are listed below and are available, upon application, at the indicated websites.
-
The GENEVA Genes and Environment Initiatives in T2D, available from the database of Genotypes and Phenotypes (dbGaP; Tryka et al. 2014) under phs000091.v2.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000091.v2.p1).
-
The Finland–United States Investigation of NIDDM Genetics (FUSION) Study, available from dbGaP under phs000867.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000867.v1.p1).
-
Welcome Trust Case Control Consortium (WTCCC; Welcome Trust Case Control Consoritum 2007; https://www.wtccc.org.uk) T2D and Control data sets EGAD00000000001, EGAD00000000002, and EGAD00000000009.
Imputation We used PLINK v1.9 (https://www.cog-genomics.org/plink2/), bcftools (http://www.sanger.ac.uk/science/tools/samtools-bcftools-htslib) and vcftools (Delaneau et al. 2012) to manipulate and filter our data sets. For all data sets, our starting points were the plink files made available from the respective source. In the case of GENEVA, we started from the dbGaP ‘zero-out’ plink files (where a set of specific SNPs in specific samples had been set to 0, because a chromosome anomaly or quality problem was detected) and merged the NHS and HPFS data after removal of the duplicate markers and duplicated/related individuals annotated in the provided sample files. For WTCCC, we merged the data sets from the three accessions cited above. In all cases, we mapped coordinates to hg19, and then applied the following filters in the listed order:
-
1.
Individuals failing the PLINK ‘--check-sex’ were removed.
-
2.
Markers with missing-call rate exceeding 0.05 were removed.
-
3.
Markers with Minor Allele Frequency (MAF) below 0.01 were removed.
-
4.
Markers with Hardy–Weinberg equilibrium exact test p value below 0.00001 were removed.
This filtering lead to the following counts:
GENEVA: 724,194 markers for 5866 individuals (2738 cases and 3128 controls; 2498 males and 3368 females).
FUSION: 314,276 markers for 1699 individuals (916 cases and 783 controls; 942 males and 757 females).
WTCCC: 422,927 markers for 4966 individuals (1973 cases and 2993 controls; 2608 males and 2358 females).
These data were recoded to vcf format and prepared for imputation according to the Sanger Imputation Server (McCarthy et al. 2016) instructions. The Sanger Imputation Server EAGLE2 + PBWT pipeline (Loh et al. 2016; Durbin 2014) was used to impute the data with UK10K + 1000 Genomes Phase 3 (Huang et al. 2015; 1000; Genomes Project Consortium 2015) as reference panel.
Principal components (PCs) PCs were computed using genotyped data only. For each data set, we applied a second (more stringent) round of filtering to the data used as inputs to the Sanger Imputation Server in the following order:
-
1.
Individuals failing the PLINK ‘--check-sex’ were removed.
-
2.
Markers with missing-call rate exceeding 0.01 were removed.
-
3.
Individuals with missing-call rate exceeding 0.01 were removed.
-
4.
Markers with MAF below 0.05 were removed.
-
5.
Markers with Hardy–Weinberg equilibrium exact test p value below 0.00001 were removed.
-
6.
Individuals were filtered based on relatedness as detailed below.
-
7.
Steps 2–5 were then repeated.
The first ten PCs were then obtained using the PLINK -pca command after Linkage Disequilibrium (LD) pruning (-indep-pairwise 50 5 0.2).
Individual filtering based on relatedness A quick and commonly used method to filter individuals based on relatedness in a given GWAS data set is described in Anderson et al. (2010). First, identity by state (IBS) is calculated (with PLINK) for each pair of individuals based on the average proportion of alleles shared in common at independent genotyped markers, where markers are pruned so that no pair within a given window (window size = 50) is correlated (typically taken as r2 > r0 for a specified r0). Then, a custom script runs through the pairs as output by PLINK and one individual is removed from each pair with an IBS greater than a specified threshold I0, where the individual removed is the one with the greater number of missing data. We applied this approach, i.e., steps 11–13 of Anderson et al. (2010), with I0 = 0.125 and the r2 threshold for LD-pruning selected to get a number of markers close to 100,000 (0.2 for GENEVA, 0.3 for FUSION and WTCCC). This removed 29 individuals for GENEVA and 7 for FUSION. However, this approach dropped a substantial number of individuals for WTCCC; upon closer examination we observed that this was due to the presence of a small set of individuals with high IBS with the majority of the other subjects in the WTCCC data set (possibly an artifact). In practice, the approach in Anderson et al. (2010) works well in many situations but in cases like the WTCCC it does not remove a reasonably small number of individuals. If we consider the problem at hand in a graph-theoretic manner, where the individuals are nodes and there is an edge between two nodes if the corresponding individuals have IBS > I0, then the goal is to remove the smallest number of nodes so that the resulting graph has no edges. Thus, the problem is reduced to the known ‘minimum vertex problem’ in graph theory. This is an NP-hard problem which, however, has an approximation algorithm. The approximation algorithm never finds a vertex cover whose size is more than twice the size of a minimum possible vertex cover. When we applied the approximation algorithm directly to our WTCCC with a custom script exploiting the python Networkx library (Hagberg et al. 2008), this suggested the removal of 148 individuals. Aware that this may not be optimal, we proceeded in a two-step way to reduce the number of removed individuals. First, we used the approach (Anderson et al. 2010) with I0 set to a higher value (0.3), which removed 64 individuals. Then we applied the minimum vertex cover approximation approach to the remaining individuals, which removed 36 additional individuals. With this two-step approach, we therefore removed 100 individuals overall in the WTCCC to achieve unrelatedness.
Post-imputation quality control (QC) For each tissue and for each GWAS data set, we considered the imputed data for the individuals remaining after the filtering used for PCs and all bi-allelic SNPs from the pairs obtained for that tissue passing all imputation filters and with an info score > 0.7. We then applied filtering in the following order:
-
1.
Markers with missing-call rate exceeding 0.01 were removed.
-
2.
Individuals with missing-call rate exceeding 0.01 were removed. For GENEVA, ancestry annotation was provided and we retained only those individuals annotated as European ancestry.
-
3.
Markers with MAF below 0.05 were removed.
-
4.
Markers with Hardy–Weinberg equilibrium exact test p value below 0.00001 were removed.
-
5.
Markers with different genotype call rates between cases and controls according to steps 24–25 of Anderson et al. (2010) were removed (actually no markers after 1–4 failed this filter).
Among the SNP pairs corresponding to each tissue we retained those where both SNPs were present in all three post-imputation QC-ed GWAS data sets. These pairs were LD-pruned using PLINK and based on the post-imputation QC-ed GENEVA data set as follows. Any pair with r2 ≥ 0.6 was removed. Moreover, pairs were sequentially removed to ensure that the final collection did not contain any two pairs (eSNP1, pSNP1), (eSNP2, pSNP2) with the r2 between eSNP1 and eSNP2 ≥ 0.9 and the r2 between pSNP1 and pSNP2 ≥ 0.9. The final number of SNP pairs to analyze for each tissue and the numbers of individuals and SNPs involved in these analyses for each of the three GWAS data sets are reported in Table 3.
2-SNP haplotype analyses
For each tissue we analyzed the corresponding SNP pair collection using UNPHASED (Dudbridge 2008) on the 2-SNP haplotypes, both with the ‘full’ model option and with the ‘gxg’ model option. In the full model, there is an odds ratio parameter for each observed haplotype. One of the haplotypes (the first one when haplotypes are sorted) is chosen as reference (with an odds ratio of 1) and the odds ratios for the other haplotypes are computed against this. The gxg test compares the haplotype main effects model to the full model. The haplotype main effects model (‘haplomain’) uses allele coding (where haplotype risks are represented in terms of main effects of each locus and interactions between them) and defines a full model for one marker, and another full model for the other marker, with no interaction terms between the two markers. More details are available in the user manual (https://sites.google.com/site/fdudbridge/software/unphased-3-1).
Upon testing gender and age for association with phenotype in each GWAS (for age, we had detailed information only for GENEVA and FUSION), we determined to only adjust for gender in the WTCCC and FUSION analyses. After assessing inflation of the UNPHASED p values on each data set, we determined that the best option was to not adjust for any PC. For each of the two analysis models, we combined the p values with the Stouffer’s method across the three GWAS using the sumz function in the R package ‘metap’ (https://cran.r-project.org/web/packages/metap/) with weights equal to square root of the number of individuals in each GWAS. We then adjusted the combined p values by applying the BH multiple testing correction (Benjamini and Hochberg 1995), implemented in R. We used a threshold of 0.1 on our adjusted combined p values.
For each GWAS and for each SNP pair with no main effects we have extracted all UNPHASED haplotype odds ratios and 95% confidence intervals and combined these across the three studies using GWAMA v2.2.2 (Mägi and Morris 2010).
Main effects
We used PLINK v1.9 to perform logistic regression analyses for main effects, with the same covariate adjustments as in UNPHASED. We combined p values across data sets and adjusted them as done for the SNP pair analyses, using the same significance threshold. To mark SNPs for well-known T2D signals we started with a manually curated list of about 60 sentinel loci and compiled the list of all SNPs having r2 ≥ 0.4 with at least one of these loci using http://raggr.usc.edu/ and the European population from 1000 Genomes phase 3. The SNP list resulting from the latter is in Online Resource 5.
Enhancer annotation
In Online Resource 1 sheet 1, we annotated enhancers by the presence of predicted transcription factor (TF) binding motifs within their region. To obtain the latter, we scanned the enhancer sequences with the FIMO (v4.11.3) software from the MEME suite (Bailey et al. 2009; Grant et al. 2011) using the (519) JASPAR CORE 2016 vertebrate motifs (Mathelier et al. 2016), with a q value threshold of 0.05 for hits.
GTEx queries
We used the GTEx v7 (https://www.gtexportal.org) query ‘Test your own eQTL’ for each specified SNP, gene and tissue and reported the resulting eQTL p value, without additional multiple testing corrections, as we were merely using this in a confirmatory fashion for pre-specified hypotheses obtained from our analyses.
Capture C data
Triplicate libraries were generated from HEPG2 cells using methods described in Xia et al. (2016). We used Agilent SureSelect oligonucleotide probes to capture hybrid reads involving regions of interest (baits). The baits relevant to this work are two DpnII fragments, respectively, in the promoters of ARID3B and HECW2. The locations of these baits and of the oligonucleotides selected within each are provided in Online Resource 6.
Data availability
The accessions for the publicly available ENCODE (http://www.encodeproject.org) data sets used in this work are listed in Online Resource 4. The EnhancerAtlas data are publicly available at http://www.enhanceratlas.org/.
The three GWAS data sets used for this work are available, upon application, at the websites listed in “Materials and methods”. One of these data sets consists of data generated by the Wellcome Trust Case-Control Consortium (WTCCC). A full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk. Funding for the WTCCC project was provided by the Wellcome Trust under awards 076113, 085475 and 090355. The Consortium and/or Individual Investigators bear no responsibility for the further analysis or interpretation of these data, over and above that published by the Consortium.
All remaining relevant data are in the Online Resources. In addition, bam files for the aligned valid hybrid read pairs corresponding to Online Resource 2 and Fig. 2 are available from the corresponding authors on reasonable request.
References
Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT (2010) Data quality control in genetic case-control association studies. Nat Protoc 5(9):1564–1573
Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M et al (2014) An atlas of active enhancers across human cell types and tissues. Nature 507(7493):455–461
Bailey TL, Bodén M, Buske FA, Frith M, Grant CE, Clementi L et al (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodological) 57(1):289–300
Boyle EA, Li YI, Pritchard JK (2017) An expanded view of complex traits: from polygenic to omnigenic. Cell 169(7):1177–1186
Brem RB, Storey JD, Whittle J, Kruglyak L (2005) Genetic interactions between polymorphisms that affect gene expression in yeast. Nature 436:701–703
Bush WS, Dudek SM, Ritchie MD (2009) Biofilter: a knowledge-integration system for the multi-locus analysis of genome-wide association studies. Pac Symp Biocomput 2009:368–379
Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P et al (2011) Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet 43(11):1131–1138
Cowper-Sal Iari R, Cole MD, Karagas MR, Lupien M, Moore JH (2011) Layers of epistasis: genome-wide regulatory networks and network approaches to genome-wide association studies. Wiley Interdiscip Rev Syst Biol Med 3(5):513–526
Delaneau O, Marchini J, Zagury JF (2012) A linear complexity phasing method for thousands of genomes. Nat Methods 9(2):179–181
DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium, Mahajan A (2014) Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 46(3):234–244
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y et al (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380
Dudbridge F (2008) Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered 66:87–98
Durbin R (2014) Efficient haplotype matching and storage using the positional Burrows–Wheeler transform (PBWT). Bioinformatics 30(9):1266–1272
Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH et al (2010) Missing heritability and strategies for finding the underlying causes of complex diseases. Nat Rev Genet 11:446–450
ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57–74
FANTOM Consortium and the RIKEN PMI and CLST (DGT), Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ et al (2014) A promoter-level mammalian expression atlas. Nature 507(7493):462–470
Gao T, He B, Liu S, Zhu H, Tan K, Qian J (2016) EnhancerAtlas: a resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics 32(23):3543–3551
Genomes Project Consortium (2015) A global reference for human genetic variation. Nature 526(7571):68–74
Grant CE, Bailey TL, Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27(7):1017–1018
Greene CS, Penrod NM, Williams SM, Moore JH (2009) Failure to replicate a genetic association may provide important clues about genetic architecture. PLoS One 4(6):e5639. https://doi.org/10.1371/journal.pone.0005639
Hagberg AA, Schult DA, Swart PJ (2008) Exploring network structure, dynamics, and function using NetworkX. In: Proceedings of the 7th Python in science conference (SciPy2008), pp 11–15
Hall MA, Wallace J, Lucas A, Kim D, Basile AO, Verma SS et al (2017) PLATO software provides analytic framework for investigating complexity beyond genome-wide association studies. Nat Commun 8(1):1167. https://doi.org/10.1038/s41467-017-00802-2
Hemani G, Shakhbazov K, Westra HJ, Esko T, Henders AK, McRae AF et al (2014) Detection and replication of epistasis influencing transcription in humans. Nature 508(7495):249–253
Hill WG, Goddard ME, Visscher PM (2008) Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet 4(2):e1000008. https://doi.org/10.1371/journal.pgen.1000008
Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H et al (2006) The UCSC genome browser database: update. Nucleic Acids Res 34(Database issue):D590-D598
Huang J, Howie B, McCarthy S, Memari Y, Walter K, Min JL et al (2015) Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nature Commun 6:8111
Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S et al (2016) Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167(5):1369–1384
Lau W, Andrew T, Maniatis N (2017) High-resolution genetic maps identify multiple type 2 diabetes loci at regulatory hotspots in African Americans and Europeans. Am J Hum Genet 100:803–816
Loh P-R, Danecek P, Palamara PF, Fuchsberger C, Reshef YA, Finucane HK et al (2016) Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 48(11):1443–1448
MacDonald MJ, Longacre MJ, Langberg E-C, Tibell A, Kendrick MA, Fukao T et al (2009) Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia 52(6):1087–1091
Mackay TFC (2014) Epistasis and quantitative traits: using model organisms to study gene-gene interactions. Nat Rev Genet 15(1):22–23
Mägi R, Morris AP (2010) GWAMA: software for genome-wide association meta-analysis. BMC Bioinform 11:288
Manduchi E, Chesi A, Hall MA, Grant SFA, Moore JH (2018) Leveraging putative enhancer-promoter interactions to investigate two-way epistasis in Type 2 Diabetes GWAS. Pac Symp Biocomput 2018:548–558
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ et al (2009) Finding the missing heritability of complex diseases. Nature 461(7265):747–753
Mathelier A, Fornes O, Arenillas DJ, Chen C, Denay G, Lee J et al (2016) JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44:D110–D115
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A et al (2016) A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48(10):1279–1283
Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L et al (2015) Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet 47:598–606
Mitra I, Lavillareuix A, Yeh E, Traglia M, Tsang K, Bearden CE et al (2017) Reverse pathway genetic approach identifies epistasis in autism spectrum disorders. PLoS Genet 13(1):e1006516. https://doi.org/10.1371/journal.pgen.1006516
Moore JH (2015) Epistasis using ReliefF. Methods Mol Biol 1253:315–325
Moore JH, White BC (2007) Tuning ReliefF for genome-wide genetic analysis. In: Marchiori E, Moore JH, Rajapakse JC (eds) Evolutionary computation, machine learning and data mining in bioinformatics. Springer, Berlin, pp 166–175
Moore JH, Williams SM (2005) Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. Bioessays 27(6):637–646
Moore JH, Williams SM (2009) Epistasis and its implications for personal genetics. Am J Hum Genet 85:309–320
Nair AK, Muller YL, McLean NA, Abdussamad M, Piaggi P, Kobes S et al (2014) Variants associated with type 2 diabetes identified by the transethnic meta-analysis study: assessment in American Indians and evidence for a new signal in LPP. Diabetologia 57(11):2334–2338
Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK et al (2012) BEDOPS: high-performance genomic feature operations. Bioinformatics 28(14):1919–1920
Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N et al (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485:381–385
Pendergrass SA, Frase A, Wallace J, Wolfe D, Katiyar N, Moore C et al (2013) Genomic analyses with biofilter 2.0: knowledge driven filtering, annotation, and model development. BioData Min 6(1):25. https://doi.org/10.1186/1756-0381-6-25
Phillips PC (2008) Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 9(11):855–867
Qi L, van Dam RM, Asselbergs FW, Hu FB (2007) Gene-gene interactions between HNF4A and KCNJ11 in predicting Type 2 diabetes in women. Diabet Med 24:1187–1191
Qi L, Cornelis MC, Kraft P, Stanya KJ, Linda Kao WH, Pankow JS et al (2010) Genetic variants at 2q24 are associated with susceptibility to type 2 diabetes. Hum Mol Genet 19(13):2706–2715
Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF et al (2001) Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet 69:138–147
Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A et al (2015) Integrative analysis of 111 reference human epigenomes. Nature 518(7539):317–330
Robnik-Šikonja M, Kononenko I (2003) Theoretical and empirical analysis of ReliefF and RReliefF. Mach Learn 53:23–69
Tryka KA, Hao L, Sturcke A, Jin Y, Wang ZY, Ziyabari L et al (2014) NCBI’s database of genotypes and phenotypes: dbGaP. Nucleic Acids Res 42(Database issue):D975–D979
Urbanowicz RJ, Granizo-Mackenzie ALS, Kiralis J, Moore JH (2014) A classification and characterization of two-locus, pure, strict, epistatic models for simulation and detection. BioData Min 7:8
Verma SS, Cooke Bailey JN, Lucas A, Bradford Y, Linnemann JG, Hauser MA et al (2016) Epistatic gene-based interaction analyses for glaucoma in eMERGE and NEIGHBOR Consortium. PLoS Genet 12(9):e1006186. https://doi.org/10.1371/journal.pgen.1006186
Way GP, Youngstrom DW, Hankenson KD, Greene CS, Grant SFA (2017) Implicating candidate genes at GWAS signals by leveraging topologically associating domains. Eur J Hum Genet 25(11):1286–1289
Welcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447(7145):661–678
Xia Q, Chesi A, Manduchi E, Johnston BT, Lu S, Leonard ME et al (2016) The type 2 diabetes presumed causal variant within TCF7L2 resides in an element that controls the expression of ACSL5. Diabetologia 59(11):2360–2368
Zhang Q, Wu KH, He JY, Zeng Y, Greenbaum J, Xia X et al (2017) Novel common variants associated with obesity and type 2 diabetes detected using a cFDR method. Sci Rep 7(1):16397. https://doi.org/10.1038/s41598-017-16722-6
Zuk O, Hechter E, Sunyaev SR, Lander ES (2012) The mystery of missing heritability: genetic interactions create phantom heritability. PNAS USA 109:1193–1198
Acknowledgements
The authors thank B. Cole, M. Hall, and D. Cousminer for useful conversations, and Kenyaita Hodge and Michelle Leonard for the HEPG2 capture C library preparation. We also thank the reviewers for their constructive feedback. Funding for this work was provided by National Institutes of Health Grants LM010098, DK112217, ES013508, R21 HD089824, and the Children’s Hospital of Philadelphia Center for Spatial and Functional Genomics and Daniel B. Burke Endowed Chair for Diabetes Research.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2018_1893_MOESM1_ESM.xlsx
Online Resource 1, sheet1. Significant UNPHASED results with a BH adjusted combined p value < 0.1 in either the full or the interaction model are reported for all tissues. In each row, SNP1 is harbored in a predicted enhancer and SNP2 in a predicted promoter (some SNPs are in multiple promoters). These are also reported together with the list of FIMO predicted motifs with hits to the enhancer. We report the main effect adjusted combined p value for each SNP, the UNPHASED p values for the SNP pair in each study, and the combined p values (unadjusted and BH adjusted). In addition, the r2 and absolute distance (in bp) between the two SNPs are reported. Online Resource 1, sheet2. GWAMA results for the significant SNP pairs with no main effect SNPs, reporting meta-analysis odds ratio results (including 95% confidence interval bounds) for each observed haplotype with respect to the indicated reference haplotype. The Cochran’s heterogeneity statistic (q) and Higgins heterogeneity index (i2) are also indicated, together with the directions of effect in the 3 studies. The ‘undirOR’ in the last 3 columns is defined as OR when OR ≥ 1 and 1/OR when OR < 1. This is provided for quick OR comparisons regardless of direction. SNP pairs where GWAMA failed and outliers are not shown (XLSX 56 KB)
439_2018_1893_MOESM2_ESM.pdf
HEPG2 capture C evidence for (rs12900028, rs8037641). The loop track indicates the ARID3B promoter bait region containing rs8037641 and the captured region containing rs12900028. The SNP track shows the location of the 2 SNPs. The location of ARID3B is indicated. Finally the tracks with the capture C reads corresponding to the ARID3B bait in 3 replicate experiments are indicated. In the latter we limited the y-axis range to [0-9] since, in capture C, peak height varies with distance from bait so including the full peaks near bait would have rendered the other peaks visually indiscernible (PDF 120 KB)
439_2018_1893_MOESM3_ESM.xlsx
Frequencies of the alternate allele (A1) for the SNPs from Table 2 in the 3 GWAS studies. The absolute differences between these frequencies are also indicated (XLSX 17 KB)
439_2018_1893_MOESM6_ESM.xlsx
Promoter DpnII bait fragments for ARID3B and HECW2 together with coordinates of subregions used for SureSelect oligonucleotides to bait them. All coordinates refer to hg19 (XLSX 10 KB)
Rights and permissions
About this article

Cite this article
Manduchi, E., Williams, S.M., Chesi, A. et al. Leveraging epigenomics and contactomics data to investigate SNP pairs in GWAS. Hum Genet 137, 413–425 (2018). https://doi.org/10.1007/s00439-018-1893-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-018-1893-0